Introduction
Gastroenteropancreatic neuroendocrine tumors
(GEP-NETs) are extremely rare tumors (<1% of stomach tumors)
that usually originate from the neuroendocrine tissues of the
digestive system. Driver mutations in MEN1, DAXX or ATRX and mTOR
pathway genes have been identified as crucial factors in GEP-NETs
tumorigenesis (1). The first case of
GEP-NETs was reported approximately one century ago. Recently, the
incidence of these tumors has greatly increased (2,3). According
to the Epidemiology and End Results (SEER) program of the National
Cancer Institute, the annual incidence rate of GEP-NETs is 3.65
cases per 100,000 individuals (4).
Gastrointestinal stromal tumors (GISTs) are the most common type of
mesenchymal tumor of the gastrointestinal system, and are derived
from the interstitial Cajal cells of the gastrointestinal tract.
GISTs are generally characterized by gain-of-function mutations in
the KIT gene (5), and less often by
PDGFRA or BRAF gene mutations (6–8). The
annual incidence of GISTs ranges between 6.8 and 19.7 individuals
per million across numerous countries, including the United States
(9–11). GEP-NETs and GISTs are malignant or
potentially malignant tumors, and are considered to have their own
specific molecular biological behavior. As the majority of patients
exhibit nonspecific symptoms, the identification of GEP-NETs and
GISTs is often incidental. At present, diagnostic techniques
include endoscopy, computed tomography (CT) and endoscopic
ultrasonography (EUS) (12) and
surgical resection remains the standard treatment for GEP-NETs and
GISTs. The present study reports a compound case of GEP-NETs and
GISTs that simultaneously occurred in the stomach.
Case report
A 56-year-old female was admitted to the Subei
People's Hospital of Jiangsu (Yangzhou, Jiangsu, China) due to
epigastric discomfort that had persisted for 4 weeks. Pre-operative
blood examinations, including tumor marker analysis, were normal.
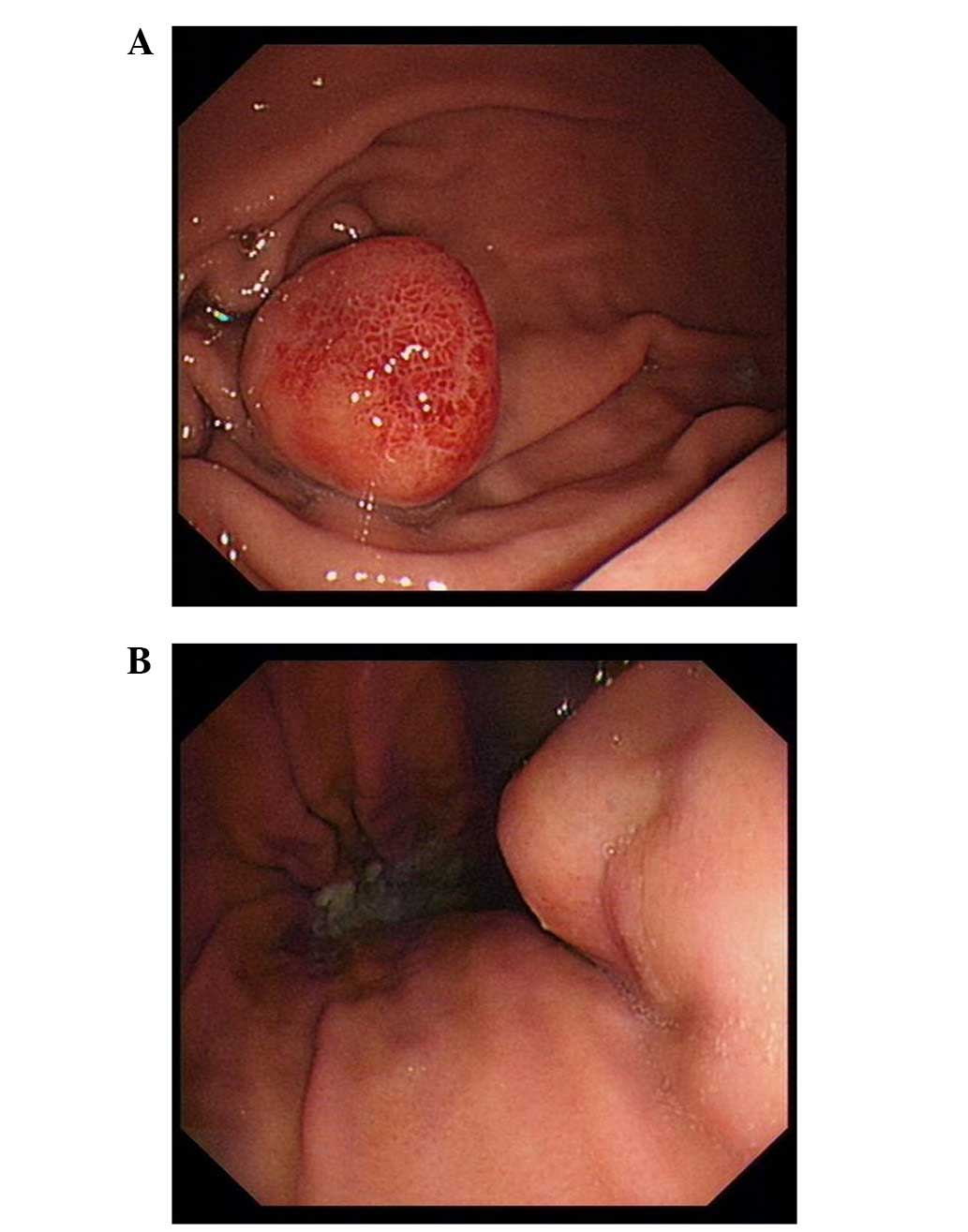
Gastrointestinal endoscopy identified two fusiform masses of
1.8×1.8 cm and 1.6×1.6 cm located at the gastric body and gastric
fundus, respectively (Fig. 1A and B).
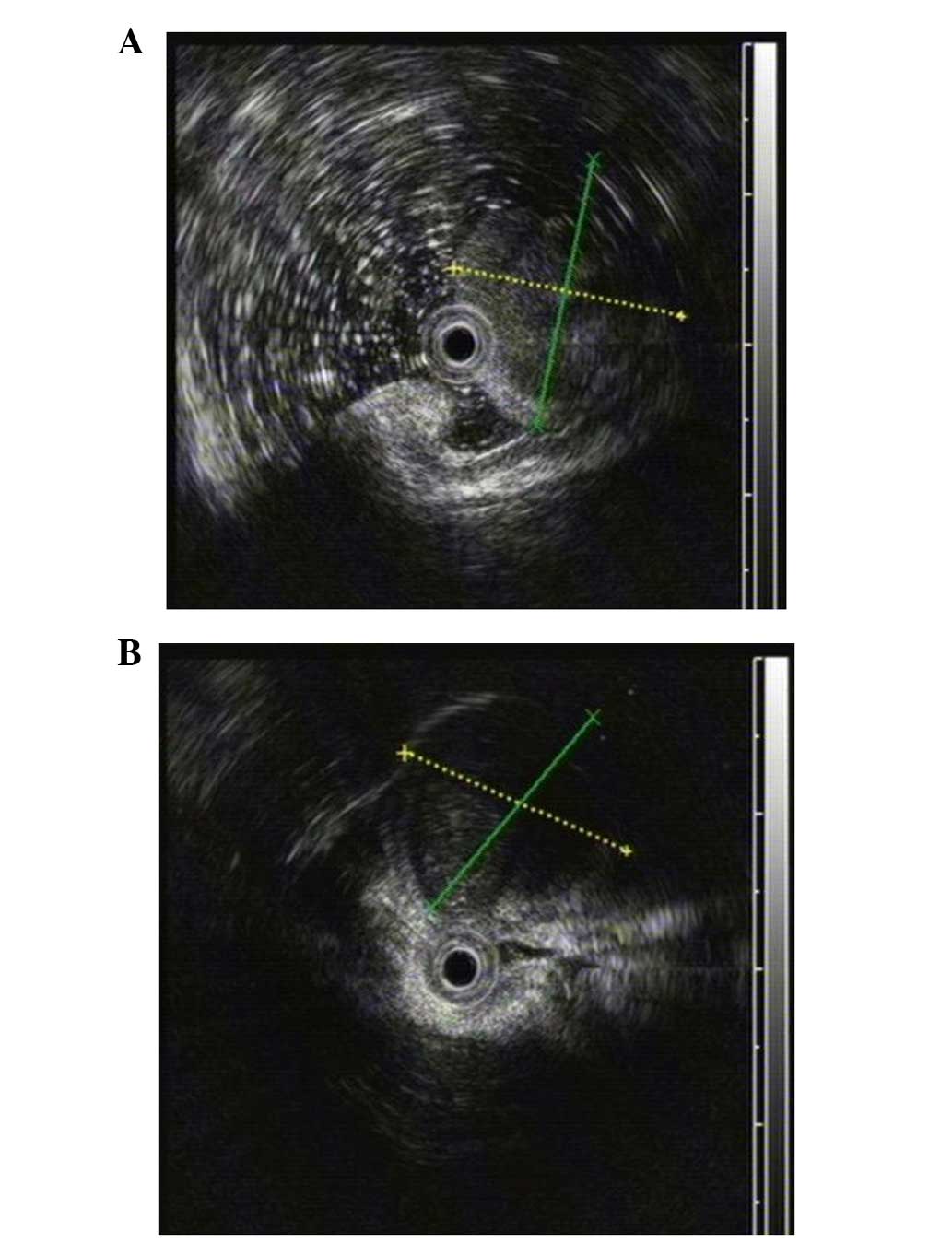
Meanwhile, EUS found that the two masses with low homogenous
echogenicity each originated from the gastric muscular layer
(Fig. 2A and B). Additionally,
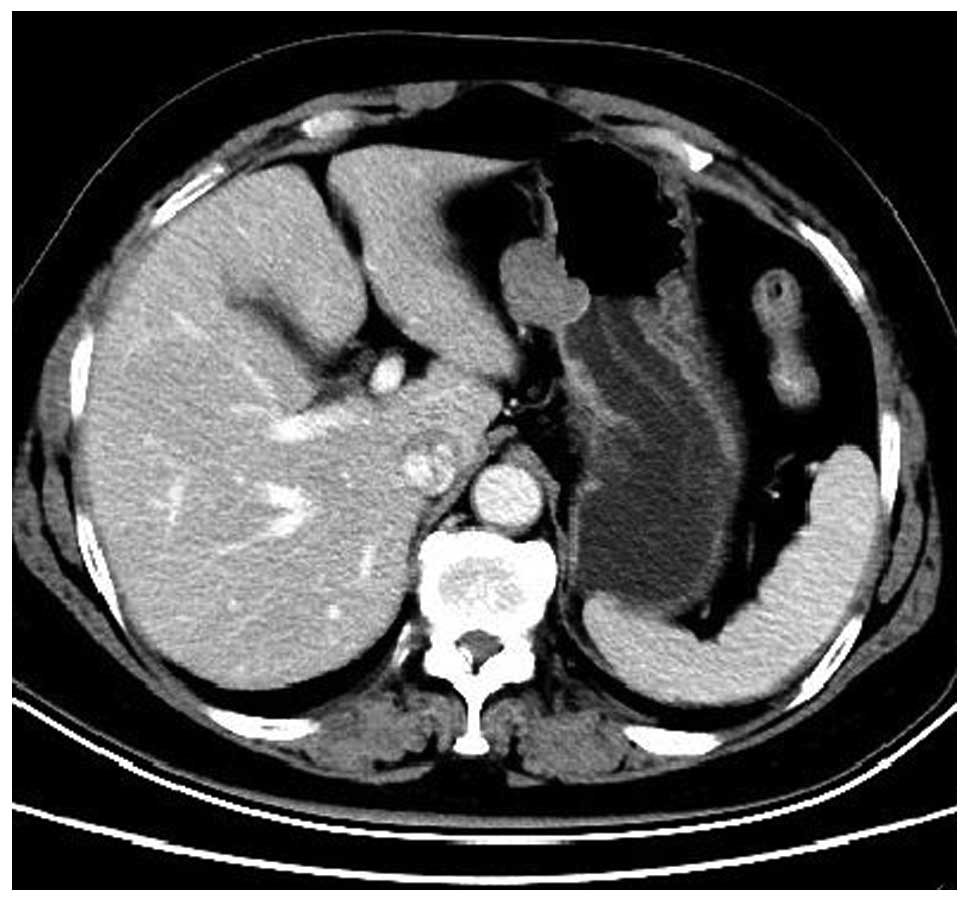
abdominal contrast-enhanced CT confirmed the presence of two
well-marginated oval-shaped masses located at the gastric fundus
and the lesser curvature of the cardia, respectively (Fig. 3). A pre-operative diagnosis of GISTs
was formed.
At laparotomy, the tumor located at the lesser
curvature of the cardia was found and measured 3×3×2.5 cm in size.
The second tumor was found in the posterior wall of the gastric
body and measured ~1×1×0.5 cm in size. A proximal gastrectomy and
Roux-en-Y reconstruction were performed.
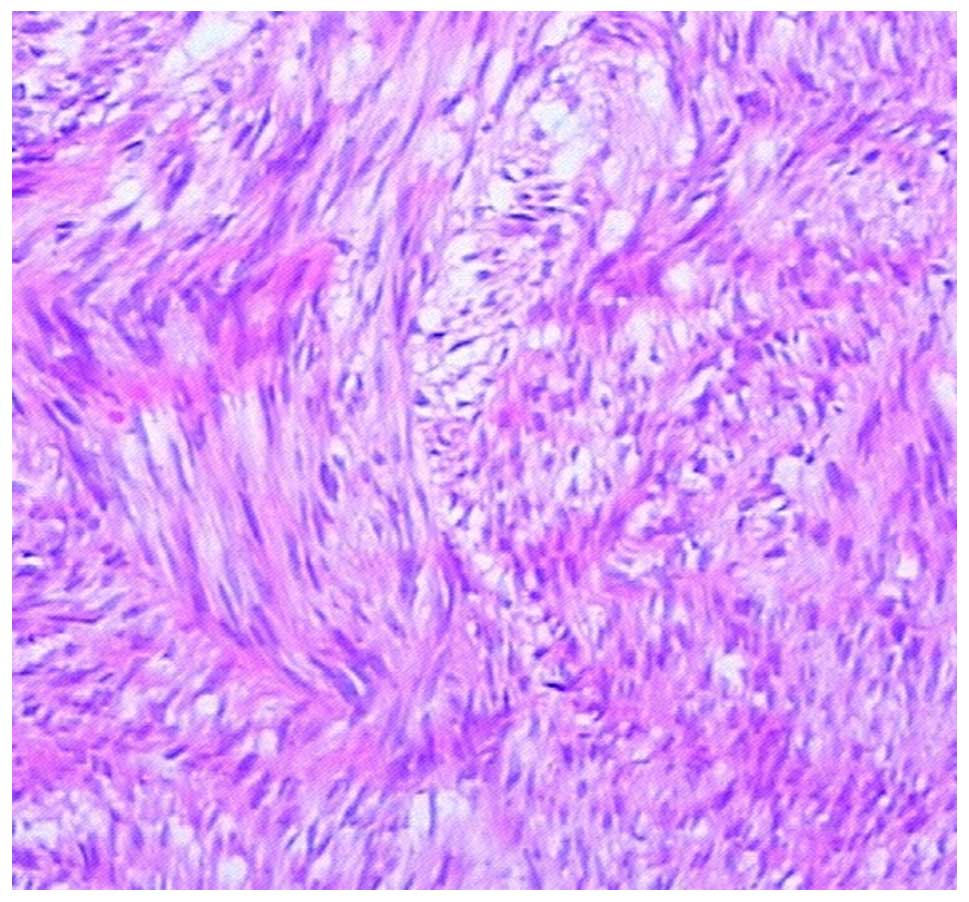
Microscopically, the first tumor was found to be
composed of spindle cells, a number of which showed high mitotic
activity (Fig.4). Immunohistochemical
staining showed that the tumor was positive for (CD)117, CD34,
discovered on GIST-1 and smooth muscle actin (SMA), but negative
for desmin and S-100, suggesting a GIST origin.
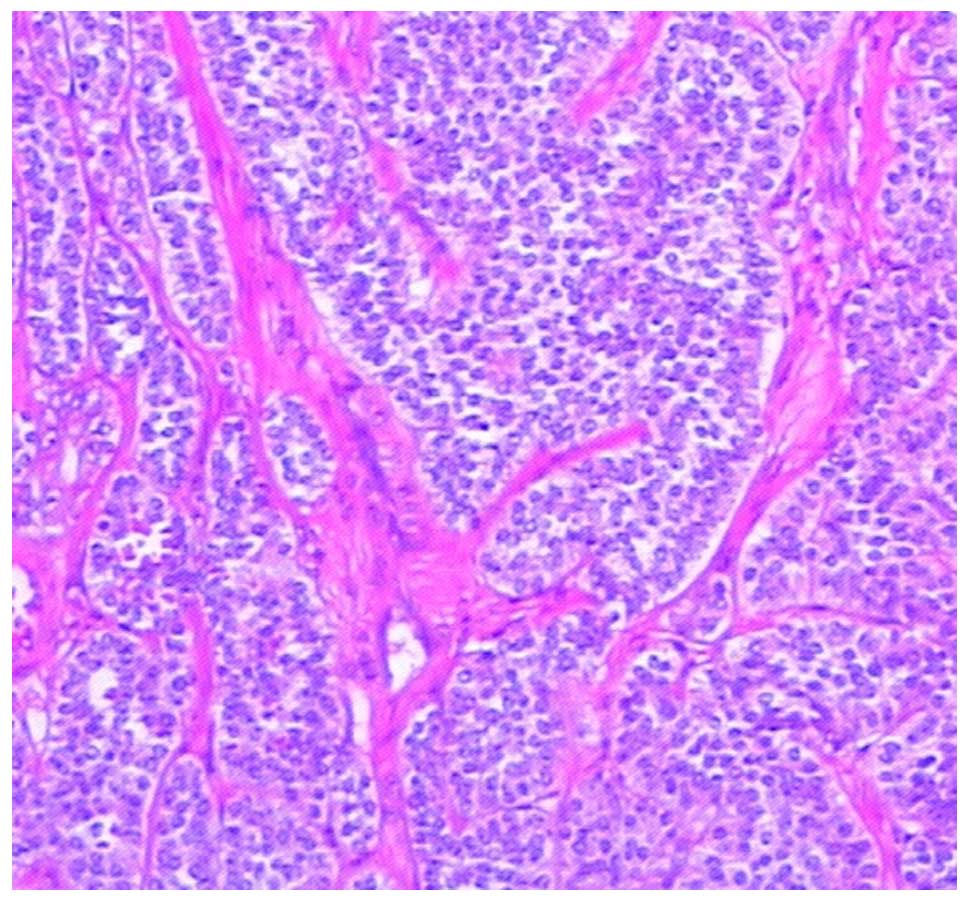
Unexpectedly, the cells of the second tumor were
organized in nests with fibrous separation (Fig. 5). The tumor cells exhibited a mitotic
index of <2/10 high-power fields. Immunohistochemical results
showed that the tumor was strongly positive for chromogranin A
(CgA; +++), synaptophysin (Syn; +++) and CD56 (+++), with a Ki-67
index of <2%. All the results suggested that this tumor was of
GEP-NET origin.
Following the surgery, there were no post-operative
complications. The patient was discharged from hospital after 10
days. After 17 months of follow-up examinations using CT and
ultrasonography, the patient showed no symptoms or signs of
recurrence. The present study was approved by the Subei People's
Hospital of Jiangsu Ethical Committee and written informed consent
was obtained from the patient.
Discussion
GEP-NETs originate from various neuroendocrine
tissues of the gastroenteropancreatic system and form the largest
subgroup of NETs (13). GEP-NETs
usually and equally distribute in the foregut (stomach or
duodenal), midgut (jejunal, ileal, appendix or proximal colon) and
hindgut (distal colon or rectum) (14). Although they are rare neoplastic
diseases in general, the incidence has greatly increased in the
last decade (15).
In 2000, the World Health Organization
classification system for NETs was published (16). Based on this system, proliferation
index (Ki-67, MIB-1), angioinvasion and mitoses are regarded as the
most important factors to determine the malignancy of NETs. NETs
can be classified into the well-differentiated (<2 cm in size,
<2% Ki-67 index), moderately-differentiated (>2 cm in size,
>2% Ki-67 index, or angioinvasive) and poorly-differentiated
(>20% Ki-67 index) subtypes (17).
In addition, the European Neuroendocrine Tumor Society proposed
another grading system (G1, G2 and G3) for the classification of
NETs (18). In general, G1 and G2
NETs refer to a well-differentiated subtype, displaying diffuse and
intense expression of CgA and Syn. G3 NET indicates a
poorly-differentiated subtype with high mitotic counts/Ki-67 index
(>20%), displaying slight staining of CgA and intense staining
of Syn (19).
GISTs are also rare tumors, but are the most common
mesenchymal tumor in the gastrointestinal tract (20). GISTs clinically present with
characteristic GI bleeding, weight loss, abdominal pain, anemia
and/or a palpable mass (21). The
highest incidence of GISTs occurs in the stomach (52–60%), followed
by the small intestine (20–30%) and colorectum (10%). Approximately
95% of GISTs are positive for c-Kit/CD117, 40–50% for CD34, 20–30%
for SMA and 10% for S100 (22,23). The
National Institutes of Health and National Comprehensive Cancer
Network systems were established to predict GIST behavior using
risk assessment (very low risk, low risk, intermediate risk and
high risk) (24).
To the best of our knowledge, the present study is
the first to demonstrate a compound case of GEP-NETs and GISTs in
the stomach. Surgical resection remains the first choice of
treatment, however, due to the malignant potency of the two tumor
types, long-term follow-up is greatly advised.
References
|
1
|
Jiao Y, Shi C, Edil BH, et al: DAXX/ATRX,
MEN1, and mTOR pathway genes are frequently altered in pancreatic
neuroendocrine tumors. Science. 331:1199–1203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Modlin IM, Lye KD and Kidd M: A 5-decade
analysis of 13,715 carcinoid tumors. Cancer. 97:934–959. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Modlin IM, Oberg K, Chung DC, Jensen RT,
de Herder WW, Thakker RV, Caplin M, Fave G Delle, Kaltsas GA,
Krenning EP, et al: Gastroenteropancreatic neuroendocrine tumours.
Lancet Oncol. 9:61–72. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lawrence B, Gustafsson BI, Chan A, Svejda
B, Kidd M and Modlin IM: The epidemiology of gastroenteropancreatic
neuroendocrine tumors. Endocrinol Metab Clin North Am. 40:1–18.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hirota S, Isozaki K, Moriyama Y, et al:
Gain-of-function mutations of c-kit in human gastrointestinal
stromal tumors. Science. 279:577–580. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heinrich MC, Corless CL, Duensing A, et
al: PDGFRA activating mutations in gastrointestinal stromal tumors.
Science. 299:708–710. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hirota S, Ohashi A, Nishida T, et al:
Gain-of-function mutations of platelet-derived growth factor
receptor alpha gene in gastrointestinal stromal tumors.
Gastroenterology. 125:660–667. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Daniels M, Lurkin I, Pauli R, et al:
Spectrum of KIT/PDGFRA/BRAF mutations and
Phosphatidylinositol-3-Kinase pathway gene alterations in
gastrointestinal stromal tumors (GIST). Cancer Lett. 312:43–54.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reddy P, Boci K and Charbonneau C: The
epidemiologic, health-related quality of life, and economic burden
of gastrointestinal stromal tumours. J Clin Pharm Ther. 32:557–565.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miettinen M and Lasota J: Histopathology
of gastrointestinal stromal tumor. J Surg Oncol. 104:865–873. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Robinson TL, Sircar K, Hewlett BR,
Chorneyko K, Riddell RH and Huizinga JD: Gastrointestinal stromal
tumors may originate from a subset of CD34-positive interstitial
cells of Cajal. Am J Pathol. 156:1157–1163. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ramage JK, Ahmed A, Ardill J, et al: UK
and Ireland Neuroendocrine Tumour Society: Guidelines for the
management of gastroenteropancreatic neuroendocrine (including
carcinoid) tumours (NETs). Gut. 61:6–32. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Oberg K: Somatostatin-receptor mediated
diagnosis and treatment in gastrointestinal neuroendocrine tumours
(GEP-NET's). Rocz Akad Med Bialymst. 50:62–68. 2005.PubMed/NCBI
|
|
14
|
Klöppel G and Anlauf M: Epidemiology,
tumour biology and histopathological classification of
neuroendocrine tumours of the gastrointestinal tract. Best Pract
Res Clin Gastroenterol. 19:507–517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schimmack S, Svejda B, Lawrence B, Kidd M
and Modlin IM: The diversity and commonalities of
gastroenteropancreatic neuroendocrine tumors. Langenbecks Arch
Surg. 396:273–298. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hamilton SR and Aaltonen LA: World Health
Organization Classification of Tumors. Pathology and Genetics of
Tumours of the Digestive System. IARC Press. (Lyon). 2000.
|
|
17
|
Modlin IM, Moss SF, Oberg K, Padbury R,
Hicks RJ, Gustafsson BI, Wright NA and Kidd M: Gastrointestinal
neuroendocrine (carcinoid) tumours: Current diagnosis and
management. Med J Aust. 193:46–52. 2010.PubMed/NCBI
|
|
18
|
Klöppel GI, Couvelard A, Perren A, et al:
Mallorca Consensus Conference participants; European Neuroendocrine
Tumor Society: ENETS Consensus Guidelines for the Standards of Care
in Neuroendocrine Tumors: Towards a standardized approach to the
diagnosis of gastroenteropancreatic neuroendocrine tumors and their
prognostic stratification. Neuroendocrinology. 90:162–166. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oberg K and Castellano D: Current
knowledge on diagnosis and staging of neuroendocrine tumors. Cancer
Metastasis Rev. 30(Suppl 1): 3–7. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
George S and Desai J: Management of
gastrointestinal stromal tumors in the era of tyrosine kinase
inhibitors. Curr Treat Options Oncol. 3:489–496. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rabin I, Chikman B, Lavy R, Sandbank J,
Maklakovsky M, Gold-Deutch R, Halpren Z, Wassermann I and Halevy A:
Gastrointestinal stromal tumors: A 19 year experience. Isr Med
Assoc J. 11:98–102. 2009.PubMed/NCBI
|
|
22
|
Corless CL, Fletcher JA and Heinrich MC:
Biology of gastrointestinal stromal tumors. J Clin Oncol.
22:3813–3825. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kang YN, Jung HR and Hwang I:
Clinicopathological and immunohistochemical features of
gastointestinal stromal tumors. Cancer Res Treat. 42:135–143. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
National Comprehensive Cancer Network
(NCCN): NCCN Clinical Practice Guidelines in Oncology. Soft Tissue
Sarcoma. v.2.2009 [Internet]. National Comprehensive Cancer Network
(Fort Washington, PA). 2009.
|