Introduction
Extra-abdominal fibromatosis, also known as a
desmoid tumor, is a rarely observed lesion of benign biological
significance, characterized as a non-metastatic lesion with local
invasiveness (1). Extra-abdominal
fibromatosis represents <0.03% of all neoplasms, with an annual
incidence of 2–4 cases/1,000,000 individuals, worldwide (1). The most common locations are the
shoulder and upper limb (33%), gluteus and lower extremities (30%),
chest wall and spine (17%), and head and neck (10%) (2). Extra-abdominal fibromatosis occurs more
often in females and has a higher incidence between puberty and the
fourth decade of life. The tumor originates from the muscle-fascial
connective tissue and is therefore also known as deep aponeurotic
fibromatosis (1). The desmoid tumor
may be primary or secondary to trauma, including surgery, or
hormonal stimuli (3). It typically
presents as a mass of a hard consistency, with a poorly demarcated
margin and poor vascularization, and is commonly infiltrating and
adherent to the surrounding tissue. The internal structure of the
tumor is composed of abundant collagen material mixed with spindle
cells and fibroblasts with abundant eosinophilic cytoplasm
(4). Magnetic resonance imaging (MRI)
is the gold standard technique for diagnosis (2). The selection of optimal treatment is not
standardized but, when possible, radical and aggressive surgical
resection with margins free of disease (R0) remains, to date, the
preferred treatment for desmoid tumor. However, this treatment
still has a fair rate of local recurrence (5).
The present study reports the case of a 34-year-old
female with a voluminous mass diagnosed as an extra-abdominal
fibromatosis. Written informed consent was obtained from the
patient for inclusion in the present study.
Case report
A 34-year-old female patient was admitted to the
Department of Surgery at San Martino Hospital (Genova, Italy) due
to a voluminous mass in the right subcostal region that had
appeared 4–5 months previously and was associated with gravative
pain. The patient's remote medical history reported an
endometriosis initially treated with hormonal therapy and then with
surgery; in addition, the lesion occurred soon after the birth of
the patient's first child. Ultrasonography (US) and
thoracoabdominal computed tomography (CT) were performed. US, which
was performed due to the difficult accessibility of the anatomical
site, identified the presence of a solid mass without central
necrosis, that appeared hypoechoic and confounding. Upon CT of the
chest, a coarse solid expansive formation was observed. The mass
was markedly inhomogeneous with the presence of hyperdense areas
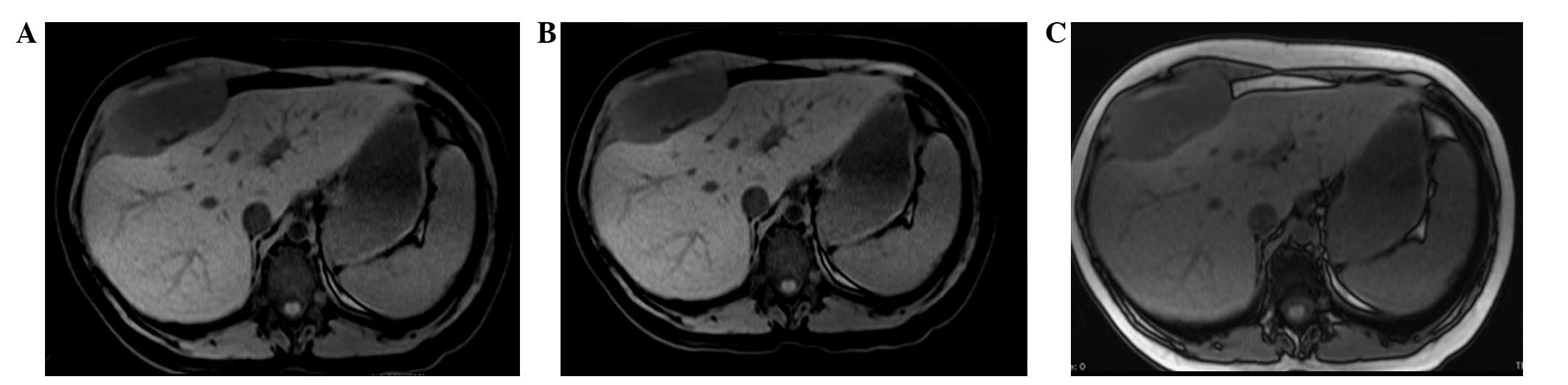
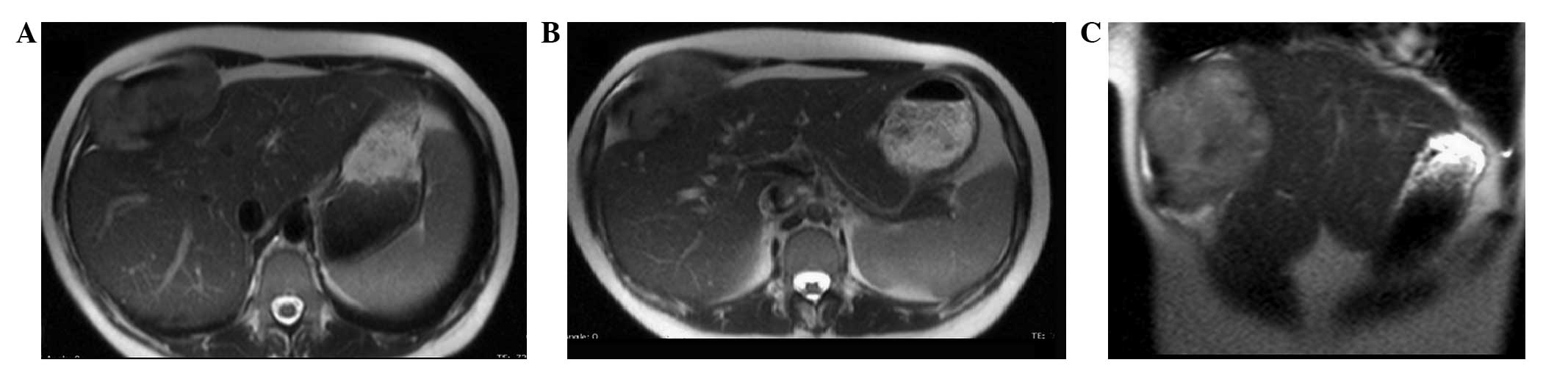
indistinguishable from the muscle and bone. MRI of the upper
abdomen, with and without contrast medium, confirmed the presence
of a 95×45×94-mm neoplasm. This was localized between the anterior
hepatic margin (dislocated backwards) and the right costal plane,
with a strict anatomical association with the diaphragm, to the
right rectus abdominis muscle and to the external oblique muscle
without a clear cleavage plane. Furthermore, the lesion tended to
herniate in the space between the costal cartilages. The lesion
appeared to be tenuously hypointense in T1-weighted sequences and
unevenly hyperintense in T2-weighted sequences, which was not
indicative of hematic content, nor pathognomonic of a specific
nature (Figs. 1 and 2). The patient therefore underwent a
surgical resection. Through a right subcostal laparotomy, the
voluminous neoplasm of the right subdiaphragmatic thoracic wall was
excised. The mass had developed in the context of the
antero-inferior thoracic wall, involving the peritoneum, the
diaphragm and the osteomuscular plane of the last four ribs. Once
the mass was isolated from the antero-lateral surface of the liver
we proceeded with resection of the last four ribs, from the sternum
to the posterior axillary line. The thoraco-abdominal wall
reconstruction was performed and an intrabdominal-diaphragmatic
prosthesis (Dual-Mesh) was positioned to restore the continuity.
The intraoperative histological examination evidenced the
mesenchymal nature of the lesion and showed that the resection
margins were free from disease (R0). The post-operative recovery
was regular, and the patient was discharged on day 8. The
definitive histological examination showed evidence consistent with
extra-abdominal fibromatosis: The neoplasm was lacking a capsule,
with infiltrative-like growth, spindle cells arranged in interlaced
bundles and focal mixoid areas, without any significant cytological
atypia, a low proliferative index (<3% Ki-67-positive cells) and
a very low mitotic index (<1 mitosis/10 high-power fields). No
necrotic outbreaks were identified. The immunophenotypic profile
showed variable positivity to actin and negativity to desmin,
cluster of differentiation (CD)34, CD99, B-cell lymphoma 2, CD117
(c-kit) and S-100 protein (Fig. 3).
Although the margins of resection were free from disease (R0), due
to the high percentage of recurrences, it was decided to subject
the patient to a course of adjuvant radiotherapy (50 Gy in 20
fractions). The use of adjuvant chemotherapy treatment was excluded
due to the lack of literature in this regard. The patient, who has
been monitored by strict follow-up examinations using CT/MRI at 6,
12, 24 and 48 months, has shown no signs of disease recurrence for
4 years since the surgery.
Discussion
Extra-abdominal fibromatosis, also known as a
desmoid tumor, is a rare, non-metastatic and locally invasive
lesion that is characterized by a high percentage of local
recurrences. The tumor originates from the connective
muscle-fascial tissue. Representing <0.03% of all neoplasms, the
tumor occurs more often in females and can occur in all age ranges,
with a peak incidence between puberty and the fourth decade of life
(1). Histologically similar desmoid
tumors have different prognostic outcomes in relation to age, being
locally more aggressive in young subjects, with a higher percentage
of recurrences (6). The most common
tumor locations are the shoulders and upper limbs (33%), the
gluteus and lower limbs (30%), the thoracic wall and spinal column
(17%), and the head and neck (10%) (2). Desmoid tumors can either be primitive or
secondary to trauma, including surgical procedures, or hormonal
stimulation. Frequently, desmoid tumors occur in young females
during or after pregnancy and can regress spontaneously in
menopause or after hormonal therapy with tamoxifen (3). In the present case, the patient was
suffering from endometriosis and underwent hormonal treatment as
well as surgery; in addition to this, the lesion occurred soon
after the birth of the patient's first child.
In a fair percentage of cases, fibromatosis is
associated with familial adenomatous polyposis (FAP) or Gardner's
syndrome (1,6). Despite the multifactorial etiology,
certain genetic factors have been identified. For example, in
patients affected by FAP or Gardner's syndrome, a mutation in the
APC gene has been identified, while in the sporadic forms, there
appears to be a mutation in the gene that codes for the β-catenins.
The two mutations cause an increase in the concentration of
intracellular β-catenins, which induces a boost in fibroblastic
proliferation (7).
Generally, desmoid tumors present as a mass of hard
consistency that is not well-defined, and is typically infiltrative
and adherent to the surrounding tissue. The lesion shows poor
vascularization and a typical light-brown color, which aids in the
differentiation from the adjacent tissue. However, it does not
usually exhibit necrotic nor hemorrhagic areas. In the majority of
cases, extra-abdominal desmoid tumors are asymptomatic and
therefore, at the time of diagnosis, which is mostly incidental,
their dimensions usually exceed 10 cm in diameter (4).
The dimensions of the tumor, its extension and the
anatomical association with the surrounding structures can be
evaluated with the use of US, CT and MRI, which always must be
performed prior to the surgical treatment. Upon US the lesion
appears to be hypoechogenic and homogeneous, with variable
vascularization. CT shows a soft-tissue mass with different
attenuation and enhancement due to the presence of intratumoral
hemorrhagic or degenerative areas. The margins often appear to be
unclearly defined due to the infiltration of the surrounding
tissues (2,7). MRI represents the gold standard to
identify the fibromatous lesion and its anatomical association with
the adjacent tissues. The technique is also useful in the
post-operative follow-up evaluation of the patient (2).
The selection of the optimal treatment is not
standardized, above all due to the small number of patients
suffering from this disease and due to the few clinical trials that
have been conducted. Asymptomatic lesions can be monitored over
time, particularly if stable, while treatment is always to be
considered in symptomatic patients presenting with lesions either
of a large size or that are compressing important vital structures
(1). Aggressive surgical resection
with safe margins (2–4 cm) (R0) is the prime treatment. An optimal
resection of the tumor involving the thoracic wall, as in the
present case, is required to include the excision of disease-free
ribs, one above and one underneath the lesion (5). It is not always possible to obtain
disease-free resection margins, particularly if the tumor involves
noble structures such as the spinal column, brachial plexus, major
vessels or structures of the fascia of the neck (5). In these cases, a surgical approach is
still recommended, even if reductive, to reduce the compressive
symptomatology.
In cases where the tumor cannot be operated on and
in cases where negative resection margins cannot be obtained, the
patient is treated with multimodal therapy with the purpose of
reducing the incidence of local recurrences. These treatments
include radiotherapy, chemotherapy (anthracycline, vinblastine and
methotrexate), hormonal therapy (tamoxifen), non-steroidal
anti-inflammatory drugs (NSAIDs), interferon and imatinib mesylate
(8). National Comprenhensive Cancer
Network guidelines suggest the use of post-operative radiotherapy
only for tumors of large dimensions and with positive resection
margins (9). Maximal doses of
radiotherapy are not required to obtain longer periods of
disease-free survival (10). There
are ongoing studies evaluating the effectiveness of pre-operative
radiotherapy in reducing the dimensions of the tumors and in
affecting the risk of local recurrence (11). Chemotherapy is used in those patients
in whom the tumor exhibits rapid growth or in those who are heavily
symptomatic, and it is associated with the use of NSAIDs or COX2
inhibitors. Tamoxifen, an anti-estrogenic agent, is administered in
cases where the tumor is inoperable at the same dosages used to
treat mammary carcinoma, in virtue of the fact that desmoid tumors
are frequently hormone-sensitive. Interferon has been shown to be
effective in increasing the period of disease-free survival in
certain patients (1). Studies have
been conducted on the use of imatinib mesylate (a selective
inhibitor of the tyrosine kinase receptor) in the inoperable forms
of the tumor and in those patients who refused chemo-radiotherapy
or hormonal therapy. At present, study results remain discordant;
this is due to the fact that biological factors predictive of
response to the drug have not yet been identified (12).
In conclusion, the present study determined that
radical resection with margins free of disease still remains the
optimal treatment strategy for patients with extra-abdominal
fibromatosis. As desmoid tumors are burdened with a high rate of
local recurrence, each case should be carefully assessed to
evaluate the possibility of using multimodal therapies. In
addition, a screening program should be immediately organized to
allow careful monitoring of the patient.
References
|
1
|
Escobar C, Munker R, Thoma JO, Li BD and
Burton GV: Update on desmoid tumors. Ann Oncol. 23:562–569. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shinagare AB, Ramaiya NH, Jagannathan JP,
Krajewski KM, Giardino AA, Butrynski JE and Raut CP: A to Z of
desmoid tumors. AJR Am J Roentgenol. 197:W1008–W1014. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ioannou M, Demertzis N, Iakovidou I and
Kottakis S: The role of imatinib mesylate in adjuvant therapy of
extra-abdominal desmoid tumors. Anticancer Res. 27:1143–1147.
2007.PubMed/NCBI
|
|
4
|
Ibrahim M, Sandogji H and Allam A: Huge
intrathoracic desmoid tumor. Ann Thorac Med. 4:146–148. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mátrai Z, Tóth L, Szentirmay Z, Vámos FR,
Klepetko W, Vadász P, Kenessey I and Kásler M: Sporadic desmoid
tumors of the chest: Long-term follow-up of 28 multimodally treated
patients. Eur J Cardiothorac Surg. 40:1170–1176. 2011.PubMed/NCBI
|
|
6
|
Romero JA, Kim EE, Kim CG, Chung WK and
Isiklar I: Different biologic features of desmoid tumors in adult
and juvenile patients: MR demonstration. J Comput Assist Tomogr.
19:782–787. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lamboley JL, Le Moigne F, Proust C,
Thivolet-Bejui F, Tronc F, Revel D and Douek P: Desmoid tumour of
the chest wall. Diagn Interv Imaging. 93:635–638. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Melis M, Zager JS and Sondak VK:
Multimodality management of desmoid tumors: How important is a
negative surgical margin? J Surg Oncol. 98:594–602. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
National Comprehensive Cancer Network
(NCCN). Guidelines for soft tissue sarcomas, Version 1.
2011.http://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdfAccessed.
March 13–2011
|
|
10
|
Ballo MT, Zagars GK and Pollack A:
Radiation therapy in the management of desmoid tumors. Int J Radiat
Oncol Biol Phys. 42:1007–1014. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mankin HJ, Hornicek FJ and Springfield DS:
Extra-abdominal desmoid tumors: A report of 234 cases. J Surg
Oncol. 102:380–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dufresne A, Bertucci F, Penel N, Le Cesne
A, Bui B, Tubiana-Hulin M, Ray-Coquard I, Cupissol D, Chevreau C,
Perol D, et al: Identification of biological factors predictive of
response to imatinib mesylate in aggressive fibromatosis. Br J
Cancer. 103:482–485. 2010. View Article : Google Scholar : PubMed/NCBI
|