Introduction
Lung cancer is one of the most commonly occurring
cancers and is associated with a high rate of mortality (1). The disease is responsible for 31% of
cancer-associated mortalities in males and 25% of cancer-associated
mortalities in females (2).
Non-small-cell lung carcinoma (NSCLC) constitutes >85% of all
lung cancers (3). For early-stage
NSCLC patients, surgery is the preferred treatment, while
radiotherapy and/or chemotherapy is preferred for late-stage
patients or patients for whom surgery is not possible (2). The overall five-year survival rate of
NSCLC patients is <10% (4).
Studies of various neoplastic diseases, primarily
breast and endometrial carcinoma, have demonstrated the prognostic
significance of estrogen receptors (5). In addition, epidemiological studies have
indicated that the incidence of lung cancer is lower in females
compared with that of males of the same age (6). It has also been hypothesized that
increased parity in females is associated with a decreased risk of
lung cancer development, and that hormone replacement treatment
suppresses lung cancer progression (7).
Estrogen predominantly binds to two receptors,
estrogen receptors (ERs) α and β, and regulates a number of
biological processes, including cell proliferation,
differentiation, apoptosis, inflammation and metabolism (8). ERα and ERβ proteins have been shown to
be expressed in primary lung tumors (9). Furthermore, these receptors are
expressed in NSCLCs and have been demonstrated to mediate the
transcriptional effects of estrogen (10). Until recently, only canonical ERs were
considered to mediate the effect of estrogens. Following their
activation, these canonical receptors are transported into the
nucleus to produce genomic or non-genomic responses (11). However, the inhibition or knockdown of
ERα and ERβ does not eliminate estrogen responses in various
tissues (12). More recent studies
have revealed that estrogen may mediate fast signal responses or
transcriptional events via G protein-coupled estrogen receptor 1
(GPER1) (13). GPER1 is expressed in
human brain, liver, heart, kidney, pancreatic, placental, blood
vessel, bone, lymphoid, endometrial, ovarian, breast and lung
cancer tissues (14). This receptor
may be localized on the cell membrane, nucleus, endoplasmic
reticulum, mitochondria or Golgi apparatus, and its effects vary
depending on this specific intracellular localization (14). It has been suggested that G-1
(chemical name,
1-[4-(6-bromo-1,3-benzodioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone)
acts a specific GPER1 agonist (15).
GPER1-dependent antiproliferative and proapoptotic effects of G-1
have been observed in prostate cancer (PC-3) and breast cancer
(MCF-7) cells (16,17). However, GPER1-independent
antiproliferative and proapoptotic effects of G-1 have been
observed in ovarian cancer (KGN) and breast cancer (MDA-MB 231)
cells (18).
Antioxidants have an essential role in cell
protection against reactive oxygen species (19). The oxidation of antioxidant enzymes
reduces the capacity of cells to eliminate free radicals (19). Therefore, an important approach in
antitumor therapeutic strategies is to inhibit antioxidant systems,
such catalase, superoxide dismutase (SOD) and glutathione
peroxidase (GPx), which are the primary defense lines of the cell
(19). A number of studies have shown
the influence of estrogen and GPER1 on antioxidant enzymes and
cytokine production (20,21).
Previous studies have revealed that GPER1 expression
is higher in various types of lung cancer tissues, including
adenocarcinoma, squamous-cell carcinoma and giant-cell carcinoma,
compared with normal lung tissue (22). Overexpression of GPER1 in lung cancer
may reflect a defense mechanism against the hyperproliferation of
cancer cells, with activation of this receptor leading to
antiproliferative or proapoptotic effects (22). However, the antiproliferative or
proapoptotic effects of GPER1 agonists in lung cancer cells have
not yet been demonstrated. Therefore, the present study aimed to
investigate the oxidant and antioxidant enzyme-mediated
antiproliferative and apoptotic effects of the GPER1 agonist G-1 on
lung cancer cells.
Materials and methods
Cell culture and chemicals
A549 human lung cancer cells were obtained from the
American Type Cell Collection (Manassas, VA, USA). Cells were
cultured in RPMI-1640 medium (Biochrom, Ltd., Cambridge, UK)
supplemented with 10% fetal bovine serum (FBS; Gibco Life
Technologies, Carlsbad, CA, USA) and 100 U/ml penicillin
(Sigma-Aldrich, St. Louis, MO, USA) at 37°C in a humidified
atmosphere of 5% CO2. For 3 days prior to the
experiment, A549 cells were cultured with phenol red-free RPMI-1640
containing 10% dextran-coated charcoal-FBS (Gibco Life
Technologies). The negative control condition for all assays was
untreated medium containing vehicle [0.1% dimethyl sulfoxide
(DMSO)]. The DMSO and estrogen 17β-estradiol were purchased from
Sigma-Aldrich, and G-1 and G-15 were purchased from Merck Millipore
(Darmstadt, Germany).
MTT and WST-8 assay
A549 human lung cancer cells were treated with
various concentrations (10−8, 10−7,
10−6, 10−5 and 10−4 M) of
17β-estradiol and G-1 in 96-well plates and incubated for 48 or 72
h. Following incubation, MTT solution (Sigma-Aldrich) was added to
each well at a concentration of 0.5 mg/ml, and incubated for 4 h at
37°C. At the end of this period, 100 µl DMSO solvent was added to
each well. The absorbance values [optical density (OD)] at 570 nm
of the solution in each well were read using a spectrophotometer
(ELx800 Absorbance Reader; BioTek Instruments, Inc., Winooski, VT,
USA). The antiproliferative potential of the G-1 was expressed as a
half maximal inhibitory concentration (IC50) value in
A549 lung cancer cells; only the IC50 (2×10−5
M) was subsequently used in the present study. The cells
(5×104/well) were treated with G-1 (2×10−5
M), either alone or in combination with G-15 (5×10−5 M),
at 37°C in an atmosphere of 5% CO2 for 48 and 72 h. A
WST-8 assay, Cell Counting Kit-8 (Sıgma-Aldrich), was also used to
assess cell proliferation, according to the manufacturer's
instructions, and absorbance at 450 nm was measured using an ELx800
microplate reader.
Acridine orange/ethidium bromide
staining
Acridine orange/ethidium bromide staining was
conducted to detect morphological evidence of apoptosis. A549 cells
were treated with G-1 for 72 h. The cells were washed with
phosphate-buffered saline (PBS; Merck Millipore) and incubated for
5 min with a solution of 10 µl of acridine orange/ethidium bromide
(Sigma-Aldrich) made up to 100 µl using PBS. The cells were then
washed with PBS and six microscopic fields were examined under an
Axio Vert.A1 inverted fluorescence microscope (Zeiss, Oberkochen,
Germany). The percentage of apoptotic cells was calculated using
the following formula: Apoptotic rate (%) = number of apoptotic
cells / total number of cells counted (23).
Biochemical measurements
Cells at 70–80% confluence were washed twice with
PBS (pH 7.2) and treated with trypsin/EDTA (0.25/0.02%; Merck
Millipore) in PBS for 10 min. The cell suspension was centrifuged
for 10 min at 400 × g. Cell pellets were then lysed in 50 mM
phosphate buffer solution (0.1 M, pH 7.0; Sigma-Aldrich), followed
by sonication for 2 min on ice. The mixture was then centrifuged
for 10 min at 14,000 × g and the supernatant was assayed for
protein concentration and enzymatic activities.
Catalase (CAT) activity was determined as described
by Aebi (24). Briefly, 100 µl
supernatant was incubated with an equal volume of absolute alcohol
for 30 min at 0°C, followed by the addition of 100 µl Triton X-100
(Sigma-Aldrich). A known volume of this mixture (200 µl) was mixed
with an equal volume of 0.066 M hydrogen peroxidase
(H2O2) in phosphate buffer solution, and
absorbance was measured at 240 nm for 30 sec using a Shimatzu
UV-1201 spectrophotometer (Shimadzu Corp., Kyoto, Japan). Protein
levels were estimated as described by Lowry et al (25) and activity was expressed in units of
GPx per mg protein.
SOD activity was determined as described by
Fridovich (26). The principle of the
method is based on the inhibition of nitro blue tetrazolium
chloride (NBT) reduction by the xanthine-xanthine oxidase system, a
superoxide generator. Xanthine (Sigma-Aldrich) and xanthine oxidase
(Sigma-Aldrich) generate superoxide radicals, which react with NBT
(Sigma-Aldrich) to form a red formazan dye. SOD activity was then
determined according to the degree of inhibition of this reaction.
One unit of SOD was defined as the quantity of enzyme (mg) causing
50% inhibition in the NBT reduction rate. SOD activity was
expressed as units of SOD per mg protein.
The GPx assay was based on the oxidation of nicotine
adenosine dinucleotide phosphate (NADPH; Sigma-Aldrich) to
NADP+, which is accompanied by a decrease in absorbance
at 340 nm. The rate of this decrease is directly proportional to
the GPx activity in the sample (27).
Therefore, GPx activity was measured by the enzymatic reaction that
was initiated by adding H2O2 to a reaction
mixture containing reduced glutathione, NADPH and glutathione
reductase (Sigma-Aldrich). The change in the absorbance at 340 nm
was monitored using a Shimatzu UV-1201 spectrophotometer (Shimadzu
Corp.). Protein levels were estimated as described by Lowry et
al (25) and activity was
expressed in units of GPx per mg protein.
The determination of concentration levels of
nitrite, which is the stable end product of nitric oxide (NO)
radicals, was used as a measure of NO production. Nitrite
concentration was determined using a classic colorimetric Griess
reaction. Briefly, equal volumes of samples and Griess reagent
(Sigma-Aldrich) were mixed at room temperature. After 5 min, the
absorbance was measured at 540 nm using a spectrophotometer (UV
1201; Shimadzu Scientific Instruments, Inc., Columbia, MD, USA).
The concentration of nitrite was determined using a standard curve
prepared with sodium nitrite (28).
Data and statistical analysis
Statistical significance was determined by one-way
analysis of variance followed by Bonferroni's multiple comparisons
test. P<0.05 was considered to indicate statistical
significance. All statistical analyses were performed using
GraphPad Prism software version 5.0, (GraphPad Software, Inc., La
Jolla, CA, USA).
Results
G-1 inhibits A549 cell
proliferation
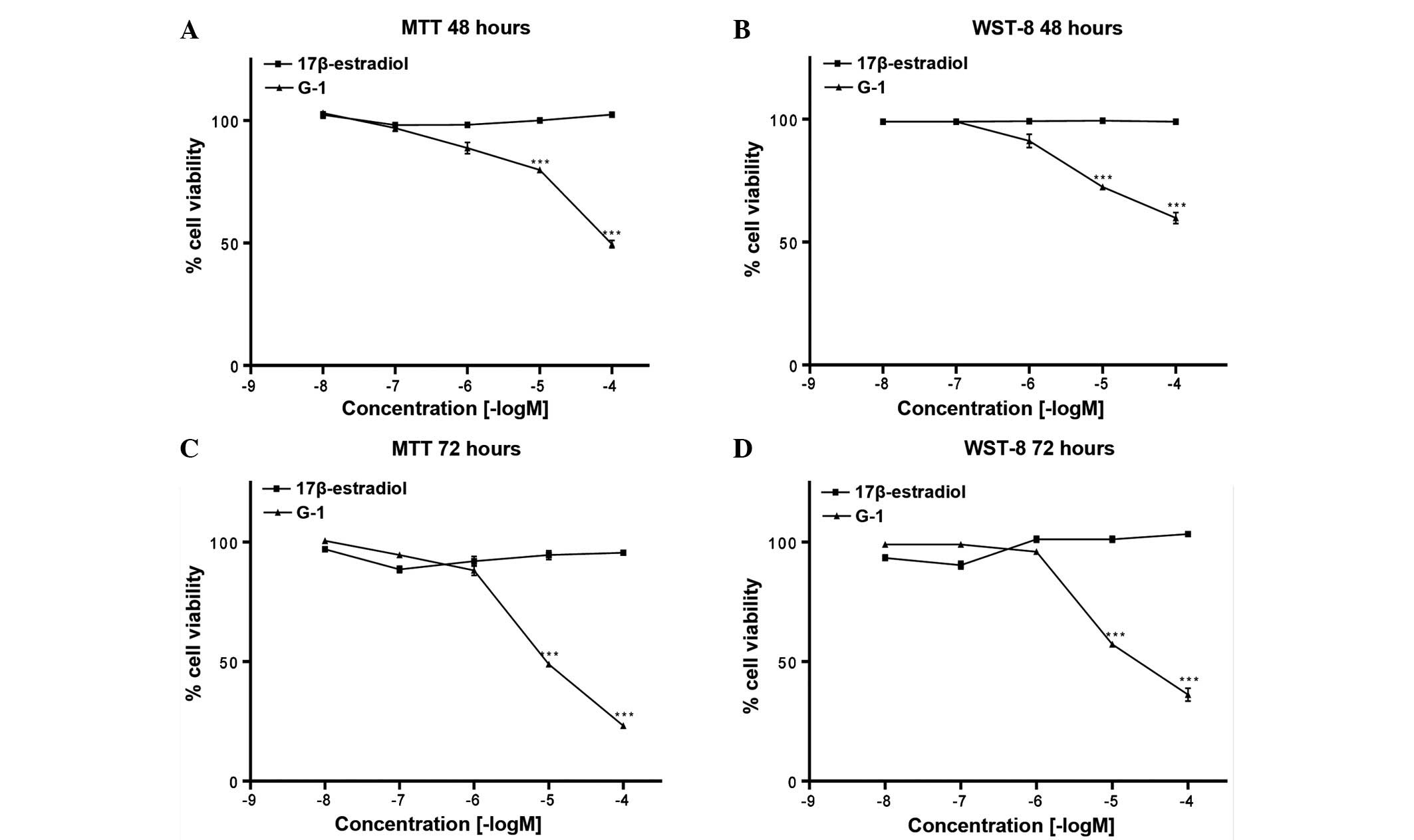
Treatment for 48 and 72 h with 17β-estradiol in A549
cells had no significant effect on cell proliferation (Fig. 1). However, treatment with G-1
(10−5 and 10−4 M) for 48 and 72 h
significantly decreased cell proliferation (P<0.001; Fig. 1).
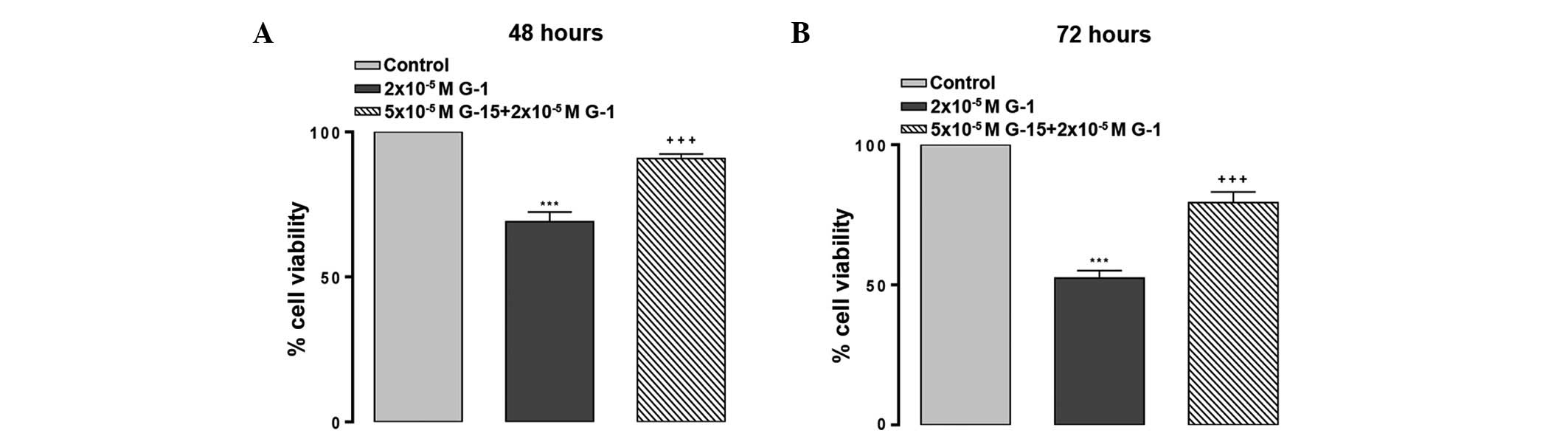
G-15, a selective GPER1 antagonist,
blocks G-1-induced suppression of A549 cell proliferation
At 72 h, the IC50 value for G-1 was
calculated to be 2×10−5 M. Treatment with
5×10−5 M of the GPER1 antagonist G-15 suppressed the
effect on cell proliferation of 2×10−5 M G-1 treatment
for 48 h (P<0.001; Fig. 2A) and 72
h (P<0.001; Fig. 2B).
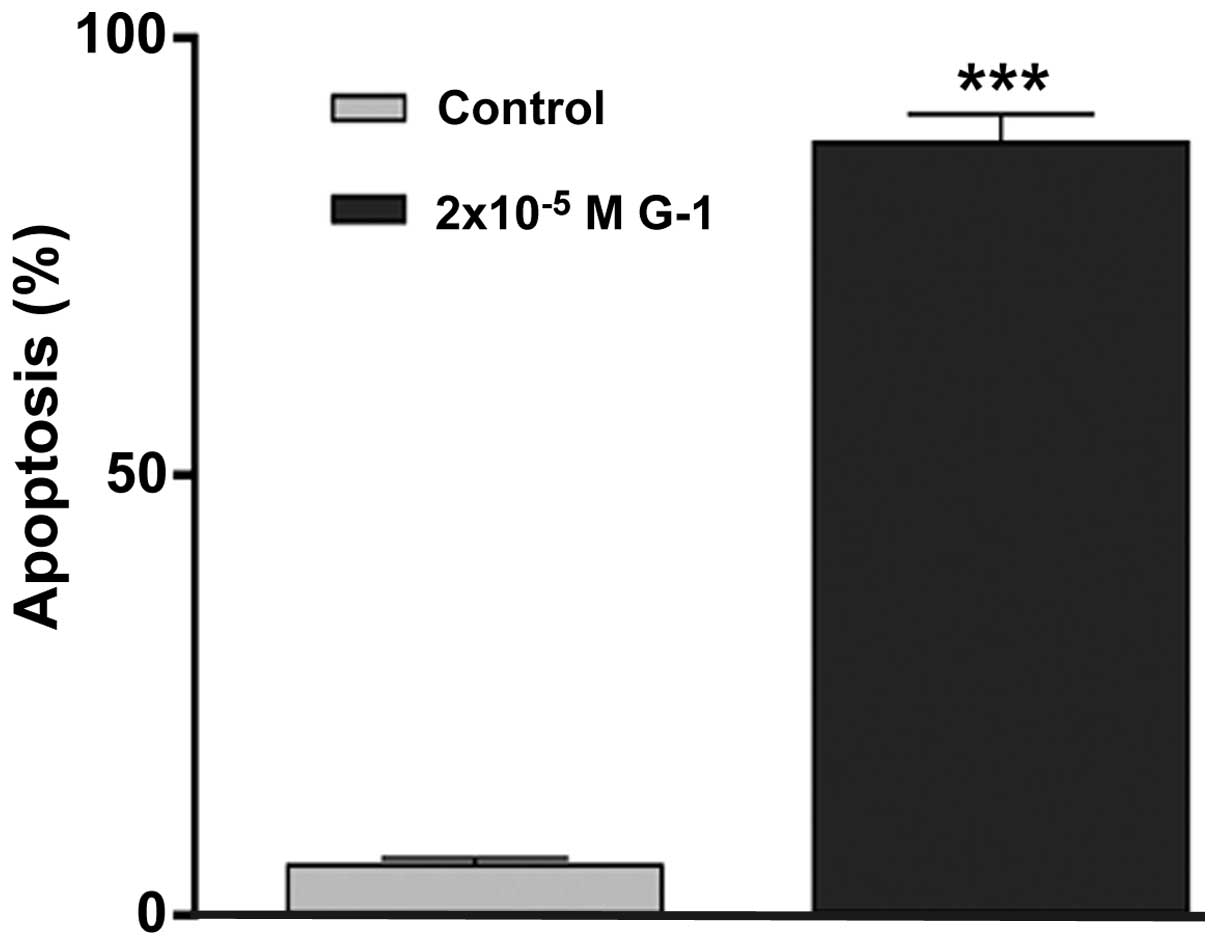
G-1 induces apoptotic cell death
Treatment of A549 cells with G-1 at a concentration
of 2×10−5 M revealed a significant increase in
apoptosis, consistent with its antiproliferative effect
(P<0.001; Fig. 3).
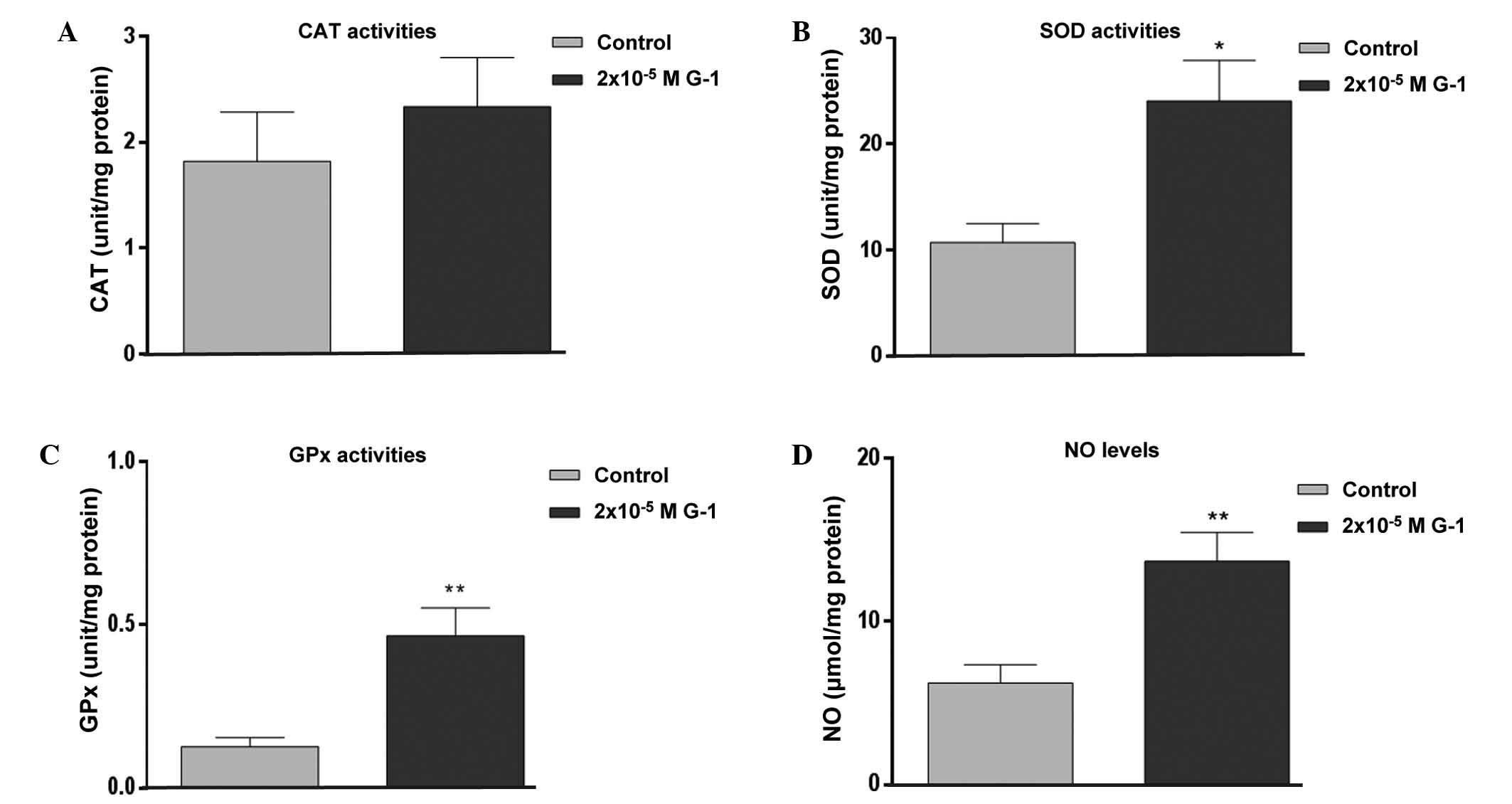
G-1 increases oxidant levels and
antioxidant enzyme activities
G-1 treatment at a concentration of
2×10−5 M had no significant effect on CAT activity
(Fig. 4A). However, this treatment
led to a significant increase in SOD activity (P<0.05; Fig. 4B), GPx activity (P<0.01; Fig. 4C) and NO level (P<0.01; Fig. 4D).
Discussion
The present study investigated the antiproliferative
and proapoptotic effects of the GPER1 agonist G-1 in lung cancer
cells, and attempted to understand how oxidant and antioxidant
molecules may mediate this effect. Using two different tetrazolium
salt (MTT and WST) assays, G-1 treatment of various concentrations
and for different times was revealed to decrease A549 cell
proliferation and viability. These two similar and alternative kits
were used to determine the IC50 value for G-1. The
current results demonstrating the antiproliferative and
proapoptotic effects of G-1 on A549 lung cancer cells are similar
to those of previous studies, which showed that G-1 suppresses
ovarian (KGN), breast cancer (MCF-7, MDA-MB 231) and prostate
cancer (PC-3) cell proliferation and induces cell apoptosis
(16–18). Furthermore, the receptor-dependent
antiproliferative and proapoptotic effects of G-1 were similar to
the receptor-dependent effects previously observed in PC-3 prostate
and MCF-7 breast cancer cells (16,17). To
date, the information established regarding apoptotic signal
transduction indicates that certain molecules and enzymes that are
responsible for intracellular signaling pathways are also
responsible for the signal transduction events during apoptosis
(29). For example, calcium ions
(Ca2+), which are widely used in intracellular
signaling, also play a role in apoptosis: An increase in the
intracellular concentration of Ca2+ can lead to the
induction apoptosis (29). It has
been demonstrated that G-1 increases phosphoinositide
3-kinase-mediated calcium mobilization in MCF-7 and SKBR-3 in
breast cancer cells (30). Following
G-1 treatment, the increase in cytoplasmic Ca2+ ions in
A549 cells may induce mRNA expression levels of phosphorylated
mitogen-activated protein kinases and proto-oncogene c-jun, which
can trigger apoptosis (31).
The present study aimed to investigate whether
oxidative and antioxidative enzymes were able to mediate the
proapoptotic and antiproliferative effects of G-1. SOD catalyzes
the conversion of superoxide molecules into
H2O2 and molecular oxygen (O2).
Then, H2O2 molecules are converted into water
and oxygen by the activity of GPx and CAT enzymes (32). These reactions protect the cells from
damage under normal conditions (33).
In the present study, an increase in the antioxidant defense system
(i.e. SOD and GPx enzyme activities) was observed in A549 cells.
H2O2 accumulation can occur as a result of
this increase (34,35). H2O2 is a
reactive oxygen species, and also an important signaling molecule.
Various studies have shown that mitochondrial
H2O2 is a direct trigger of apoptosis
(35). The administration of 100 and
150 µM resveratrol increased apoptosis rates to maximum values of
50 and 20%, respectively, for the androgen-sensitive LNCaP cancer
line and the androgen-insensitive PC-3 cancer line (36). Therefore, resveratrol may serve as an
effective agent in early, mid and late stages of cancer (36). Khan et al (37) demonstrated that resveratrol causes an
increase in SOD, CAT, and GPx enzyme activities and apoptosis in
PC-3 prostate carcinoma and HepG2 liver carcinoma cells, and that
H2O2 mediates these events. Resveratrol has
been demonstrated to inhibit potassium channels via GPER1 in
various cell lines, and this inhibition may induce apoptosis
(38).
Thus far, the role of NO in tumor biology has not
been elucidated; however, it has been hypothesized that NO plays a
role in various physiological and pathophysiological processes,
including vasodilation, nerve conduction, immune system processes
and cancer (39). The results of the
present study demonstrated that G-1 treatment significantly
increased cell death, and was associated with significantly
increased NO levels. NO may be involved in this process in two
different ways, primarily by the interaction of NO with superoxides
and peroxynitrite, resulting in toxic effects such as DNA damage,
protein thiol exchange and inactivation of mitochondrial enzymes in
the respiratory chain or citric acid cycle. All of these reactions
may be linked to NO-induced apoptosis (39). Furthermore, the secondary cause of the
significant increase in NO levels may be associated with nitrite
accumulation in the culture medium due to apoptotic cell death.
In conclusion, the present study demonstrated that
the GPER agonist G-1 had proapoptotic and antiproliferative effects
on A549 NSCLC cells, which may be mediated through oxidative and
antioxidative enzymes, including SOD and GPx. A better
understanding of GPER1's role in lung cancer will contribute
significantly to disease management and prevention. GPER1 may serve
as a novel target for lung cancer therapies, and the regulation of
GPER1 signaling in cancer treatment may prove beneficial for female
and male lung cancer patients. Thus, future studies which
investigate the use of GPER1-based therapies involving G-1 and/or
its derivatives are required.
Acknowledgements
This study was supported by a grant from
Kahramanmaraş Sütçü İmam University (BAP-2013/1-27M).
References
|
1
|
Lortet TJ, Soerjomataram I, Ferlay J,
Rutherford M, Weiderpass E and Bray F: International trends in lung
cancer incidence by histological subtype: Adenocarcinoma
stabilizing in men but still increasing in women. Lung Cancer.
84:13–22. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Siegel R, Ward E, Murray T, Xu J,
Smigal C and Thun MJ: Cancer statistics, 2006. CA Cancer J Clin.
56:106–130. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment and survivorship. Mayo Clin Proc. 83:584–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang T, Nelson RA, Bogardus A and Grannis
FW Jr: Five-year lung cancer survival: Which advanced stage
nonsmall cell lung cancer patients attain long-term survival?
Cancer. 116:1518–1525. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Deroo BJ and Korach KS: Estrogen receptors
and human disease. J Clin Invest. 6:561–570. 2006. View Article : Google Scholar
|
|
6
|
Donington JS and Colson YL: Sex and gender
differences in non-small cell lung cancer. Semin Thorac Cardiovasc
Surg. 23:137–145. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meinhold CL, de González Berrington A,
Bowman ED, Brenner AV, Jones RT, Lacey JV Jr, Loffredo CA,
Perlmutter D, Schonfeld SJ, Trivers GE and Harris CC: Reproductive
and hormonal factors and the risk of nonsmall cell lung cancer. Int
J Cancer. 128:1404–1413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cheskis BJ, Greger JG, Nagpal S and
Freedman LP: Signaling by estrogens. J Cell Physiol. 213:610–617.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dubey S, Siegfried JM and Traynor AM:
Non-small-cell lung cancer and breast carcinoma: Chemotherapy and
beyond. Lancet Oncol. 7:416–424. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pietras RJ, Marquez DC, Chen HW, Tsai E,
Weinberg O and Fishbein M: Estrogen and growth factor receptor
interactions in human breast and non-small cell lung cancer cells.
Steroids. 70:372–381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Luksha L and Kublickiene K: The role of
estrogen receptor subtypes for vascular maintenance. Gynecol
Endocrinol. 25:82–95. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ullrich ND, Krust A, Collins P and MacLeod
KT: Genomic deletion of estrogen receptors ERalpha and ERbeta does
not alter estrogen-mediated inhibition of Ca2+ influx and
contraction in murine cardiomyocytes. Am J Physiol Heart Circ
Physiol. 294:H2421–H2427. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Prossnitz ER, Arterburn JB, Smith HO,
Oprea TI, Sklar LA and Hathaway HJ: Estrogen signaling through the
transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol.
70:165–190. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mizukami Y: In vivo functions of
GPR30/GPER-1, a membrane receptor for estrogen: From discovery to
functions in vivo. Endocr J. 57:101–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bologa CG, Revankar CM, Young SM, Edwards
BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck
NP, Sklar LA, et al: Virtual and biomolecular screening converge on
a selective agonist for GPR30. Nat Chem Biol. 2:207–212. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chan QK, Lam HM, Ng CF, Lee AY, Chan ES,
Ng HK, Ho SM and Lau KM: Activation of GPR30 inhibits the growth of
prostate cancer cells through sustained activation of Erk1/2,
c-jun/c-fos-dependent upregulation of p21 and induction of G(2)
cell-cycle arrest. Cell Death Differ. 17:1511–1523. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ariazi EA, Brailoiu E, Yerrum S, Shupp HA,
Slifker MJ, Cunliffe HE, Black MA, Donato AL, Arterburn JB, Oprea
TI, et al: The G Protein-coupled receptor GPR30 inhibits
proliferation of estrogen receptor-positive breast cancer cells.
Cancer Res. 1:1184–1194. 2010. View Article : Google Scholar
|
|
18
|
Wang C, Lv X, Jiang C and Davis JS: The
putative G-protein coupled estrogen receptor agonist G-1 suppresses
proliferation of ovarian and breast cancer cells in a
GPER-independent manner. Am J Transl Res. 4:390–402.
2012.PubMed/NCBI
|
|
19
|
Tong L, Chuang CC, Wu S and Zuo L:
Reactive oxygen species in redox cancer therapy. Cancer Lett.
367:18–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kurt AH and Buyukafsar K: Vasoconstriction
induced by G1, a G-protein-coupled oestrogen receptor1 (GPER-1)
agonist, in the isolated perfused rat kidney. Eur J Pharmacol.
702:71–78. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pektas M, Kurt AH, Un I, Tiftik RN and
Buyukafsar K: Effects of 17β-estradiol and progesterone on the
production of adipokines in differentiating 3T3-L1 adipocytes: Role
of Rho-kinase. Cytokine. 72:130–134. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jala VR, Radde BN, Haribabu B and Klinge
CM: Enhanced expression of G-protein coupled estrogen receptor
(GPER/GPR30) in lung cancer. BMC Cancer. 28:612–624. 2012.
|
|
23
|
Ribble D, Goldstein NB, Norris DA and
Shellman YG: A simple technique for quantifying apoptosis in
96-well plates. BMC Biotechnol. 5:122005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Aebi H: Catalase in vitro. Methods
Enzymol. 105:121–126. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lowry OH, Rosebrough NJ, Farr AL and
Randall RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
26
|
Fridovich I: Superoxide dismutase. Adv
Enzymol. 41:35–97. 1974.PubMed/NCBI
|
|
27
|
Levander OA, DeLoach DP, Morris VC and
Moser PB: Platelet glutathione peroxidase activity as an index of
selenium status in rats. J Nutr. 113:55–63. 1983.PubMed/NCBI
|
|
28
|
Griess JP: Comments on the discussion of
the HH. Weselsky and Benedict ‘on some azo compounds’. Ber Deutsch
Chem Ges. 12:426–428. 1879.(In German). View Article : Google Scholar
|
|
29
|
Cohen JJ: Apoptosis: The physiological
pathway of cell death. Hosp Pract (Off Ed). 15:35–43. 1993.
|
|
30
|
Wang C, Lv X, He C, Hua G, Tsai MY and
Davis JS: The G-protein-coupled estrogen receptor agonist G-1
suppresses proliferation of ovarian cancer cells by blocking
tubulin polymerization. Cell Death Dis. 4:e8692013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chan QK, Lam HM, Ng CF, Lee AY, Chan ES,
Ng HK, Ho SM and Lau KM: Activation of GPR30 inhibits the growth of
prostate cancer cells through sustained activation of Erk1/2,
c-jun/c-fos-dependent upregulation of p21 and induction of G(2)
cell-cycle arrest. Cell Death Differ. 17:1511–1523. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Young IS and Woodside JV: Antioxidants in
health and disease. J Clin Pathol. 54:176–186. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Khan MA, Tania M, Zhang DZ and Chen HC:
Antioxidant enzymes and cancer. Chin J Cancer Res. 22:87–92. 2010.
View Article : Google Scholar
|
|
34
|
Pallepati P and Averill-Bates DA:
Activation of ER stress and apoptosis by hydrogen peroxide in HeLa
cells: Protective role of mild heat preconditioning at 40°C.
Biochim Biophys Acta. 1813:1987–1999. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Giorgio M, Migliaccio E, Orsini F,
Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S,
Marcaccio M, et al: Electron transfer between cytochrome c and
p66Shc generates reactive oxygen species that trigger mitochondrial
apoptosis. Cell. 122:221–233. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Benitez DA, Pozo-Guisado E,
Alvarez-Barrientos A, Fernandez-Salguero PM and Castellón EA:
Mechanisms involved in resveratrol-induced apoptosis and cell cycle
arrest in prostate cancer-derived cell lines. J Androl. 28:282–293.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Khan MA, Chen HC, Wan XX, Tania M, Xu AH,
Chen FZ and Zhang DZ: Regulatory effects of resveratrol on
antioxidant enzymes: A mechanism of growth inhibition and apoptosis
induction in cancer cells. Mol Cells. 35:219–225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dong WH, Chen JC, He YL, Xu JJ and Mei YA:
Resveratrol inhibits K(v)2.2 currents through the estrogen receptor
GPR30-mediated PKC pathway. Am J Physiol Cell Physiol. 305:547–557.
2013. View Article : Google Scholar
|
|
39
|
Pacher P, Beckman JS and Liaudet L: Nitric
oxide and peroxynitrite in health and disease. Physiol Rev.
87:315–424. 2007. View Article : Google Scholar : PubMed/NCBI
|