Introduction
B-cell malignancies are a heterogeneous group of
disorders and treatment of these conditions has essentially
remained the same for >30 years, with the exception of the
inclusion of monoclonal anti-cluster of differentiation (CD)20
agents in combination strategies (1,2). A pivotal
moment in the development of novel drugs for this group of
disorders arose with the introduction of the first biologic
targeted agent, the anti-CD20 monoclonal antibody rituximab, with
improved outcomes in almost every B-cell disorder in which it was
applied (3,4). However, although improved survival was
obtained for the B-cell lymphoma patients, the majority continued
to relapse following standard chemo-immunotherapy, and currently,
>15,000 patients still succumb to B-cell malignancies each year
in the United States. Over the past few years, marked progress has
been made in understanding a number of the key pathways that drive
proliferation, survival and resistance in lymphoma and leukemia
(5–7).
Our understanding of oncogenic mechanisms has recently been greatly
accelerated by the advent of functional and structural genomics.
The identification and validation of a number of essential pathways
that drive malignant B-cell lymphoma progression have allowed
medical chemists to create highly specific small-molecule
inhibitors (8,9). In addition, the effect that the tumor
microenvironment (TME) has on tumor survival, proliferation and
therapy resistance is being increasingly understood and appreciated
(10).
Among the most widely studied oncogenic pathways in
B-cell lymphomas is the B-cell receptor (BCR) signaling pathway,
which has emerged as a crucial player in the survival,
proliferation and trafficking of malignant B cells (11,12).
Inhibitors of the key kinases in the BCR signaling pathway,
including spleen tyrosine kinase (SYK), mammalian target of
rapamycin (mTOR), phosphoinositide 3′-kinase (PI3K) and Bruton's
tyrosine kinase (BTK), have become a focus of notable clinical
interest due to their striking clinical responses (13,14). A
certain degree of the efficacy of these agents is due to the
attenuation of BCR-dependent lymphoma-TME interactions. The present
review discusses the pivotal role of BCR signaling in the
integration of intrinsic and extrinsic determinants of TME-mediated
lymphoma survival and drug resistance, and examines the use of the
BTK inhibitor, ibrutinib, as an example of a promising targeted
therapy and future treatment strategy.
Overview of the BCR signaling pathway and
BTK
Normal B cells utilize the BCR pathway to promote
the proliferation, differentiation and function of cells, including
antibody production (15,16). A simplified version of the BCR pathway
and its role in lymphoma cell interactions with TME is illustrated
in Fig. 1. Recently, BCR signaling
has emerged as a pivotal pathway and is likely to be a key driver
of a number of B-cell lymphomas (17). Kinase inhibitors that target BCR
signaling have induced striking clinical responses (18). When it is activated, BCR recruits SYK
kinase from the cytoplasm to the perimembrane location to become
BCR signalosome (19). SRC then
phosphorylates SYK, and is auto-phosphorylated and activated prior
to interacting with and catalyzing the phosphorylation of several
other signaling molecules, including PLCγ2 (a lipase), BTK and
B-cell linker protein (an adaptor molecule) (20–22).
Phosphatidylinositol 4,5-bisphosphate is cleaved into
diacylglycerol and inositol triphosphate by activated PLCγ2, which
results in the mobilization of calcium and the activation of a
number of downstream signaling pathways, such as the AKT,
mitogen-activated protein kinase (MAPK) and nuclear factor-κB
(NF-κB) pathways (23). A number of
transcriptional factors are activated through these pathways, and
eventually, the cells undergo metabolic adaptation, resulting in
increased cell proliferation, survival and differentiation into
plasma or memory B cells, as well as antibody production (24,25). Most
recently, inhibitors of BCR signaling have become an area of
substantial clinical interest, particularly in chronic lymphocytic
leukemia (CLL), which is likely attributed to its role in BCR- and
chemokine-controlled integrin-mediated adhesion and the homing of
malignant B cells to lymph node and bone marrow microenvironments
that support growth and survival (26,27).
Collectively, these data support the fact that BCR activation not
only controls intrinsic survival pathways associated with B
lymphoma cells, but that it also regulates stroma-mediated
extrinsic lymphoma cell survival, and lymphoma cell homing and
interplay with the microenvironment. Targeting the BCR signal
pathway molecules will block the growth and survival signals
emanating from cell intrinsic mutations and the microenvironment,
and is therefore a promising therapeutic strategy for lymphoma
therapy.
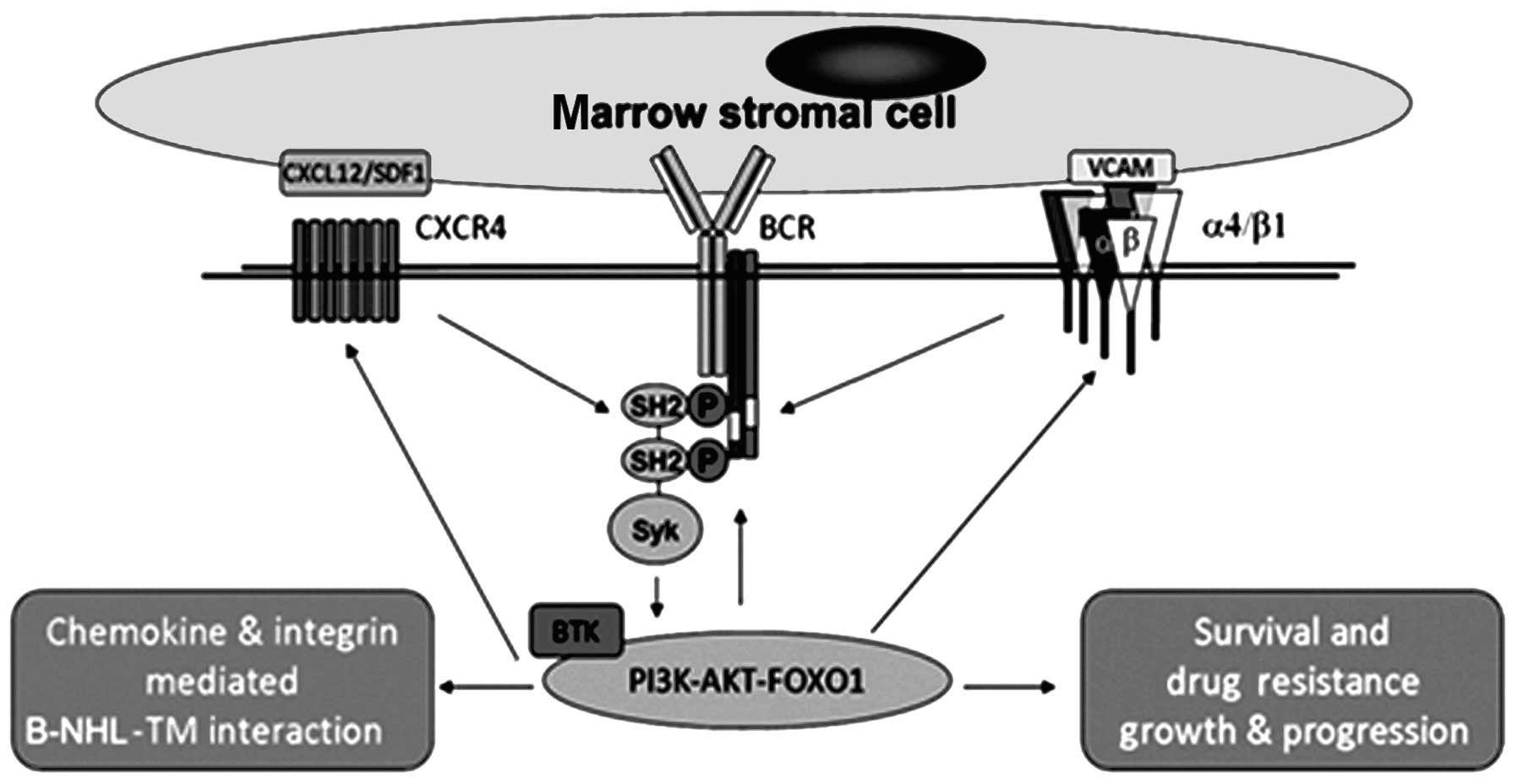 | Figure 1.BCR is a central mediator of
malignant B-cell homing, survival and microenvironment-mediated
drug resistance. BCR, B-cell receptor; SYK, spleen tyrosine kinase;
BTK, Bruton's tyrosine kinase; PI3K, phosphoinositide 3′-kinase;
SH2, Src Homology 2; CXCR4, chemokine (C-X-C motif) receptor 4;
CXCL12/SDF1, C-X-C motif chemokine ligand 12/stromal cell-derived
factor 1; VCAM, vascular cell adhesion protein 1; FOXO1, forkhead
box protein O1; P, phosphate; B-NHL-TM, B-cell-non-Hodgkin's
lymphoma and tumor microenvironment interaction. |
BTK is a member of the tyrosine-protein (Tec) family
of kinases, expressed in hematopoietic cells, particularly in B
cells, but not in T cells or normal plasma cells (28). BTK is activated by the upstream
Src-family and leads to the downstream activation of essential cell
survival pathways, such as NF-κB and MAPK. Furthermore, it plays an
important role in the signaling pathways of C-X-C motif chemokine
ligand 12-chemokine (C-X-C motif) receptor 4; (CXCL12-CXCR4),
B-cell activating factor (BAFF) receptor, Fcγ receptor (FcγR),
Toll-like receptor and receptor activator of NF-κB, which prompt
B-cell migration, adhesion, self-tolerance, inmmune activation and
cytokine secretion (29–32). Deficient BTK is associated with its
loss of function and reduced mature B cell numbers, and results in
X-linked agammaglobulinemia. Ibrutinib, an oral irreversible BTK
inhibitor, is designed to bond to a Cys481 residue within the BTK
active site, preventing the Tyr223 phosphorylation required for its
activation (33). The drug was
approved by the Food and Drug Administration in February 2013 for
several clinical trials on various forms of B-cell malignancies.
Notably, marked efficacy for ibrutinib has been noted thus far,
particularly in CLL/small lymphocytic lymphoma (SLL) and mantel
cell lymphoma (MCL).
Preclinical data on ibrutinib in the
treatment of B-cell malignancies
In vitro experiments revealed that ibrutinib
significantly inhibited CLL cell survival, DNA synthesis and
migration in response to tissue homing chemokines (CXCL12 and
CXCL13), and that it effectively blocked survival signals, which
are provided externally to CLL cells from the microenvironment
[CD40L, BAFF, interleukin (IL)-6, IL-4 and tumor necrosis factor-α]
(34,35). Ibrutinib also downregulated the
BCR-dependent chemokines [chemokine (C-C motif) ligand 3 (CCL3) and
CCL4] by CLL cell secretion (36,37).
Ibrutinib strongly inhibited the survival of malignant cells,
including CLL, MCL, diffuse large B-cell lymphoma (DLBCL),
follicular lymphoma (FL) and multiple myeloma (MM) cells, in in
vivo and in vitro experiments (38–40). In
addition, ibrutinib and bortezomib synergistically kill activated B
cell-like-DLBCL (ABC-DLBCL) or germinal-center B-cell-like DLBCL
(GC-DLBCL) cells and MCL cells, including those highly resistant to
bortezomib, but not normal cells (41). Furthermore, Rushworth et al
(42) reported for the first time
that ibrutinib treatment significantly augments the cytotoxic
activity of bortezomib and lenalidomide chemotherapies by
inhibiting the NF-κB pathway in malignant plasma cells from
patients with MM, which may provide a theoretical basis for future
combination therapy. Notably, ibrutinib was also shown to disrupt
the chemokine-induced adhesion and migration of primary chronic
leukemia B cells (43).
In a mouse xenograft in vivo model, ibrutinib
inhibited CLL progression. As in humans, the mice exhibited
transient lymphocytosis at day 4 and a reduction in tumor size,
demonstrating that CLL cells can be mobilized into peripheral blood
from lymphoid tissue by ibrutinib (33). This is likely as the protection from
the TME is prevented and indicates that ibrutinib may overcome the
drug resistance mediated by the microenvironment. As canine
non-Hodgkin's lymphoma (NHL) shares a number of characteristics
with human NHL, including diagnostic classifications and response
to cyclophosphamide, doxorubicin, vincristine and
prednisone/prednisolone regimen-based chemotherapy (44), Honigberg et al (45) treated treatment-naïve (TN) and
relapsed dogs with ibrutinib using the capsule formulation prepared
for human clinical trials. The study showed that ibrutinib led to
objective clinical responses [3 partial response (PR) and 3 stable
disease (SD)] in spontaneous canine B-cell lymphomas by response
evaluation criteria. Total Btk levels varied significantly across
samples, which may suggest heterogeneity in biopsy sampling in
tumor cells.
Taken together, these ex vivo and in
vivo studies demonstrated that BCR/BTK is a central mediator of
malignant B-cell homing, survival and microenvironment-mediated
drug resistance. Further support for this conclusion may be
observed in the marked responses of B-cell disorders treated with
ibrutinib alone or in combination with cytotoxic agents.
Clinical trials of ibrutinib in the
treatment of B-cell malignancies using monotherapy and combination
regimens
In a multi-cohort phase Ib/II trial of ibrutinib in
TN or relapsed/refractory (RR) CLL/SLL patients, 83% of patients
with RR disease and 96% of treatment-naïve patients, including
those with high-risk disease, were estimated to experience overall
survival (OS) times of 26 months (46). The study demonstrated that ibrutinib
may be the first-line treatment for previously untreated CLL
patients. Notably, the high- and low-dose groups achieved the same
efficacy in this trial, which suggested that the efficacy was not
positively correlated with the dose of ibrutinib but may be
associated with the complete suppression of BTK. Advani et
al (47) confirmed this
hypothesis and presented a study in which patients with RR B-cell
lymphoma and CLL received escalating oral doses of ibrutinib
(1.25–12.5 mg/kg per day). BTK occupancy >95% was observed in
dose level cohorts II to V (2.5–12.5 mg/kg per day), and each of
these cohorts experienced similar response rates, consistent with
the efficacy derived from BTK inhibition. Notably, during the first
treatment cycle, all CLL patients experienced rapid reductions in
lymphadenopathy accompanied by an increase in absolute lymphocyte
count, indicating that the malignant cells were moving from the
lymph nodes into the peripheral blood. The results suggested that
old response criteria for progressive disease based on
lymphocytosis may have to be modified, since lymphocytosis
associated with inhibitors targeting the BCR pathway is clearly not
a sign of disease progression. To this end, National Comprehensive
Cancer Network guidelines have eliminated progressive lymphocytosis
as a sign of disease progression when spleen and lymph node sizes
are reduced (48).
Following this, to confirm the efficacy of ibrutinib
in patients with RR MCL, Wang et al (49) conducted a phase II study, with 109 RR
MCL patients (63 bortezomib-naïve and 46 bortezomib-exposed). The
overall response rate (ORR) is 68% according to the International
Working Group response criteria (49), and in the study by Wang et al,
the ORR was 65% in the bortezomib-naive cohort and 72% in the
bortezomib-exposed cohort. The data from this phase II trial showed
that the single agent ibrutinib is highly active in R/R MCL.
Moreover, patients who received ibrutinib therapy following
bortezomib treatment experienced improved treatment efficacy,
suggesting that combining the drug with other therapies deserves
further study, which may be of great patient benefit. Staudt et
al (50) reported a study in
which 8 patients with RR ABC DLBCL were enrolled, with a complete
response (CR) recorded in 2 patients (25%), SD in 3 patients (37%)
and progressive disease in 3 patients (38%). Notably, 1 patient
with primary refractory disease achieved SD with ibrutinib,
associated with a 25% tumor reduction, and is currently in CR
following allogeneic bone marrow transplantation. CD79B mutations,
which cause chronic BCR signaling in ABC DLBCL, were revealed in 2
patients; 1 patient with SD who achieved a 25% tumor response and
another who achieved a CR. However, ibrutinib was shown to
significantly improve survival only in ABC DLBCL but not GC DLBCL
patients. Another phase 2 study also indicated that ibrutinib
showed preferential response activity in ABC versus GCB DLBCL
(Table I) (51). Thus, chronic active BCR signaling
maybe a significant therapeutic target in ABC DLBCL or other B-cell
malignancies. In FL, when ibrutinib was administered orally with
dose escalation in 16 patients with FL (52), the ORR was 54.5% (3 CR and 3 PR), the
duration of response was 12.3 months and the median
progression-free survival time was 13.4 months, showing promising
treatment efficacy for this disease. These clinical experiments
showed that the side effects of ibrutinib are well tolerated. Grade
1 or 2 diarrhea, fatigue and nausea have been the most frequently
reported adverse events (AEs). Grade 3 AEs included anemia,
anxiety, hypersensitivity, hypokalemia, hypophosphatemia and
decreased neutrophil count, while grade 4 hypokalemia was also
considered to be associated with ibrutinib (53,54). As
more experience is gained with this agent, the patients who will
benefit the most will be chosen. Further research is required not
only to identify the response biomarkers and the mechanism of
resistance, but also to understand how these agents may be
rationally combined.
 | Table I.Basic characteristics of lymphoma and
efficacy of ibrutinib. |
Table I.
Basic characteristics of lymphoma and
efficacy of ibrutinib.
| Diseases | Patients, n | Age, years | Doses | AE | ORR | Study phase |
|---|
| CLL | 61 | ≥65 | 420 or 840 mg | Diarrhea, fatigue,
rash; serious AEs occurred in 10% of patients | 70% (420-mg cohort)
63% (840-mg cohort) | Ib/II |
| MCL | 109 | 68
(40–84)a | 560 mg (po qd) | Diarrhea, fatigue,
upper respiratory tract infection, dyspneaoedema peripheral, grade
5 pneumonia | 65%
(bortezomib-naïve) 67% (bortezomib-exposed) | II |
| DLBCL | 70 | 60
(41–71)a | 560 mg (po qd) | AEs consistent with
that reported in other ibrutinib studies | 40% (ABC subtype)
5% (GCB subype) | II |
| FL | 16 | 60
(41–71)a | 1.25–12.5
mg/kg | Diarrhea, fatigue
nausea, coughing | 54.5% | I |
The most comprehensive study of ibrutinib has been
performed in CLL. Results from a phase 1/2 trial suggested that
high- and low-risk CLL patients respond equally as well to
ibrutinib (55). CLL patients treated
with single-agent ibrutinib therapy characteristically exhibit
delayed responses or SD. To accelerate and improve these responses,
a phase 2 single-center clinical trial of ibrutinib plus rituximab
was conducted, which accrued 40 patients. In total, 32 patients
with unmutated immunoglobulin heavy chain variable, 20 patients
with del17p or tumor protein p53 mutation (4 without prior therapy)
and 13 patients with del11q were enrolled. From the 20 patients in
whom an early response assessment could be evaluated at 3 months,
17 patients achieved a partial remission for an ORR of 85%, and 3
achieved a PR. However, in this combination trial, it was noted
that the re-distribution lymphocytosis peaked earlier and with a
shorter duration compared with single-agent ibrutinib (42), which was possibly due to the addition
of rituximab. Treatment was well tolerated, with diarrhea, bone
pain and fatigue as the most frequent side effects. In addition,
another study (56) showed that
ibrutinib in combination with ofatumumab is well-tolerated and
highly active in patients with RR CLL (ORR, 100%) irrespective of
prognostic markers (Table II). A
further study (57) was performed to
test ibrutinib in combination with a bendamustine and rituximab
regimen (BR) or a fludarabine/Cytoxan/Rituxan regimen (FCR) in
patients with RR CLL. The results indicated that at a median
follow-up time of 4.9 months, 16 patients had completed BR therapy
and 14 patients were still receiving BR therapy, with an ORR of 93%
(28/30 patients; CR, 13% and PR 80%). The responses appeared to be
independent of high-risk clinical or genomic features. The majority
of patients (77%) remained a part of the study at the time the data
was presented. While only 3 patients were included in the
FCR+ibrutinib cohort, the therapy was well tolerated, with only one
serious AE, which was neutropenic fever. The ORR was 100% (3/3) and
the 2 confirmed minimal residual disease (MRD)-negative cases
achieved CRs (57).
| Table II.Comparison of the efficacy of
ibrutinib in monotherapy and combination therapy. |
Table II.
Comparison of the efficacy of
ibrutinib in monotherapy and combination therapy.
| Study | n | ORR, % | CR, % | PFS |
|---|
| Single agent |
|
|
|
|
| RR | 61 | 67 | 4 | 88% at 18
months |
| TN | 31 | 74 | 10 | 96% at 15
months |
| Combination |
|
|
|
|
|
PCI+BR | 30 | 93 | 13 | 90% at 11
months |
|
PCI+FCR | 3 | 100 | 67 | 100% at
11 monthsa |
|
PCI+ofatumumab | 27 | 100 | 4 | 89% at 11
monthsa |
Although the number of patients receiving
combination therapy and the number of associated clinical
experiments is small, the benefits that CLL patients have gained is
clear. Next, the optimal combination strategies must be chosen
according to the accurate molecular classification of patients or
other criteria, such as disease progression, leading to
personalized therapy. The clinical data on combination therapy
using ibrutinib in patients with MCL, DLBCL, MM and FL is limited;
however, these pre-clinical experiments on CLL provide the
rationale for using a combination strategy for other types of B
cell malignancies.
Conclusions
Over the last three years, the progress in the study
of pathological BCR signaling in lymphoma has resulted in the rapid
development of BCR pathway inhibitors. The compounds that have been
developed furthest in the clinic are the inhibitors of SYK, mTOR,
PI3K-δ and BTK. To date, marked success has been achieved by these
therapeutic agents in the treatment of patients with B cell
malignancies who, in a number of cases, were resistant to
conventional chemotherapeutic agents. Furthermore, the side effect
profile of BCR-targeted therapies appears to be easily manageable.
However, despite high ORRs for BCR pathway inhibitors, a
substantial minority of patients is unresponsive or shows
progression relatively soon after the commencement of therapy.
Therefore, the identification of molecular mechanisms that may
predict and sustain the response to BCR pathway inhibitors would be
beneficial in order to overcome drug resistance. The BCR
inhibitors, as discussed for the aforementioned BTK inhibitor,
target the extracellular and intracellular determinants of the bone
marrow niche. The aim of associated studies is to identify the
appropriate factors to target within the complex network of the
tumor cell microenvironment. To this end, therapies that are able
to overcome the coordinated effort between lymphoma cells and the
microenvironment are required. In this way, the sequence of events
(de novo and acquired) facilitating MRD and ending in
therapy resistance may be interrupted. Going forward, it is
important to note that with the significant heterogeneity of
signaling factors and transduction pathways within the TME (bone
marrow and lymph node) niche, the designing of combination
therapies with targeted agents is required. To this end, the
targeting of multiple pathways, either simultaneously or in
sequence, may be the only measure by which to overcome the
sanctuary of the TME milieu, since these pathways act in concert in
lymphomagenesis. It will be critical to merit the combined
targeting of downstream (BCR) signaling pathways to maintain the
advantage of direct modulation of the cell survival and
proliferation machinery, with targeting of upstream or parallel
pathways to circumvent the compensatory survival pathway. As a
result, the success of BCR inhibitor therapy in B-cell lymphoma
therapy will be dependent on the use of rational combinations of
targeted agents, and good knowledge of the nature of signaling
pathways and their interactions with the TME.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 30672208 and
81270603).
References
|
1
|
Younes A: Beyond chemotherapy: New agents
for targeted treatment of lymphoma. Nat Rev Clin Oncol. 8:85–96.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Manzur S, Cohen S, Haimovich J and
Hollander N: Enhanced therapeutic effect of B cell-depleting
anti-CD20 antibodies upon combination with in-situ dendritic cell
vaccination in advanced lymphoma. Clin Exp Immunol. 170:291–299.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Friedberg JW: New strategies in diffuse
large B-cell lymphoma: Translating findings from gene expression
analyses into clinical practice. Clin Cancer Res. 17:6112–6117.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Timmerman JM, Byrd JC, Andorsky DJ, et al:
A phase I dose-finding trial of recombinant interleukin-21 and
rituximab in relapsed and refractory low grade B-cell
lymphoproliferative disorders. Clin Cancer Res. 18:5752–5760. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fowler N and Oki Y: Developing novel
strategies to target B-cell malignancies. Am Soc Clin Oncol Educ
Book. 366–372. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ramsay AD and Rodriguez-Justo M: Chronic
lymphocytic leukaemia-the role of the microenvironment pathogenesis
and therapy. Br J Haematol. 162:15–24. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gupta P, Goldenberg DM, Rossi EA and Chang
CH: Multiple signaling pathways induced by hexavalent,
monospecific, anti-CD20 and hexavalent, bispecific, anti-CD20/CD22
humanized antibodies correlate with enhanced toxicity to B-cell
lymphomas and leukemias. Blood. 116:3258–3267. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roschewski M, Dunleavy K and Wilson WH:
Diffuse large B cell lymphoma: Molecular targeted therapy. Int J
Hematol. 96:552–561. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kenkre VP and Kahl BS: The future of
B-cell lymphoma therapy: The B-cell receptor and its downstream
pathways. Curr Hematol Malig Rep. 7:216–220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sison EA, Rau RE, McIntyre E, Li L, Small
D and Brown P: MLL-rearranged acute lymphoblastic leukaemia stem
cell interactions with bone marrow stroma promote survival and
therapeutic resistance that can be overcome with CXCR4 antagonism.
Br J Haematol. 160:785–797. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen L, Monti S, Juszczynski P, et al: SYK
inhibition modulates distinct PI3K/AKT-dependent survival pathways
and cholesterol biosynthesis in diffuse large B cell lymphomas.
Cancer Cell. 23:826–838. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Irish JM, Myklebust JH, Alizadeh AA, et
al: B-cell signaling networks reveal a negative prognostic human
lymphoma cell subset that emerges during tumor progression. Proc
Natl Acad Sci USA. 107:12747–12754. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Young RM and Staudt LM: Targeting
pathological B cell receptor signalling in lymphoid malignancies.
Nat Rev Drug Discov. 12:229–243. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shain KH and Tao J: The B-cell receptor
orchestrates environment-mediated lymphoma survival and drug
resistance in B-cell malignancies. Oncogene. 33:4107–4113. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rickert RC: New insights into pre-BCR and
BCR signalling with relevance to B cell malignancies. Nat Rev
Immunol. 13:578–591. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Choi MY and Kipps TJ: Inhibitors of B-cell
receptor signaling for patients with B-cell malignancies. Cancer J.
18:404–410. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stevenson FK, Krysov S, Davies AJ, et al:
B-cell receptor signaling in chronic lymphocytic leukemia. Blood.
118:4313–4320. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen L, Huynh L, Apgar J, et al: ZAP-70
enhances IgM signaling independent of its kinase activity in
chronic lymphocytic leukemia. Blood. 111:2685–2692. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Depoil D, Fleire S, Treanor BL, et al:
CD19 is essential for B cell activation by promoting B cell
receptor-antigen microcluster formation in response to
membrane-bound ligand. Nat Immunol. 9:63–72. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Niiro H and Clark EA: Regulation of B-cell
fate by antigen-receptor signals. Nat Rev Immunol. 2:945–956. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Humphries LA, Dangelmaier C, Sommer K, et
al: Tec kinases mediate sustained calcium influx via site-specific
tyrosine phosphorylation of the phospholipase Cgamma Src homology
2-Src homology 3 linker. J Biol Chem. 279:37651–37661. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hashimoto A, Okada H, Jiang A, et al:
Involvement of guanosine triphosphatases and phospholipase C-gamma2
in extracellular signal-regulated kinase, c-Jun NH2-terminal
kinase, and p38 mitogen-activated protein kinase activation by the
B cell antigen receptor. J Exp Med. 188:1287–1295. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shinohara M, Koga T, Okamoto K, et al:
Tyrosine kinases Btk and Tec regulate osteoclast differentiation by
linking RANK and ITAM signals. Cell. 132:794–806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Davis RE, Ngo VN, Lenz G, et al: Chronic
active B-cell-receptor signalling in diffuse large B-cell lymphoma.
Nature. 463:88–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Khan WN: Regulation of B lymphocyte
development and activation by Bruton's tyrosine kinase. Immunol
Res. 23:147–156. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hamblin TJ, Davis Z, Gardiner A, Oscier DG
and Stevenson FK: Unmutated Ig V(H) genes are associated with a
more aggressive form of chronic lymphocytic leukemia. Blood.
94:1848–1854. 1999.PubMed/NCBI
|
|
27
|
Gobessi S, Laurenti L, Longo PG, et al:
ZAP-70 enhances B-cell-receptor signaling despite absent or
inefficient tyrosine kinase activation in chronic lymphocytic
leukemia and lymphoma B cells. Blood. 109:2032–2039. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Genevier HC, Hinshelwood S, Gaspar HB, et
al: Expression of Bruton's tyrosine kinase protein within the B
cell lineage. Eur J Immunol. 24:3100–3105. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vargas L, Hamasy A, Nore BF and Smith CI:
Inhibitors of BTK and ITK: State of the new drugs for cancer,
autoimmunity and inflammatory diseases. Scand J Immunol.
78:130–139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
O'Hayre M, Salanga CL, Kipps TJ, et al:
Elucidating the CXCL12/CXCR4 signaling network in chronic
lymphocytic leukemia through phosphoproteomics analysis. PLoS One.
5:e117162010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Redondo-Muñoz J, Escobar-Díaz E, Samaniego
R, et al: MMP-9 in B-cell chronic lymphocytic leukemia is
up-regulated by alpha4beta1 integrin or CXCR4 engagement via
distinct signaling pathways, localizes to podosomes, and is
involved in cell invasion and migration. Blood. 108:3143–3151.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Richards KL, Motsinger-Reif AA, Chen HW,
et al: Gene profiling of canine B-cell lymphoma reveals germinal
center and postgerminal center subtypes with different survival
times, modeling human DLBCL. Cancer Res. 73:5029–5039. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Herman SE, Sun X, McAuley EM, et al:
Modeling tumor-host interactions of chronic lymphocytic leukemia in
xenografted mice to study tumor biology and evaluate targeted
therapy. Leukemia. 27:2311–2321. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chang BY, Huang MM, Francesco M, et al:
The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates
autoimmune arthritis by inhibition of multiple effector cells.
Arthritis Res Ther. 13:R1152011. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee KG, Xu S, Wong ET, Tergaonkar V and
Lam KP: Bruton's tyrosine kinase separately regulates NFkappaB
p65RelA activation and cytokine interleukin (IL)-10/IL-12
production in TLR9-stimulated B cells. J Biol Chem.
283:11189–11198. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mráz M, Doubek M and Mayer J: Inhibition
of B cell receptor signaling: A first targeted therapeutic approach
for chronic lymphocytic leukemia and other B cell lymphomas. Klin
Onkol. 26:179–185. 2013.(In Czech). View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tai YT, Chang BY, Kong SY, et al: Bruton
tyrosine kinase inhibition is a novel therapeutic strategy
targeting tumor in the bone marrow microenvironment in multiple
myeloma. Blood. 120:1877–1887. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Herman SE, Gordon AL, Hertlein E, et al:
Bruton tyrosine kinase represents a promising therapeutic target
for treatment of chronic lymphocytic leukemia and is effectively
targeted by PCI-32765. Blood. 117:6287–6296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schwamb J, Feldhaus V, Baumann M, et al:
B-cell receptor triggers drug sensitivity of primary CLL cells by
controlling glucosylation of ceramides. Blood. 120:3978–3985. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang Y, Shaffer AL III, Emre NC, et al:
Exploiting synthetic lethality for the therapy of ABC diffuse large
B cell lymphoma. Cancer Cell. 21:723–737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dasmahapatra G, Patel H, Dent P, et al:
The Bruton tyrosine kinase (BTK) inhibitor PCI-32765
synergistically increases proteasome inhibitor activity in diffuse
large-B cell lymphoma (DLBCL) and mantle cell lymphoma (MCL) cells
sensitive or resistant to bortezomib. Br J Haematol. 161:43–56.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rushworth SA, Bowles KM, Barrera LN, et
al: BTK inhibitor ibrutinib is cytotoxic to myeloma and potently
enhances bortezomib and lenalidomide activities through NF-κB. Cell
Signal. 25:106–112. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
de Rooij MF, Kuil A, Geest CR, et al: The
clinically active BTK inhibitor PCI-32765 targets B-cell receptor-
and chemokine-controlled adhesion and migration in chronic
lymphocytic leukemia. Blood. 119:2590–2594. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sabine P, Balasubramanian S, Pham LV, et
al: Activity of Bruton's tyrosine kinase (Btk) inhibitor PCI-32765
in mantle cell lymphoma (MCL) identifies Btk as a novel therapeutic
target. Blood (ASH Annual Meeting Abstracts). 118:36882011.
|
|
45
|
Honigberg LA, Smith AM, Sirisawad M, et
al: The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell
activation and is efficacious in models of autoimmune disease and
B-cell malignancy. Proc Natl Acad Sci USA. 107:13075–13080. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
O'Brien SMD, Furman RR, Coutre SE, et al:
Ibrutinib as initial therapy for elderly patients with chronic
lymphocytic leukaemia or small lymphocytic lymphoma: An open-label,
multicentre, phase 1b/2 trial. Lancet Oncol. 15:48–58. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Advani RH, Buggy JJ, Sharman JP, et al:
Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has
significant activity in patients with relapsed/refractory B-cell
malignancies. J Clin Oncol. 31:88–94. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Byrd JC, Furman RR, Coutre SE, et al:
Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic
Leukemia. N Engl J Med. 369:32–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang L, Martin P, Blum KA, et al: The
Bruton's tyrosine kinase inhibitor PCI-32765 is highly active as
single-agent therapy in previously-treated mantle cell lymphoma
(MCL): Preliminary results of a phase II trial. Blood (ASH Annual
Meeting Abstracts). 118:4422011.
|
|
50
|
Staudt LM, Dunleavy K, Buggy JJ, et al:
The Bruton's tyrosine kinase (BTK) inhibitor PCI-32765 modulates
chronic active BCR signaling and induces tumor regression in
relapsed/refractory ABC DLBCL. Blood (ASH Annual Meeting
Abstracts). 118:31742011.
|
|
51
|
Wilson WH, Gerecitano JF, Goy A, et al:
The Bruton's tyrosine kinase (BTK) inhibitor, Ibrutinib
(PCI-32765), Has Preferential activity in the ABC subtype of
relapsed/refractory de novo diffuse large B-cell lymphoma (DLBCL):
Interim results of a multicenter, open-label, phase 2 study. Blood
(ASH Annual Meeting Abstracts). 120:6862012.
|
|
52
|
Fowler NH, Advani RH, Sharman JP, et al:
The Bruton's tyrosine kinase inhibitor ibrutinib (PCI-32765) is
active and tolerated in relapsed follicular lymphoma. Blood (ASH
Annual Meeting Abstracts). 120:1562012.
|
|
53
|
Jaglowski SM, Jones JA, Flynn JM, et al: A
phase Ib/II study evaluating activity and tolerability of BTK
inhibitor PCI-32765 and ofatumumab in patients with chronic
lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and
related diseases. J Clin Oncol (2012 ASCO Annual Meeting).
30:65082012.
|
|
54
|
O'Brien S, Burger JA, Blum KA, et al: The
Bruton's tyrosine kinase (BTK) inhibitor PCI-32765 induces durable
responses in relapsed or refractory (R/R) chronic lymphocytic
leukemia/small lymphocytic lymphoma (CLL/SLL): Follow-up of a phase
Ib/II study. Blood (ASH Annual Meeting Abstracts). 120:9832011.
|
|
55
|
Burger JA, Keating MJ, Wierda WG, et al:
Safety and activity of ibrutinib plus rituximab for patients with
high-risk chronic lymphocytic leukaemia: A single-arm, phase 2
study. Lancet. 15:1090–1099. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
O'Brien SM, Barrientos JC, Flinn IW, et
al: Combination of the Bruton's tyrosine kinase (BTK) inhibitor
PCI-32765 with bendamustine(B)/rituximab (R) (BR) in patients (pts)
with relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL):
Interim results of a phase Ib/IIstudy. J Clin Oncol (2012 ASCO
Annual Meeting). 30:65152012.
|
|
57
|
Brown JRBJ, Flinn I, et al: The Bruton's
tyrosine kinase (BTK) inhibitor ibrutinib combined with
bendamustine and rituximab is active and tolerable in patients with
relapsed/refractory CLL, interim results of a phase Ib/II study.
EHA Meeting Abstracts. 97:2012.
|














