Introduction
Breast cancer is the leading cause of
cancer-associated mortality in women worldwide (1). Breast cancer is a heterogeneous disease
that is characterized by various molecular subtypes, which exhibit
distinct molecular characteristics and clinical behaviors (2,3). Although
improvements in the early detection and treatment of breast cancer
have decreased the associated mortality rates in previous years,
clarification of the molecular and cellular mechanisms underlying
the development and progression of breast cancer continues to be
required (2).
An increasing number of studies have revealed that
malignant tumor progression involves multiple genetic and
epigenetic changes, each of which may result in the deregulation of
important etiology-specific pathways involved in complex cellular
processes, such as proliferation, migration, invasion and apoptosis
(4–6).
The accumulation of these genetic and epigenetic alterations
confers a malignant phenotype (7).
microRNAs (miRNA) are a class of small, endogenously
expressed non-coding RNAs comprised of 18–25 nucleotides. miRNAs
induce the silencing of the cognate target genes by either
degrading the target messenger RNA (mRNA) or repressing translation
(8). Previous studies have revealed
that miRNA plays a critical role in the regulation of various
cellular processes, including cell growth and metastasis, which
indicated that miRNA may function either as an oncogene or as a
tumor suppressor (9,10).
The cylindromatosis gene CYLD was initially
identified as a mutated gene in familial cylindromatosis (11). CYLD contains an ubiquitin C-terminal
hydrolase domain, which allows the protein to function as a
deubiquitinating enzyme (12). CYLD
has been revealed to play a central role in regulating various
signaling pathways, including transforming growth factor-β,
Wnt/β-catenin, c-Jun N-terminal kinase and nuclear factor-κB
(NF-κB) signaling, and thus regulates the promotion of cancer
development and progression (13–16).
Furthermore, the downregulation of CYLD has been reported in
several malignancies (17–20). Previous studies have revealed that
CYLD expression is downregulated in breast cancer tissues compared
with normal breast tissues (2,17). In
addition, the downregulation of CYLD promotes cell survival and
migratory activities through NF-κB activation (21). However, the precise molecular
mechanisms underlying the deregulation of CYLD expression in breast
cancer are not fully understood.
In the present study, miR-362-5p was investigated as
it is known to be overexpressed in breast cancer cells, and CYLD
was revealed to be a target of miRNA-362-5p (miR-362-5p) by
TargetScan. To the best of our knowledge, the roles of miR-362-5p
and the targets of miR-362-5p in breast cancer have not yet been
reported. In this study, the inhibition of miR-362-5p was revealed
to reduce the proliferation, migration and invasion of breast
cancer cells. Therefore, the present results suggest a potential
medical value of the miR-362-5p/CYLD axis in effective breast
cancer therapy.
Materials and methods
Cell lines and cell culture
The human breast cancer MDA-MB-231 and MCF7, normal
breast fibroblast CCD-1095Sk and human embryonic kidney HEK293 cell
line were purchased from the American Type Culture Collection
(Manassas, VA, USA). The MCF-7 cells were cultured in RPMI-1640
medium, and the MDA-MB-231 and HEK293 cells were cultured in
Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (Hyclone; GE Healthcare Life Sciences, Little
Chalfont, UK).
RNA isolation and reverse
transcription-polymerase chain reaction (RT-PCR) analysis
Total RNA was isolated with TRIzol reagent
(Invitrogen, Carlsbad, CA, USA), according to the manufacturer's
protocol. For miRNA analysis, equal amounts of RNA were reverse
transcribed using a miRNA-specific primer (Hsa-miR-362-5p; Qiagen,
Venlo, Limburg, Netherlands) and then subjected to RT-PCR,
according to the manufacturer's protocol for the miScript Reverse
Transcription and miScript SYBR Green PCR kits (Qiagen). RNU6B
small nuclear RNA was used as an internal control. For CYLD mRNA
analysis, RNA was reverse-transcribed using Moloney murine leukemia
virus reverse transcriptase (Invitrogen) and random primers (Roche,
Basel, Switzerland). RT-PCR was performed using SYBR Premix Ex Taq
II (Takara Biotechnology Co., Ltd., Dalian, Liaoning, China). The
PCR primers for CYLD were as follows: Forward,
5′-TCAGGCTTATGGAGCCAAGAA-3′; and reverse,
5′-ACTTCCCTTCGGTACTTTAAGGA-3′. GAPDH mRNA levels were used as an
internal normalization control. The cycle threshold method was used
to analyze the expression of miR-362-5p and CYLD relative to GAPDH
expression.
Cell proliferation, cell cycle,
apoptosis analysis and colony formation assays
The cell proliferation was measured using an MTT
assay. The cells were seeded in 96-well plates at a density of
4,000 cells per well and maintained in a culture medium containing
5% fetal bovine serum for 48 h. The absorbance was then measured at
490 nm. The cell cycle analysis was conducted with a FACSCalibur
flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) using a
propidium iodide cell cycle detection kit (Beyotime Institute of
Biotechnology, Beijing, China). The apoptosis assay was performed
using the Annexin V-phycoerythrin (PE) Apoptosis Detection kit I
(BD Biosciences), according to the manufacturer's instructions, and
apoptosis was analyzed using a FACSCalibur flow cytometer. The
apoptotic cells were indicated by high Annexin V-PE fluorescence
signals. For the colony formation assays, 100 cells were placed
into each well of a 6-well plate and cultured for two weeks at
37°C. The numbers of colonies per dish were counted subsequent
staining with 0.1% crystal violet. All experiments were conducted
with three replicates.
Wound-healing migration and invasion
assays
For the wound-healing migration assay, the cells
were seeded onto 35-mm dishes coated with fibronectin. Once the
cells reached 100% confluency, a sterile p200 pipette tip was used
to create a scratch (~500 µm) through the confluent monolayer. The
medium was changed to fresh serum-free medium to remove the
cellular debris, and the cells were cultured for another 48 h.
Serial images were obtained at 0, 24 and 48 h. For the invasion
assay, 1×104 cells were placed into the upper chamber of
the Matrigel Transwell chamber (BD Biosciences) in serum-free
medium. Medium containing 10% FBS in the lower chamber acted as the
chemoattractant. Subsequent to 64 h of incubation at 37°C, the
invasive cells attached to the lower membrane of the inserts were
fixed, stained and then counted using a counting chamber and
microscope (CX31; Olympus, Tokyo, Japan).
Western blotting
The cell cytoplasm or nucleus lysates subsequent to
transfection with the miR-362-5p mimic, inhibitor or negative
control miRNA (Shanghai GenePharma Co., Ltd., Shanghai, China) were
separated by SDS-PAGE and then transferred onto a polyvinylidene
fluoride membrane (Millipore, Bedford, MA, USA). The blotted
membranes were incubated with rabbit anti-human polyclonal CYLD
(cat. no. 11110-1-AP; dilution, 1:1,000; ProteinTech Group, Inc.,
Chicago, IL, USA), rabbit anti-human polyclonal NF-κB P65 (cat. no.
sc-372; dilution, 1:1,000; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA), rabbit anti-human monoclonal anti-Lamin B1 (cat. no.
12586; dilution, 1:1,500; Cell Signaling Technology, Inc., Danvers,
MA, USA) and monoclonal mouse anti-human β-actin (cat. no. A5316;
dilution, 1:5,000; Sigma-Aldrich, St. Louis, MO, USA) antibodies.
The membrane was then incubated with the secondary goat anti-rabbit
horseradish peroxidase-conjugated antibody (cat. no. sc-2004;
dilution, 1:5,000; Santa Cruz Biotechnology, Inc.). The
immunoreactive protein bands were developed using the Enhanced
Chemiluminescence System (Pierce Biotechnology, Inc., Rockford, IL,
USA).
Dual-luciferase reporter assay
A fragment of the 3′-UTR of the CYLD gene that
contained the miR-362-5p target sequence (CAAGGAT) was inserted
into XhoI/NotI-digested psiCHECK-2 vectors (Promega,
Madison, WI, USA). The insertion of the sequences was confirmed by
sequencing. The psiCHECK-2 constructs and miR-362-5p mimic or
negative control miRNA were cotransfected into HEK293 cells using
the Lipofectamine 2000 transfection reagent (Invitrogen), according
to the manufacturer's instructions. Subsequent to 48 h, the cells
were lysed, and the luciferase reporter assay was performed using
the Dual-Luciferase Reporter Assay kit (Promega), according to the
manufacturer's instructions. The results were expressed as the
ratio of Renilla luciferase activity to firefly luciferase
activity.
Statistical analysis
The data are expressed as the mean ± standard error
of the mean from at least three independent experiments. Unless
otherwise noted, the differences between groups were analyzed using
two-sided Student's t-tests for two groups or by one-way analysis
of variance (ANOVA) when more than two groups were compared.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Inhibitory effects of anti-miR-362-5p
on the proliferation of MCF7 cells
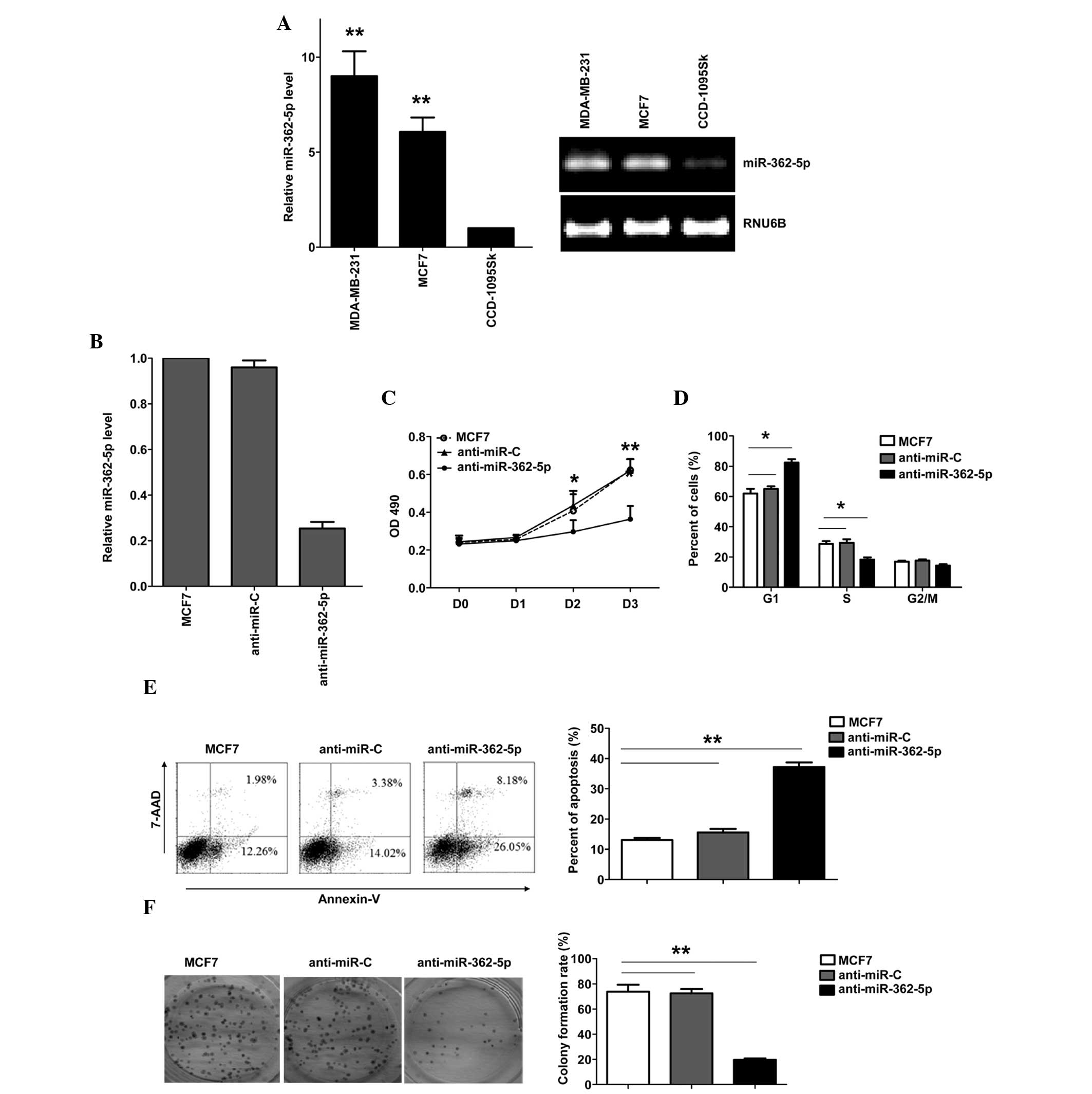
To evaluate the expression and importance of
miR-362-5p in breast cancer, the expression of miR-362-5p was
identified in human breast cancer MDA-MB-231 and MCF7 cell lines,
and normal breast fibroblast CCD-1095Sk cell line. miR-362-5p was
upregulated in breast cancer cell lines compared with CCD-1095Sk
cells (Fig. 1A). To explore the
biological significance of miR-26a in breast cancer cells, a
miR-362-5p inhibitor (anti-miR-362-5p) was transfected into the
human breast cancer MCF7 cell line. The expression of miR-362-5p
was verified by RT-qPCR (Fig. 1B).
Downregulation of miR-362-5p in the MCF7 cells resulted in a
significant suppression of cell proliferation, G1 arrest and
apoptosis induction. As shown in Fig.
1C, the results of the MTT assay revealed that downregulation
of miR-362-5p significantly suppressed MCF7 cell proliferation
(Fig. 1C). The cell cycle was
arrested in the G1 phase, with 82.3% of the
anti-miR-362-5p-transfected cells in the G0/G1 phase, compared with
65% of the control cells (Fig. 1D).
In addition, the transfection of anti-miR-362-5p induced apoptosis,
with 37.2% of anti-miR-362-5p-transfected cells demonstrating
apoptosis, compared with 15.5% of the cells in the control group
(Fig. 1E). Additional analysis of the
effects of anti-miR-362-5p on the clonogenicity of breast cancer
cells was performed, and it was found that the inhibition of
miR-362-5p significantly decreased the colony formation of MCF7
cells (Fig. 1F). These results
demonstrate that miR-362-5p regulates the proliferation of MCF7
cells.
Inhibition of miR-362-5p reduces the
migration and invasion of MCF7 cells
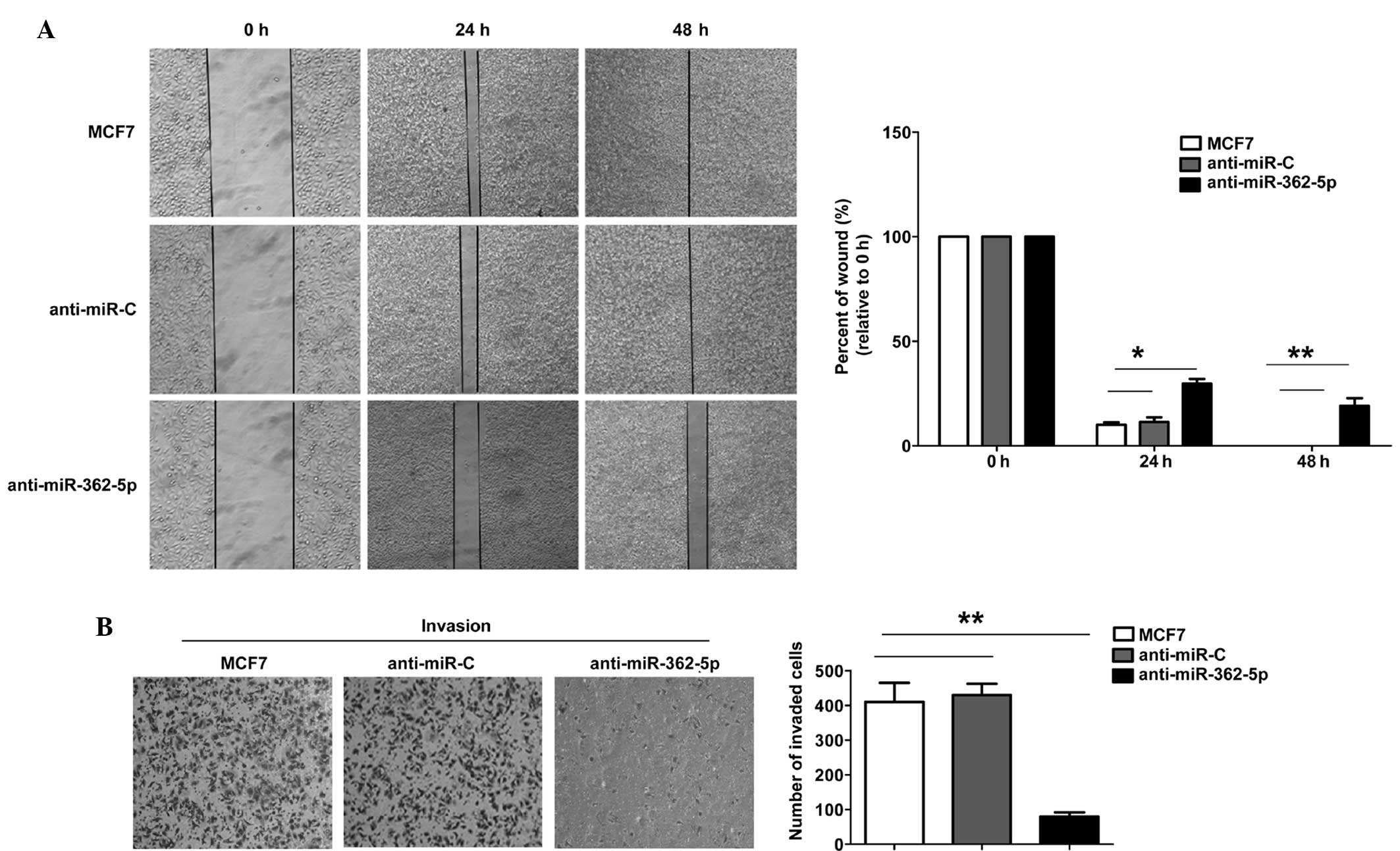
To further determine the biological significance of
miR-362-5p in breast cancer cell metastasis, wound-healing
migration and Transwell assays were performed using the MCF7 cells.
The mobility of the MCF7 cells in the wound-healing assay was
significantly decreased subsequent to transfection with
anti-miR-362-6p miRNA (Fig. 2A). The
Transwell assay with Matrigel demonstrated that the inhibition of
miR-362-5p markedly inhibited the invasive capacity of MCF7 cells
compared with the control cells (Fig.
2B). These results suggest that anti-miR-362-5p significantly
inhibits the migration and invasion of MCF7 cells.
CYLD is the direct downstream target
of miR-362-5p
The aforementioned phenotypic data indicate that the
inhibition of miR-362-5p reduces the growth and migration of breast
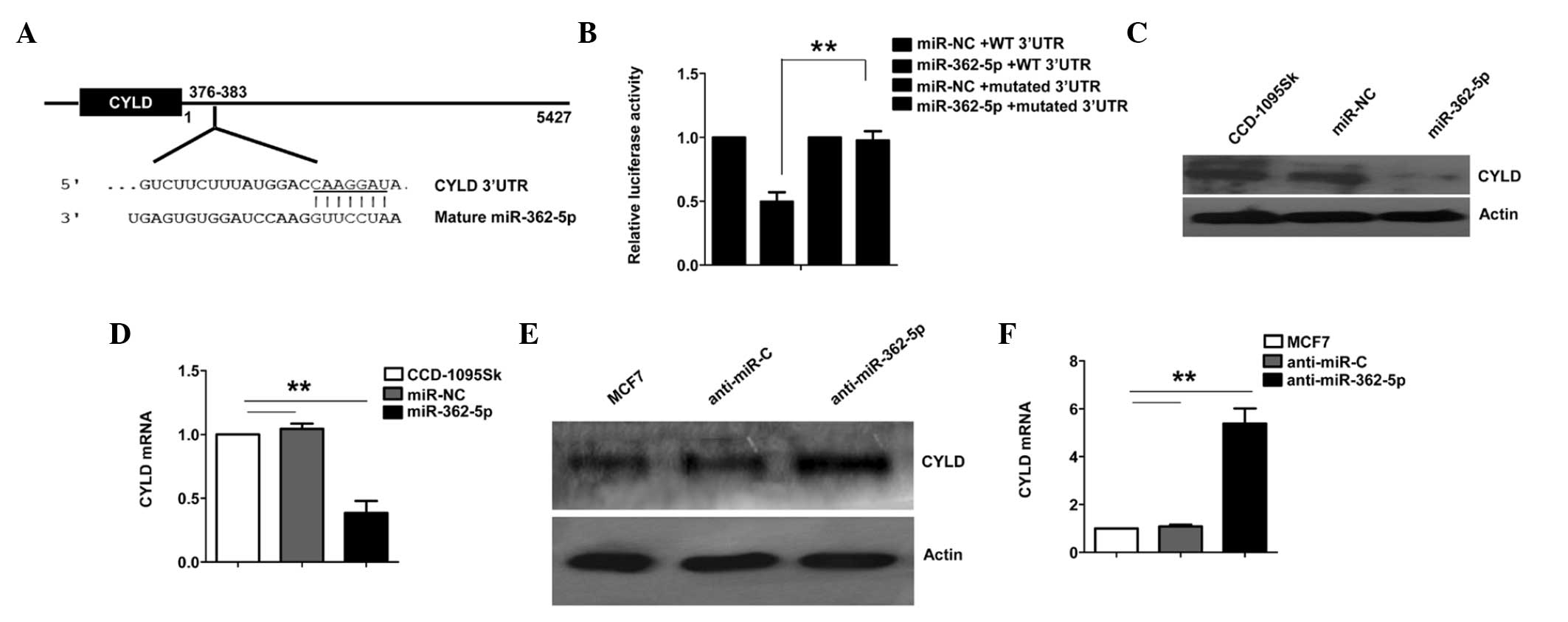
cancer cells. TargetScan was used to predict the mRNA targets of
miR-362-5p, to identify potential mRNA targets that may contribute
to its tumor-associated function. The present analysis revealed
that CYLD was a potential target of miR-362-5p. The 3′-UTR of CYLD
mRNA was found to contain a complementary site for the seed region
of miR-362-5p (Fig. 3A). To determine
whether CYLD is a direct target of miR-362-5p, a human CYLD 3′-UTR
fragment containing the wild-type (WT 3′-UTR) or mutant (mutated
3′-UTR) miR-362-5p-binding sequences was cloned into the psiCHEK-2
vector (Fig. 3A). As shown in
Fig. 3B, the relative luciferase
activity of the reporter containing WT 3′-UTR was significantly
suppressed following miR-362-5p transfection. However,
site-directed deletion of the miR-362-5p-binding site within the
3′-UTR of CYLD completely abolished this suppression (Fig. 3B), suggesting that miR-362-5p directly
binds to this site.
Furthermore, the ability of miR-362-5p to regulate
CYLD expression in human breast cells was tested. Normal breast
fibroblast CCD-1095Sk cells, which demonstrate a low miR-362-5p
expression level, were transfected with the miR-362-5p mimic or
control miRNA and were cultured for 48 h. The results demonstrated
that the CYLD mRNA and protein levels were decreased in CCD-1095Sk
cells transfected with the miR-362-5p mimic (Fig. 3C and D). The ability of endogenous
miR-362-5p to regulate CYLD expression was then assessed in breast
cancer cells. Compared with the miRNA control, the CYLD mRNA and
protein levels were upregulated when miR-362-5p was knocked down
using anti-miR-362-5p in MCF7 cells (Fig.
3E and F). Overall, these results strongly indicate that CYLD
is a direct target of miR-362-5p in breast cancer cells.
miR-362-5p promotes cell
proliferation, migration and invasion through the NF-κB signaling
pathway
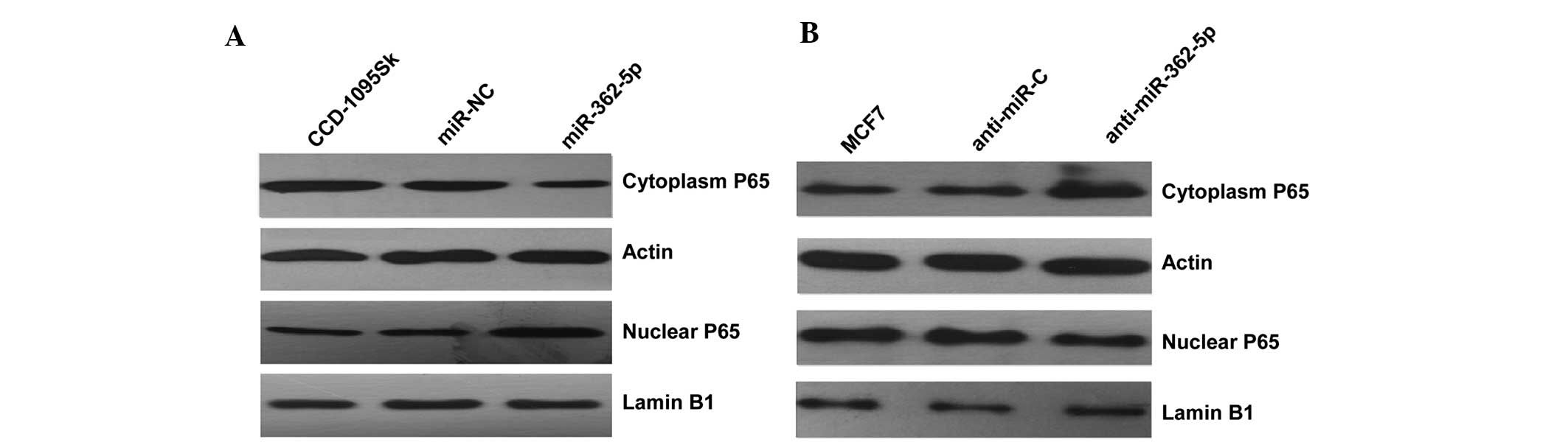
Since CYLD plays a predominant role in the negative
regulation of NF-κB (21), the
mechanism underlying the observed cellular phenotypic changes was
explored by evaluating the impact of CYLD on NF-κB activation by
detecting the level of nucleus NF-κB P65 protein in the two cell
lines transfected with miR-362-5p mimics/inhibitors. Subsequent to
transfection with miR-362-5p mimics, the results indicated that the
level of nucleus NF-κB P65 protein was clearly increased compared
with the negative control (Fig. 4A).
By contrast, the expression level of nuclear P65 was clearly
decreased in miR-362-5p-suppressed MCF7 cells (Fig. 4B). Overall, the present findings
indicate that miR-362-5p likely promotes proliferation, migration
and invasion through the NF-κB signaling pathway.
Discussion
An increasing number of studies have demonstrated
that the dysregulation of miRNA contributes to tumorigenesis
(22). Numerous studies have
confirmed that miRNA plays a critical role in tumor cell survival,
invasion and metastasis. Xia et al identified that miR-362
induces cell proliferation and apoptosis resistance in gastric
cancer (23), but the impact on
breast cancer cell growth and metastasis remains unclear. In the
present study, miR-362-5p was focused on, and it was found that
miR-362-5p was clearly upregulated in human breast cancer cell
lines. To the best of our knowledge, the present study is the first
to report the upregulation of miR-362-5p in breast cancer cells.
Since the upregulation of miR-362-5p in breast cancer has not been
previously described, the role of miR-362-5p in breast cancer cells
was analyzed. The present study provides the first evidence that
the downregulation of miR-362-5p significantly inhibits cell
proliferation, migration and invasion, induces cell cycle arrest,
and promotes apoptosis in MCF7 cells. These results indicate that
miR-362-5p may be a novel oncogene that plays an important role in
the regulation of breast cancer cell growth and metastasis.
The function of miRNA is to regulate the target
genes by direct cleavage of the mRNA or the inhibition of protein
synthesis (24). To further explore
the molecular mechanism underlying miR-362-5p function, direct
target genes of miR-362-5p were identified through bioinformatics
analysis. It was found that CYLD possesses a putative
miR-362-5p-binding site within its 3′-UTR. CYLD was identified as a
direct target of miR-362-5p in breast cancer cells, and this
conclusion was supported by the following findings. Firstly, the
complementary sequence of miR-362-5p was identified in the 3′-UTR
of CYLD mRNA. In addition, miR-362-5p overexpression was found to
suppress CYLD 3′-UTR luciferase reporter activity, and this effect
was abolished by the deletion of the miR-362-5p seed binding site.
The overexpression of miR-362-5p also led to a significant
reduction in CYLD at the mRNA and protein levels. Finally, the
inhibition of miR-362-5p increased the CYLD expression level in
breast cancer cells.
CYLD is known to be a tumor suppressor gene in
various types of cancer (25,26). Previous studies have revealed that
CYLD expression is downregulated in breast cancer tissues. In
addition, the downregulation of CYLD promoted cell survival and
migration through the NF-κB signaling pathway (21). However, the exact regulatory mechanism
responsible for the decrease of CYLD expression in breast cancer
cells remains unclear. Notably, in the present study, miR-362-5p
was revealed to be associated with CYLD and the downstream NF-κB
signaling pathway. The current results revealed that miR-362-5p was
likely to repress the expression of CYLD, which, in turn, promoted
the proliferation, migration and invasion of breast cancer cells by
regulating the NF-κB pathway.
In conclusion, the present study provides novel
evidence that downregulation of miR-362-5p expression suppresses
the proliferation, migration and invasion of human breast cancer
cells. These data suggest that miR-362-5p and its downstream
targets may be potential therapeutic targets in human breast
cancer.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81272258 and 31300715),
Anhui Province Natural Science Foundation (grant no. 1308085QH136)
and Anhui Medical University Training Program of National
Outstanding Youth Foundation (grant no. GJYQ-1401). The study was
also supported by a grant from the Grants for Scientific Research
of bo shi ke yan of Anhui Medical University (grant no. XJ201316)
awarded to Dr Fang Ni.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goldhirsch A, Wood WC, Coates AS, Gelber
RD, Thürlimann B and Senn HJ: Panel members: Strategies for
subtypes - dealing with the diversity of breast cancer: Highlights
of the St. Gallen International Expert Consensus on the Primary
Therapy of Early Breast Cancer 2011. Ann Oncol. 22:1736–1747. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sørlie T, Perou CM, Tibshirani R, Aas T,
Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey
SS, et al: Gene expression patterns of breast carcinomas
distinguish tumor subclasses with clinical implications. Proc Natl
Acad Sci USA. 98:10869–10874. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Giordano S and Columbano A: MicroRNAs: New
tools for diagnosis prognosis, and therapy in HCC? Hepatology.
57:840–847. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sanyal AJ, Yoon SK and Lencioni R: The
etiology of hepatocellular carcinoma and consequences for
treatment. Oncologist. 15(Suppl 4): 14–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
El-Serag HB: Epidemiology of viral
hepatitis and hepatocellular carcinoma. Gastroenterology.
142:1264–1273.e1. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kasinski AL and Slack FJ: Epigenetics and
genetics. Histopathology. Nat Rev Cancer. 11:849–864. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartel DP: MicroRNAs: genomics biogenesis,
mechanism, and function. Cell. 116:281–297. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dvinge H, Git A, Gräf S, Salmon-Divon M,
Curtis C, Sottoriva A, Zhao Y, Hirst M, Armisen J, Miska EA, et al:
The shaping and functional consequences of the microRNA landscape
in breast cancer. Nature. 497:378–382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ebert MS and Sharp PA: Roles for microRNAs
in conferring robustness to biological processes. Cell.
149:515–524. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bignell GR, Warren W, Seal S, Takahashi M,
Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, et al:
Identification of the familial cylindromatosis tumour-suppressor
gene. Nat Genet. 25:160–165. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kovalenko A, Chable-Bessia C, Cantarella
G, Israël A, Wallach D and Courtois G: The tumour suppressor CYLD
negatively regulates NF-kappaB signalling by deubiquitination.
Nature. 424:801–805. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lim JH, Jono H, Komatsu K, Woo CH, Lee J,
Miyata M, Matsuno T, Xu X, Huang Y, Zhang W, et al: CYLD negatively
regulates transforming growth factor-beta-signalling via
deubiquitinating Akt. Nat Commun. 3:7712012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tauriello DV, Haegebarth A, Kuper I,
Edelmann MJ, Henraat M, Canninga-van Dijk MR, Kessler BM, Clevers H
and Maurice MM: Loss of the tumor suppressor CYLD enhances
Wnt/betacatenin signaling through K63-linked ubiquitination of Dvl.
Mol Cell. 37:607–619. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pannem RR, Dorn C, Ahlqvist K, Bosserhoff
AK, Hellerbrand C and Massoumi R: CYLD controls c-MYC expression
through the JNK-dependent signaling pathway in hepatocellular
carcinoma. Carcinogenesis. 35:461–468. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang WY, Lim JH and Li JD: Synergistic and
feedback signaling mechanisms in the regulation of inflammation in
respiratory infections. Cell Mol Immunol. 9:131–135. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gautheron J and Luedde T: A novel player
in inflammation and cancer: The deubiquitinase CYLD controls HCC
development. J Hepatol. 57:937–939. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Font-Burgada J, Seki E and Karin M: CYLD
and HCC, When being too sensitive to your dirty neighbors results
in self-destruction. Cancer Cell. 21:711–712. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Urbanik T, Köhler BC, Boger RJ, et al:
Down-regulation of CYLD as a trigger for NF-κB activation and a
mechanism of apoptotic resistance in hepatocellular carcinoma
cells. Int J Oncol. 38:121–131. 2011.PubMed/NCBI
|
|
20
|
Hayashi M, Jono H, Shinriki S, et al:
Clinical significance of CYLD downregulation in breast cancer.
Breast Cancer Res Treat. 143:447–457. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun SC: CYLD: A tumor suppressor
deubiquitinase regulating NF-kappaB activation and diverse
biological processes. Cell Death Differ. 17:25–34. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang W, Liu J and Wang G: The role of
microRNAs in human breast cancer progression. Tumor Biol.
35:6235–6244. 2014. View Article : Google Scholar
|
|
23
|
Xia JT, Chen LZ, Jian WH, Wang KB, Yang
YZ, He WL, He YL, Chen D and Li W: MicroRNA-362 induces cell
proliferation and apoptosis resistance in gastric cancer by
activation of NF-κB signaling. J Transl Med. 12:332014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chekulaeva M and Filipowicz W: Mechanisms
of miRNA-mediated posttranscriptional regulation in animal cells.
Curr Opin Cell Biol. 21:452–460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deng LL, Shao YX, Lv HF, Deng HB and Lv
FZ: Over-expressing CYLD augments antitumor activity of TRAIL by
inhibiting the NF-κB survival signaling in lung cancer cells.
Neoplasma. 59:18–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Massoumi R: CYLD: A deubiquitination
enzyme with multiple roles in cancer. Future Oncol. 7:285–297.
2011. View Article : Google Scholar : PubMed/NCBI
|