Introduction
Gastric cancer accounts for 7.8% of cancers
worldwide (1). Furthermore, it is the
fourth most common type of cancer, and the second most common cause
of cancer-related mortality worldwide (2,3).
Approximately 85% of stomach cancers are adenocarcinomas (4). The gastric antrum is the most common
site of gastric carcinoma (1).
Endoscopy is useful for diagnosis as it exhibits a high level of
sensitivity and specificity (1).
Gastric carcinomas are usually asymptomatic in the early stages of
the disease (4). However, as the
carcinomas progress, symptoms including, upper abdominal
discomfort, anorexia, nausea, vomiting and weight loss may develop.
Gastric carcinomas may metastasize to lymph nodes, perigastric
tissue, pancreas, colon, liver and the ovaries (4). Gastric cancer treatment and prognosis
are dependent on tumor stage (3).
Endoscopic mucosal resection may be used for the treatment of early
stage gastric carcinoma lesions, <2 cm in size, without invasion
of the lamina propria or muscularis mucosa (3). Complete resection of tumors by subtotal
gastrectomy or total gastrectomy with lymph node dissection are the
first choices for curative treatment. However, surgery is
applicable in less than a third of patients (4). In advanced gastric cancers, additional
chemotherapy or chemoradiation treatment prior to or following
surgery is considered a good option (2,3). It has
been found that overall survival is increased by 9% after
neoadjuvant chemotherapy (3).
Furthermore, adjuvant chemotherapy with various regimens, including
5-fluorouracil, have been found to increase overall survival by 6%
and reduce the risk of mortality by 18% (3). Targeted therapies involving the
inhibition of human epidermal growth factor receptor 2, vascular
endothelial growth factor receptor 2 and epidermal growth factor
receptor (EGFR) have also been investigated (2,3). Despite
advances in treatment, the survival of advanced gastric carcinoma
patients remains poor with a 5-year survival rate of <30% in
stage III patients (1,3,4). Response
rates for various chemotherapies range between 9 and 51% (5). However, these response rates do not
correlate with survival rate. The discrepancy is associated with
drug resistance, which leads to the failure of therapy and poor
prognosis. At present, the mechanisms of anticancer drug resistance
remain unclear, despite extensive investigation.
Anticancer agents targeting specific proteins in
cancer cells have revolutionized pharmacotherapy in the field of
oncology (6). However, targeted
therapies have limitations due to the development of resistance
with long-term use, in which certain types of cancer cells acquire
resistance to drugs that inhibit specific proteins (7). Resistance to anticancer drugs results in
relapse or refractoriness to therapy for cancer patients (7). Thus, during the development of targeted
agents, the resistance mechanisms must be investigated in order to
aid in overcoming the problems associated with such resistance.
Inhibitors of c-Met kinase have been investigated as
anticancer agents and subjected to clinical trials (8). The receptor tyrosine kinase c-Met is
activated by its ligand, hepatocyte growth factor (HGF). Upon
binding to HGF, c-Met dimerizes and transduces cell signaling by
activating multiple downstream pathways, including Akt,
mitogen-activated protein kinase and focal adhesion kinase. The
physiological roles of the c-Met signaling pathway include cell
survival and migration; therefore, dysregulated c-Met activation
may lead to tumorigenesis (8).
Mutation or overexpression of the c-Met protein has been reported
in various types of cancer, including gastric cancer (9,10).
Blocking c-Met activity using small molecule inhibitors or
monoclonal antibodies may be an effective strategy for cancer
therapy, and numerous c-Met inhibitors are currently under
development for anticancer drugs (11). Among the drugs approved by the Food
and Drug Administration, cabozantinib (also known as XL-184) is an
inhibitor of c-Met, vascular endothelial growth factor receptor 2,
and Ret proto-oncogene and is used for treatment of medullary
thyroid cancer (12,13).
KRC-108 is a small molecule that was previously
identified in an effort to find inhibitors of c-Met with oral
antitumor properties (14). In order
to identify the mechanisms underlying resistance to KRC-108
treatment, the present study generated gastric cancer cell lines
resistant to KRC-108, and the phenotypic and molecular changes
associated with KRC-108 resistance were investigated.
Materials and methods
Generation of KRC-108-resistant cell
lines
MKN-45 human gastric cancer cells were purchased
from the Korean Cell Line Bank (Seoul, Korea) and cultured in
RPMI-1640 medium (GE Healthcare Life Sciences HyClone Laboratories,
Logan, UT, USA) supplemented with 10% fetal bovine serum (GE
Healthcare Life Sciences HyClone Laboratories). The cells were
incubated at 37°C in a humidified atmosphere of 5% CO2.
KRC-108-resistant clones of MKN-45 cell lines were established by
exposing the cells to increasing concentrations of KRC-108
(synthesized by Professor Jongkook Lee; Kangwon National
University, Kangwon, South Korea) for 3 months. The cells were
grown in the presence of KRC-108 at 100 nM at first for two weeks,
and the concentration of KRC-108 was increased gradually to a final
concentration of 1 µM. Replicate clones capable of proliferating in
1 µM of KRC-108 were used in this study.
Cell viability assay
The cells were plated in a 96-well plate (2,000
cells/well), and serial dilutions of KRC-108 were added. At the end
of the incubation period (72 h), cell viability was measured using
the tetrazolium-based EZ-CYTOX cell viability assay kit (DaeilLab
Service Co., Ltd., Seoul, Korea). Growth inhibition of 50%
(GI50) was calculated by non-linear regression using
GraphPad Prism software version 5.01 (GraphPad Software, Inc., La
Jolla, CA, USA).
Western blot analysis
Samples of the cell extracts prepared in sodium
dodecyl sulfate (SDS) lysis buffer (12 mM Tris-Cl, pH 6.8; 5%
glycerol; 0.4% SDS; USB Corporation, Cleveland, OH, USA) were
resolved by SDS-PAGE and transferred to a polyvinylidene fluoride
membrane (EMD Millipore, Billerica, MA, USA). The filters were
blocked in Tris-buffered saline (10 mM Tris-Cl, pH 7.4; 140 mM
NaCl) containing 0.1% Tween-20 (TBST) and 5% non-fat dry milk.
Subsequently, the filters were incubated with a blocking solution
containing the indicated primary antibodies for 2 h. After washing
three times in TBST, the membranes were incubated with a
peroxidase-conjugated secondary antibody for 1 h at room
temperature (RT). Blots were then washed three times and developed
using chemiluminescent substrate (GE Healthcare Life Sciences,
Chalfont, UK), and luminescent signals were visualized using an
ImageQuant LAS 4000 Mini (GE Healthcare Life Sciences). Primary
antibodies against human c-Met (rabbit polyclonal IgG; #sc-10;
1:1,000), phosphorylated (p-)Met (rabbit polyclonal IgG; #sc-34085;
1:1,000), E-cadherin (mouse monoclonal IgG; #sc-21791; 1:1,000),
N-cadherin (rabbit polyclonal IgG; #sc-7939; 1:1,000; all from
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and β-actin (mouse
monoclonal IgG; #A5441; 1:5,000; Sigma-Aldrich, St. Louis, MO, USA)
were used. Goat anti-rabbit IgG (#111-035-003; 1:5,000) and
anti-mouse IgG (#115-035-003; 1:5,000) secondary antibodies were
purchased from Jackson ImmunoResearch Laboratories, Inc. (West
Grove, PA, USA).
Immunoprecipitation
Cell lysates were prepared in NETN lysis buffer
[0.5% NP-40 (Abcam, Cambridge, MA, USA), 1 mM EDTA (Bioneer,
Daejeon, South Korea), 120 mM NaCl, 1 mM DTT (Sigma-Aldrich), 10 mM
NaF (Sigma-Aldrich), 2 mM Na3VO4
(Sigma-Aldrich), 50 mM Tris-Cl, pH 8.0) with a protease inhibitor
cocktail (Sigma-Aldrich). Lysates were incubated with the indicated
antibodies overnight at 4°C and Protein G Sepharose beads (GE
Healthcare Life Sciences) were added. Following incubation for 2 h,
the immune complexes were washed and released from the beads by
boiling and then analyzed by western blotting using the indicated
antibodies.
Immunofluorescence
The cells were plated on 8-chamber culture slides
and incubated for 24 h. They were then rinsed briefly in
phosphate-buffered saline (PBS), fixed in 4% formaldehyde
(Sigma-Aldrich) and subsequently permeabilized in 0.2% Triton X-100
(Sigma-Aldrich). After washing the samples twice with ice-cold PBS,
the cells were blocked with 1% bovine serum albumin (BSA; Gibco;
Thermo Fisher Scientific, Waltham, MA, USA) in PBS with Tween-20
(PBST; Anatrace Inc., Maumee, OH, USA) for 30 min and then
incubated with the primary antibody (1:50) (diluted in 1% BSA in
PBST) overnight at RT. Following three washes with PBS, the cells
were incubated with the corresponding secondary antibody [Alexa
Fluor® 488 goat anti-mouse IgG (H+L) (#A-11001; 1:500) or Alexa
Fluor® 546 goat anti-rabbit IgG (H+L) (#A-11010; 1:500); Invitrogen
Life Technologies, Carlsbad, CA, USA] for 4 h at RT in the dark.
Mounting medium (ProLong® Gold Antifade reagent with DAPI;
Invitrogen Life Technologies) was dropped onto the washed cells,
which were subsequently photographed with a confocal laser scanning
microscope (FV-1000; Olympus Corporation, Tokyo, Japan).
Tissue microarray
Tumor samples from 71 patients with primary gastric
cancer that had undergone laparoscopic distal gastrectomy, subtotal
gastrectomy or total gastrectomy at Gyeongsang National University
Hospital (Jinju, Korea) in 2010 were collected. The patient cohort
included 53 males and 18 females, with a mean age of 63.5 years
(range, 36–80 years). The pathologist reviewed hematoxylin and
eosin-stained slides from neutral buffered formalin-fixed,
paraffin-embedded tissue blocks. One representative area of
carcinoma was selected from each case. Three tissue microarray
blocks containing 71 cores of 3 mM diameter were constructed. The
process was conducted using a precision arraying instrument (Quick
Ray®; Unitma Co., Ltd., Seoul, Korea). This study was approved by
the Institutional Review Board (IRB) of Gyeongsang National
University Hospital and a waiver of written informed consent was
obtained.
Immunohistochemistry
Immunohistochemical staining of the tumor specimens
was conducted using a Benchmark XT autostainer (Ventana Medical
Systems, Inc., Tucson, AZ, USA) using anti-E-cadherin and
anti-c-Met antibodies. E-cadherin staining was assessed and cases
were divided into three groups according to membrane staining: -, +
or ++. Cases with no staining or partial cell membrane staining
were interpreted as negative (−). Cases with discontinuous staining
along the cell membrane were interpreted as positive (+) and cases
stained continuously along the cell membrane were interpreted as
strongly positive (++). c-Met was interpreted with regard to
cytoplasmic staining, with positivity classified into three groups
according to intensity; -, + or ++. Cases exhibiting no staining
were interpreted as negative (−), cases exhibiting partial staining
were interpreted as positive (+) and cases exhibiting complete
cytoplasmic staining were interpreted as strongly positive
(++).
Statsitical analysis
Data were analyzed using SPSS software version 18.0
(SPSS, Inc., Chicago, IL, USA). Correlations were assessed using
the χ2 test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Establishment of MKN-45 cells
resistant to c-Met kinase inhibitor KRC-108
The KRC-108 compound is an antitumor agent with
c-Met inhibitory activity in vitro and in vivo
(14). To study the mechanisms
associated with acquired resistance to KRC-108, the gastric cancer
cell line MKN-45, which expresses a high level of c-Met (15), was utilized to develop
KRC-108-resistant cell lines. The cells were treated with KRC-108,
initially at a low concentration, and the dose was increased
stepwise. The resulting KRC-108-resistant cells were designated
MKN-R, and three replicate clones were used: MKN-R1, MKN-R2, and
MKN-R3. The parental MKN-45 cells were sensitive to KRC-108
treatment with the GI50 concentration of 1.1 µM, whilst
the MKN-R cells did not exhibit growth inhibition with treatment of
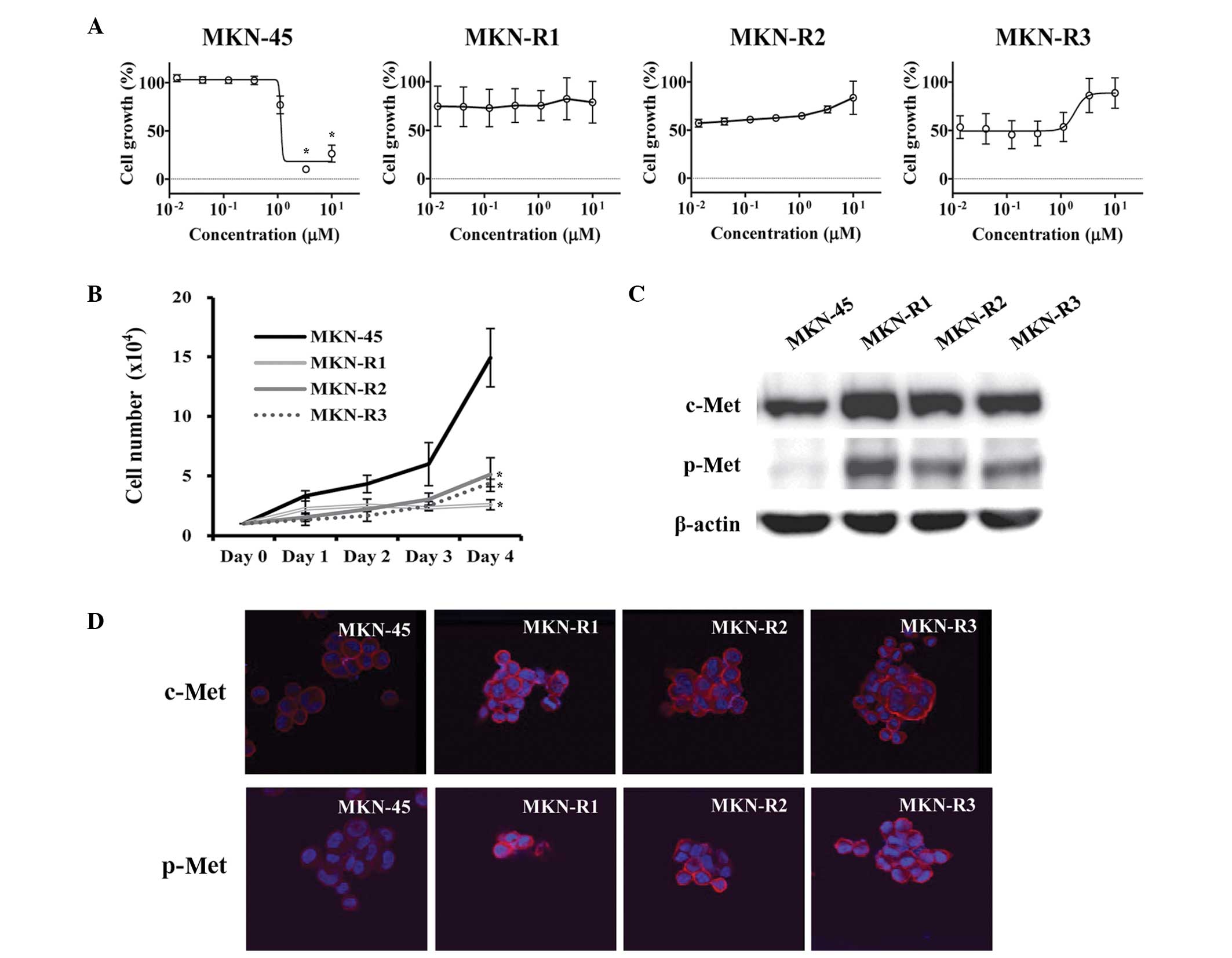
KRC-108 up to a concentration of 10 µM (Fig. 1A). The growth characteristics of the
MKN-45 and MKN-R cells were different, with the MKN-R cells growing
more slowly than the parental MKN-45 cells, as shown in Fig. 1B. Western blot analysis was conducted
to investigate the effect of KRC-108 treatment, revealing increased
expression of c-Met in the MKN-R cells compared with that of the
parental cells (Fig. 1C). Along with
the overexpression of c-Met, the phosphorylated form of c-Met
(p-Met) was increased in the MKN-R cells compared with the parental
MKN-45 cells. The increase in the expression level and the activity
of c-Met was confirmed by immunofluorescence (Fig. 1D). We hypothesized that a high level
of active c-Met (p-Met) may cause the MKN-R cells to be resistant
to KRC-108 treatment. The inhibition of c-Met kinase activity by
KRC-108 was overcome by overexpression of the c-Met protein, thus
resulting in cell survival in the presence of KRC-108.
KRC-108-resistant cells have an
epithelial cell-like phenotype
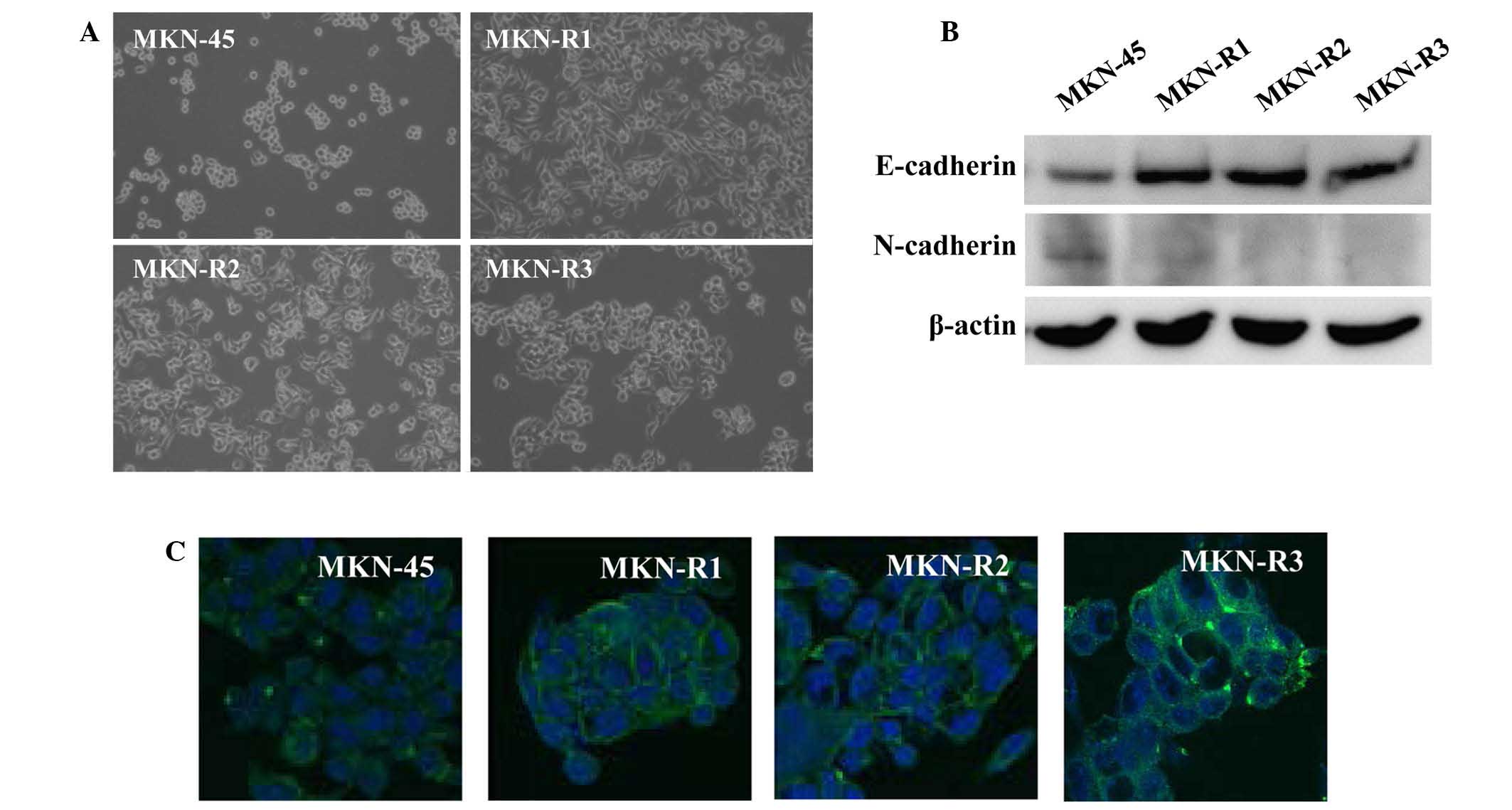
A phenotypic difference was observed between the
MKN-45 cells and the MKN-R cells. Fig.
2A displays the phase contrast images of the cells,
demonstrating morphological changes in the KRC-108-resistant cells.
The parental MKN-45 cells were round with a poorly differentiated
form, whilst the MKN-R cells exhibited a flat epithelial cell-like
phenotype. All three clones of MKN-R cells displayed similar
morphology.
Consistent with the epithelial characteristics of
the MKN-R cells, higher expression of E-cadherin in MKN-R cells
relative to the MKN-45 cells was observed (Fig. 2B). E-cadherin is an epithelial marker
and cell-surface adhesion protein (16,17). In
addition, expression levels of N-cadherin, a mesenchymal marker,
were decreased in MKN-R cells relative to MKN-45 cells. To confirm
the change in the expression of E-cadherin observed on the western
blot, immunostaining using an anti-E-cadherin antibody was
performed. Immunocytochemical analyses of E-cadherin revealed high
expression of E-cadherin in the cell surface area of the MKN-R
cells (Fig. 2C). These results
indicate that E-cadherin is expressed in cell-cell contact areas of
MKN-R cells.
c-Met associates with E-cadherin in
MKN-R cells
Next, the mechanism of morphological change and
upregulation of E-cadherin expression associated with KRC-108
resistance were investigated. As a direct interaction between c-Met
and E-cadherin has been reported in a number of studies (18–20), the
possibility of direct binding of c-Met and E-cadherin in the MKN-R
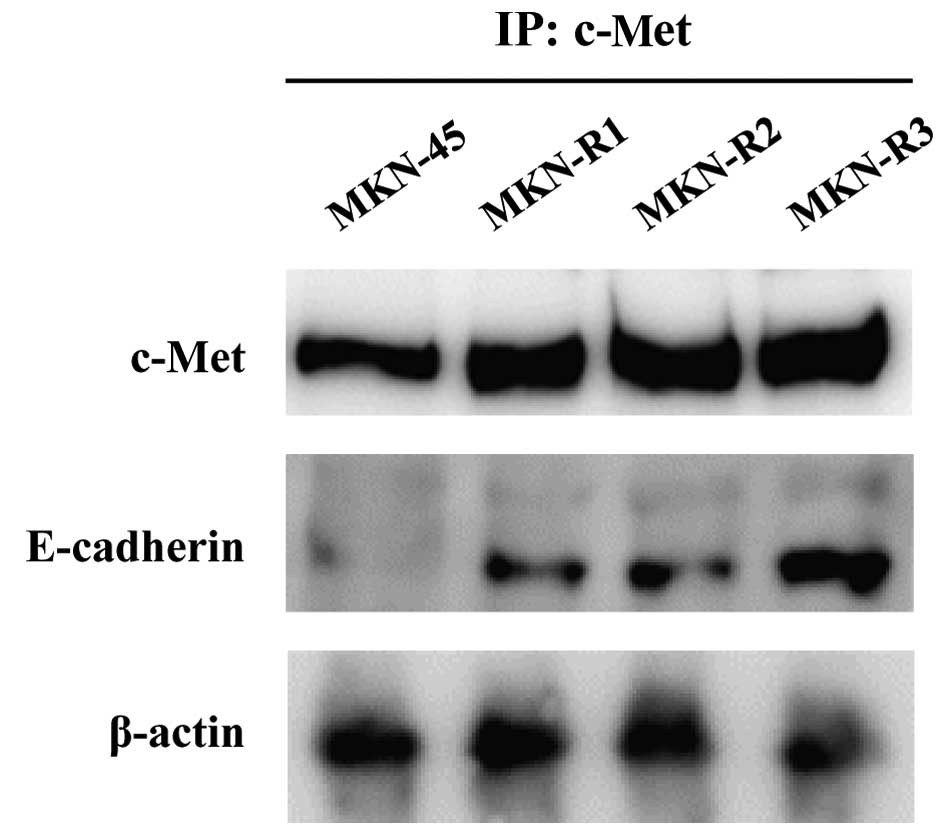
cells was explored using immunoprecipitation. Cell lysates were
immunoprecipitated with an anti-c-Met antibody, and the
immunoprecipitates were subjected to SDS-PAGE and probed with an
anti-E-cadherin antibody. As shown in Fig. 3, E-cadherin was detected in the
immunoprecipitates from the MKN-R cells. Thus, c-Met and E-cadherin
interacted in the MKN-R cells, but not in the MKN-45 cells. These
results imply that the epithelial transition in the MKN-R cells is
mediated by overexpression of c-Met, leading to the recruitment of
E-cadherin to the cell surface. The recruitment of E-cadherin by
c-Met may induce epithelial cell-like changes in MKN-R cells.
Double expression of E-cadherin and
c-Met in human gastric cancer tissues
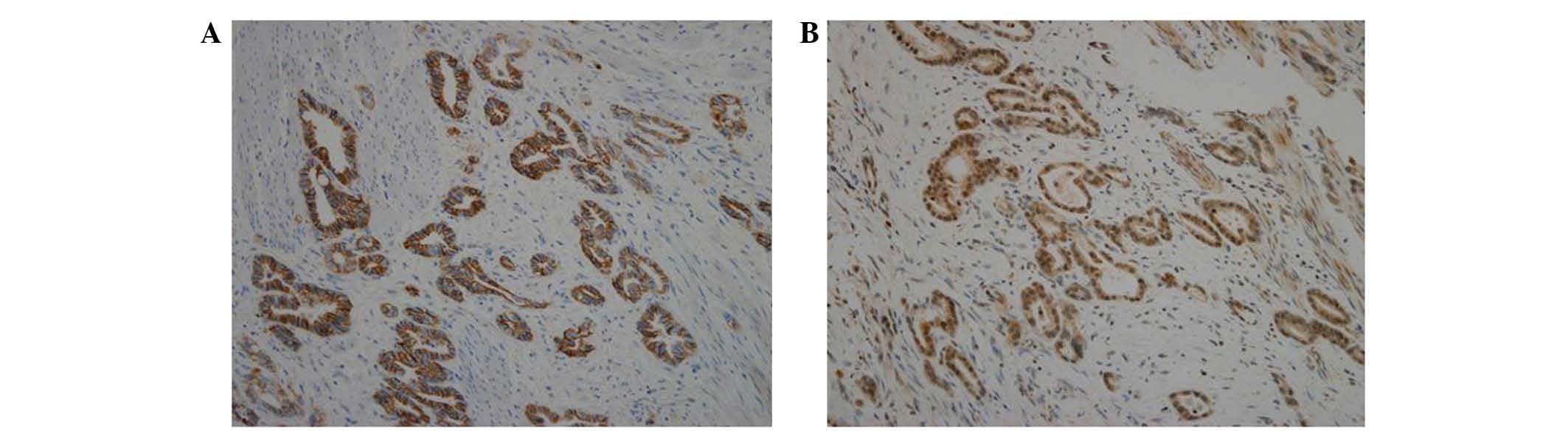
To confirm the association of c-Met and E-cadherin
in human tissue samples, a tissue microarray was performed using
specimens of human gastric carcinoma. Tissue microarray blocks were
subjected to immunohistochemical staining using antibodies against
E-cadherin and c-Met. As shown in Table
I, 48 of the 71 cores (67.6%) exhibited double expression of
E-cadherin and c-Met. Furthermore, all samples with c-Met ++
expression exhibited high expression of E-cadherin (++). As shown
in Fig. 4, staining for E-cadherin
and c-Met revealed the same pattern of expression in gastric
carcinoma. The co-expression of E-cadherin and c-Met observed in
human gastric carcinoma tissues supports the findings of an
association of c-Met with E-cadherin in gastric carcinoma cell
lines (Fig. 3).
 | Table I.Expression of E-cadherin and c-Met in
human gastric carcinoma tissues. |
Table I.
Expression of E-cadherin and c-Met in
human gastric carcinoma tissues.
|
| c-Met expression,
n |
|
|---|
|
|
|
|
|---|
| E-cadherin
expression | − | + | ++ | Total |
|---|
| − | 3 | 0 | 0 | 3 |
| + | 8 | 5 | 0 | 13 |
| ++ | 12 | 18 | 25 | 55 |
| Total | 23 | 23 | 25 | 71 |
Discussion
The present study explored the cellular changes
accompanied by resistance to the c-Met inhibitor KRC-108 and their
possible mechanisms. The results revealed an overexpression of
c-Met and phenotypic changes to more epithelial characteristics in
MKN-R cells. Mechanisms of resistance to other c-Met inhibitors
have been reported in previous studies (21–23).
McDermott et al (21)
investigated drug resistance to a c-Met inhibitor, PF-2341066, in
non-small-cell lung cancer (NSCLC) cell lines. The study utilized
NSCLC cells with c-Met amplification, which were sensitive to c-Met
inhibitors. Prolonged exposure to c-Met inhibitor PF-2341066
resulted in activation of EGFR in the cells. Resistant cells with
EGFR activation demonstrated sensitivity to combined treatment with
PF-2341066 and an EGFR inhibitor, suggesting that the EGFR pathway
compensated for c-Met signaling in resistant cells. A similar
mechanism was reported in the gastric cancer cell line SNU638
(22). The c-Met inhibitors
PHA-665752 and PF-2341066 were utilized to develop c-Met-resistant
SNU638 cells. Two mechanisms of resistance were revealed: One
involving the same mechanism observed in NSCLC cells (i.e.,
activation of EGFR), and the other involving a mutation of the
c-Met sequence, Y1230C. Y1230 is located in the activation loop of
c-Met, and its mutation results in structural changes that affect
interaction with c-Met inhibitors. In a separate study, a drug
resistance screen using other c-Met inhibitors, NVP-BYU972 and
AMG458, also demonstrated mutations in the c-Met sequence affecting
binding of c-Met inhibitors (23). In
the current paper, the mechanism of KRC-108 resistance was the
overexpression of c-Met. Overexpression of the target protein is a
frequently observed mechanism of drug resistance, as demonstrated
by Bcr-Abl inhibitors (7). Some of
leukemic cells were found to develop resistance to Bcr-Abl
inhibitor imatinib following a few years of use in leukemic
patients. Several mechanisms of resistance were revealed and
amplification of the BCR-ABL gene and subsequent
overexpression of the Bcr-Abl kinase was frequently observed
(24).
The epithelial-mesenchymal transition (EMT) is known
to be crucial in cancer progression (25). The EMT involves phenotypic changes
from an epithelial to mesenchymal cell type, with the acquisition
of migratory and invasive properties. The EMT process is
reversible, and the reverse process is termed the
mesenchymal-epithelial transition (MET). The term
‘epithelial-mesenchymal plasticity’ was previously proposed to
describe the flexible transitions observed between epithelial cells
and mesenchymal cells (26). The
changes observed in the MKN-R cells in the present study were
similar to those seen in MET. The MKN-R cells acquired epithelial
morphology, which was accompanied by upregulation of E-cadherin
(Fig. 2). We hypothesize that
epithelial plasticity may be closely related to drug resistance in
cancer cells.
Morphological changes, such as the EMT/MET
phenomena, accompanied by drug resistance have been described in a
number of studies (27–30). Many reported a positive correlation
between EMT activation and drug resistance (27–29). EMT
was observed in lung cancer cells with EGFR inhibitor resistance
(28). Thomson et al (29) reported that cells insensitive to the
EGFR inhibitor have a mesenchymal phenotype. By contrast, another
report revealed that drug-resistant cell lines exhibited epithelial
cell characteristics (30). The study
by Mahadevan et al (30) of
gastrointestinal stromal tumor (GIST) cells resistant to imatinib
reported that imatinib-resistant GIST cells changed to an
epithelial cell-like phenotype. From the available literature and
the results of the present study, it is not clear whether the EMT
confers drug resistance. However, it is clear that cellular
plasticity represented by morphological changes is frequently
observed in drug-resistant cells and thus, we hypothesize that such
cells lose or acquire epithelial characteristics. The direction of
change (EMT or MET) may be dependent on the specific cell types
used in the experiments.
Various studies of the association of c-Met with
E-cadherin in a number of tumor cell lines have been published
(18–20,31). The
co-localization of c-Met and β-catenin in a cell adhesion complex,
as well as E-cadherin, was observed in breast and colorectal cancer
cell lines (18). Forced expression
of E-cadherin in BT-549 human breast cancer cells, which do not
express E-cadherin, resulted in the recruitment of c-Met to the
cell membrane (19). A tissue
microarray study of ductal breast carcinoma in situ revealed
a correlation between c-Met and E-cadherin expression (20). The importance of the c-Met/E-cadherin
interaction has also been demonstrated in pathological states other
than cancer: Helicobacter pylori infection induced an
invasive phenotype in gastric epithelial cells (31), and E-cadherin expression suppressed
the invasive phenotype through an interaction with c-Met. In the
present study, the interaction of c-Met and E-cadherin is presumed
to have contributed to the epithelial phenotype in the MKN-R cells
(Fig. 3), consistent with previous
reports.
In conclusion, the present study revealed a
mechanism utilized by cancer cells to confer resistance to
anticancer agents. These results may aid the establishment of
therapeutic strategies in cases of c-Met kinase inhibitor
resistance. The association between tumor morphology and c-Met
inhibitor resistance requires further study. Accumulating knowledge
of resistance mechanisms will aid the development of successful
therapy with targeted agents in the future.
Acknowledgements
This work was supported by grants of the National
Research Foundation (2015R1C1A2A01053928) funded by the government
of Korea, and grants from the Ministry of Trade, Industry &
Energy and Korea Institute for Advancement of Technology through
Inter-Economic Region co-operation projects (A004500005).
Glossary
Abbreviations
Abbreviations:
|
HGF
|
hepatocyte growth factor
|
|
NSCLC
|
non-small-cell lung cancer
|
|
EGFR
|
epidermal growth factor receptor
|
|
EMT
|
epithelial-mesenchymal transition
|
|
MET
|
mesenchymal-epithelial transition
|
|
GIST
|
gastrointestinal stromal tumor
|
|
GI50
|
growth inhibition of 50%
|
|
TBST
|
Tris-buffered saline containing 0.1%
Tween-20
|
|
PBS
|
phosphate-buffered saline
|
|
BSA
|
bovine serum albumin
|
|
RT
|
room temperature
|
References
|
1
|
Bosman FT, Carneiro F, Hruban RH and
Theise ND: WHO Classification of Tumours of the Digestive System
(4th). Lyon: IARC Press. 2010.
|
|
2
|
Carcas LP: Gastric cancer review. J
Carcinog. 13:142014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marrelli D, Polom K, de Manzoni G,
Morgagni P, Baiocchi GL and Roviello F: Multimodal treatment of
gastric cancer in the west, Where are we going? World J
Gastroenterol. 21:7954–7969. 2015.PubMed/NCBI
|
|
4
|
Kasper DL, Fauci A, Hauser S, Longo D,
Jameson J and Loscalzo J: Harrison's Principles of Internal
Medicine (19th). New York, NY: McGraw-Hill Education. 2015.
|
|
5
|
Koizumi W: Chemotherapy for advanced
gastric cancer: R eview of global and Japanese status. Gastrointest
Cancer Res. 1:197–203. 2007.PubMed/NCBI
|
|
6
|
Aggarwal S: Targeted cancer therapies. Nat
Rev Drug Discov. 9:427–428. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Engelman JA and Settleman J: Acquired
resistance to tyrosine kinase inhibitors during cancer therapy.
Curr Opin Genet Dev. 18:73–79. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gherardi E, Birchmeier W, Birchmeier C and
Vande Woude G: Targeting MET in cancer. Rationale and progress. Nat
Rev Cancer. 12:89–103. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuniyasu H, Yasui W, Kitadai Y, Yokozaki
H, Ito H and Tahara E: Frequent amplification of the c-met gene in
scirrhous type stomach cancer. Biochem Biophys Res Commun.
189:227–232. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hara T, Ooi A, Kobayashi M, Mai M,
Yanagihara K and Nakanishi I: Amplification of c-myc, K-sam and
c-met in gastric cancers, Detection by fluorescence in situ
hybridization. Lab Invest. 78:1143–1153. 1998.PubMed/NCBI
|
|
11
|
Scagliotti GV, Novello S and von Pawel J:
The emerging role of MET/HGF inhibitors in oncology. Cancer Treat
Rev. 39:793–801. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Y, Guessous F, Kofman A, Schiff D
and Abounader R: XL-184, a MET, VEGFR-2 and RET kinase inhibitor
for the treatment of thyroid cancer, glioblastoma multiforme and
NSCLC. IDrugs. 13:112–121. 2010.PubMed/NCBI
|
|
13
|
Hoy SM: Cabozantinib: A review of its use
in patients with medullary thyroid cancer. Drugs. 74:1435–1444.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han SY, Lee CO, Ahn SH, Lee MO, Kang SY,
Cha HJ, Cho SY, Ha JD, Ryu JW, Jung H, et al: Evaluation of a
multi-kinase inhibitor KRC-108 as an anti-tumor agent in
vitro and in vivo. Invest New Drugs. 30:518–523. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McDermott U, Sharma SV, Dowell L,
Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A,
Lee D, et al: Identification of genotype-correlated sensitivity to
selective kinase inhibitors by using high-throughput tumor cell
line profiling. Proc Natl Acad Sci USA. 104:19936–19941. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gumbiner BM: Cell adhesion: T he molecular
basis of tissue architecture and morphogenesis. Cell. 84:345–357.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Takeichi M: Cadherin cell adhesion
receptors as a morphogenetic regulator. Science. 251:1451–1455.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hiscox S and Jiang WG: Association of the
HGF/SF receptor c-met, with the cell-surface adhesion molecule,
E-cadherin and catenins in human tumor cells. Biochem Biophys Res
Commun. 261:406–411. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reshetnikova G, Troyanovsky S and Rimm DL:
Definition of a direct extracellular interaction between Met and
E-cadherin. Cell Biol Int. 31:366–373. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Götte M, Kersting C, Radke I, Kiesel L and
Wülfing P: An expression signature of syndecan-1 (CD138),
E-cadherin and c-met is associated with factors of angiogenesis and
lymphangiogenesis in ductal breast carcinoma in situ. Breast
Cancer Res. 9:R82007. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
McDermott U, Pusapati RV, Christensen JG,
Gray NS and Settleman J: Acquired resistance of non-small cell lung
cancer cells to MET kinase inhibition is mediated by a switch to
epidermal growth factor receptor dependency. Cancer Res.
70:1625–1634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qi J, McTigue MA, Rogers A, Lifshits E,
Christensen JG, Jänne PA and Engelman JA: Multiple mutations and
bypass mechanisms can contribute to development of acquired
resistance to MET inhibitors. Cancer Res. 71:1081–1091. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tiedt R, Degenkolbe E, Furet P, Appleton
BA, Wagner S, Schoepfer J, Buck E, Ruddy DA, Monahan JE, Jones MD,
et al: A drug resistance screen using a selective met inhibitor
reveals a spectrum of mutations that partially overlap with
activating mutations found in cancer patients. Cancer Res.
71:5255–5264. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gorre ME, Mohammed M, Ellwood K, Hsu N,
Paquette R, Rao PN and Sawyers CL: Clinical resistance to STI-571
cancer therapy caused by BCR-ABL gene mutation or amplification.
Science. 293:876–880. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED and Thompson EW: Epithelial-mesenchymal
and mesenchymal-epithelial transitions in carcinoma progression. J
Cell Physiol. 213:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thompson EW and Haviv I: The social
aspects of EMT-MET plasticity. Nat Med. 17:1048–1049. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie M, Zhang L, He CS, Xu F, Liu JL, Hu
ZH, Zhao LP and Tian Y: Activation of Notch-1 enhances
epithelial-mesenchymal transition in gefitinib-acquired resistant
lung cancer cells. J Cell Biochem. 113:1501–1513. 2012.PubMed/NCBI
|
|
29
|
Thomson S, Buck E, Petti F, Griffin G,
Brown E, Ramnarine N, Iwata KK, Gibson N and Haley JD: Epithelial
to mesenchymal transition is a determinant of sensitivity of
non-small-cell lung carcinoma cell lines and xenografts to
epidermal growth factor receptor inhibition. Cancer Res.
65:9455–9462. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mahadevan D, Cooke L, Riley C, Swart R,
Simons B, Della Croce K, Wisner L, Iorio M, Shakalya K, Garewal H,
et al: A novel tyrosine kinase switch is a mechanism of imatinib
resistance in gastrointestinal stromal tumors. Oncogene.
26:3909–3919. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Oliveira MJ, Costa AM, Costa AC, Ferreira
RM, Sampaio P, Machado JC, Seruca R, Mareel M and Figueiredo C:
CagA associates with c-met, E-cadherin and p120-catenin in a
multiproteic complex that suppresses Helicobacter
pylori-induced cell-invasive phenotype. J Infect Dis.
200:745–755. 2009. View
Article : Google Scholar : PubMed/NCBI
|