Introduction
Picfeltarraenin IA (IA) is extracted from the plant
Picria fel-terrae Lour and has been used in traditional
Chinese medicine as an acetylcholinesterase inhibitor (1). However, little is understood of the
mechanisms underlying the beneficial properties of IA that are
exploited today in the treatment of respiratory diseases.
Inflammation is a defense mechanism that arises to
remove injurious stimuli. However, prolonged inflammation may lead
to various disorders, including respiratory diseases (2). Due to anti-inflammatory agents serving
as potential therapeutics of inflammatory respiratory disorders,
the potential anti-inflammatory effects of IA warrant
investigation.
The initial tissue that meets inhaled allergens is
the respiratory epithelium, which has the ability to release
mediators and cytokines (3). As a
primary interface between the lungs and pathogens, epithelial cells
lining the airways and alveoli provide a physicochemical barrier,
responding to inhaled irritants by releasing various inflammatory
mediators (4).
A number of proinflammatory cytokines and
chemokines, consisting of interleukin (IL)-1, IL-6, IL-8, eotaxin,
granulocyte-macrophage colony-stimulating factor,
macrophage-inflammatory protein, and regulate on activation, normal
T-cell expressed and secreted, as well as anti-inflammatory
mediators, including prostaglandin E2 (PGE2) and nitric oxide (NO),
are all synthesized by respiratory epithelial cells. The
aforementioned cytokines and chemokines regulate inflammation by
altering cell recruitment, activation and survival (5). It has been previously reported that the
activation of respiratory epithelial cells is associated with
respiratory disorders, including asthma, chronic obstructive
pulmonary disease and cystic fibrosis, and also with respiratory
infections. Respiratory epithelial cell stimulation via the use of
inflammatory mediators results in the increased expression and
secretion of a number of cytokines with proinflammatory functions
(6). Of the chemical mediators that
are secreted by the epithelial cells, prostaglandins serve an
important role in the inflammatory processes of the respiratory
system. Prostaglandins are synthesized from arachidonic acid
through a reaction that is catalyzed by cyclooxygenase (COX). COX2,
an isoform of COX, is an inducible enzyme and is expressed in
response to inflammatory cytokines or lipopolysaccharide (LPS), the
primary component that forms the outer membrane of gram-negative
bacteria (7). Increased COX2
expression and PGE2 production has been observed to result from
inflammatory respiratory diseases. When tracheal and pulmonary
epithelial cells are induced to express COX2, they predominantly
release PGE2 (8).
As a fundamental chemokine secreted by lung
epithelial cells, IL-8 serves a crucial function in the recruitment
of inflammatory cells into the lung. The level of IL-8 produced is
observed to correlate with the severity of lung injury (9). Several studies have suggested that the
overexpression of IL-8 is essential to the pathophysiological
changes observed in chronic inflammatory lung disease (10,11). The
level of IL-8 is reportedly a crucial prognostic factor for acute
respiratory distress syndrome-associated mortality (12). The regulation of the respiratory
epithelial cell IL-8 response is therefore necessary to prevent
excessive inflammatory reactions that are injurious to the lung. In
acute lung injury, IL-8 production is reliant on gene products
released during early inflammatory stages (13). In respiratory epithelial cells, the
NF-κB pathway is activated, resulting in the translocation of cells
to the nucleus, where they bind and activate the IL-8 promoter.
Therefore, the NF-κB pathway activates IL-8 transcription. The
maximal production of IL-8 is dependent on cooperative interactions
with the NF-κB signaling pathways (14). Furthermore, NF-κB is a ubiquitously
expressed transcription factor family that regulates the expression
of multiple genes that are involved in the inflammatory and immune
responses, and also cellular proliferation (15).
In the present study, the effects of IA on COX2
expression, and IL-8 and PGE2 production were investigated through
the selective modulation of NF-κB in the human pulmonary epithelial
A549 cell line in vitro.
Materials and methods
Cell culture and chemicals
The human pulmonary adenocarcinoma epithelial A549
cell line and the human monocytic leukemia THP-1 cell line were
purchased from the American Type Culture Collection (Manassas, VA,
USA). Each cell type was suspended in Dulbecco's modified Eagle's
medium (Gibco; Thermo Fisher Scientific, Waltham, MA, USA)
incorporated with 4 mmol/l L-glutamine, 10% fetal bovine serum, 100
U/ml penicillin and 100 µg/ml streptomycin. Growing on culture
dishes, the cells were left until they achieved a subconfluence of
80–90%. Once this level had been reached, the cells were
dissociated by treatment with 0.2% trypsin/0.02% ethylenediamine
tetraacetic acid for a total of 5 min, and were subsequently
collected by centrifugation at 300 × g for 2 min. The cells were
plated at 2×104 cells/cm2 on 24-well plates,
and were maintained for 48 h until they reached optimal confluence.
Wu et al previously noted that A549 cells may respond to LPS
when under serum-free conditions (8).
To reduce the effect of serum-derived factors, the medium was
replaced by a serum-free culture medium. The experiments were
conducted 24 h later in the A549 cells.
IA with a purity of >98% was purchased from
Shanghai Tongtian Biotechnology Co., Ltd. (Shanghai, China). The IA
was dissolved in dimethyl sulfoxide (DMSO) as a 100-mmol/l solution
for the treatment of the cells. LPS and pyrrolidine dithiocarbamate
(PDTC) were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Polyclonal goat anti-mouse COX2 (cat. no. sc-23984; 1:800) and
polyclonal rabbit anti-mouse NF-κB p65 (cat. no. sc-372; 1:200)
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Monoclonal mouse anti-human glyceraldehyde
3-phosphate dehydrogenase (GAPDH) antibodies (cat. no. kc-5G4;
1:10,000) were obtained from Kangchen Biotech Co., Ltd. (Shanghai,
China) and monoclonal mouse anti-human toll-like receptor 4 (TLR4)
antibodies (cat. no. 14-9917-80; 1:100) were purchased from
eBioscience, Inc. (San Diego, CA, USA).
Cell viability assay
Cells in the exponential growth phase were seeded in
96-well culture plates (5,000 cells per well) and treated with
various concentrations of LPS (1, 10 and 100 µg/ml) and IA (0.1, 1,
10 and 100 µmol/l) for 6, 12 and 24 h and 24 h, respectively.
Following incubation, 20 µl methylthiazol-tetrazolium (MTT)
solution (5 mg/ml) was added to each well and the cells were
incubated for a further 4 h. The supernatant was then removed, and
the resulting crystals were dissolved in DMSO. The absorbance of
each well was measured using a VICTOR™X microplate reader
(PerkinElmer Inc., Waltham, MA, USA) at 570 nm. The cell viability
was calculated using the following formula: Cell viability (%) =
(average absorbance of treated cells / average absorbance of
control cells) × 100. A lactate dehydrogenase detection kit (Reckon
Diagnostics Pvt. Ltd., Vadodara, India) was used to evaluate the
lactate dehydrogenase released from the cells following treatment
with the different concentrations of LPS and IA. The kit was used
according to the manufacturer's protocols.
Western blot analysis
Following treatment with the test agents, the cells
were washed twice in phosphate-buffered saline and lysed in 400 µl
lysis buffer [62.5 mmol/l Tris-HCl (pH 6.8), 2% SDS, 10% glycerol,
0.01% bromophenol blue and 5% 2-mercaptoethanol]. Western blot
analysis was performed as described previously (16). Following this, the protein was
separated using electrophoresis on a 10% polyacrylamide gel at 200
V for 80 min. The protein was transferred to a polyvinylidene
fluoride membrane, which was subsequently blocked and incubated
with a specific goat polyclonal antibody against COX2 (1:800), a
specific rabbit polyclonal antibody against NF-κB (1:200) and a
specific mouse polyclonal antibody against GAPDH (1:10,000).
Chemiluminescence was observed using an enhanced chemiluminescence
kit (GE Healthcare Life Sciences, Chalfont, UK). Quantitative
analysis of the western blotting was performed using AlphaEase FC
software (version 4.1.0; Alpha Innotech Corporation, San Leandro,
CA, USA).
Enzyme-linked immunosorbent assays
(ELISAs) for IL-8 and PGE2 production
Following treatment with the test agents, the cell
culture medium was collected and the levels of IL-8 and PGE2 were
measured using ELISAs; specifically, through the use of IL-8 and
PGE2 assay kits (R&D Systems Europe, Abingdon, UK),
respectively. The assay was conducted by comparing the unlabeled
IL-8/PGE2 in the sample and the fixed amount of labeled
IL-8/PGE2.
Statistical analysis
The data are expressed as the mean ± standard
deviation of at least three independent experiments. To determine
the differences between the groups, Tukey's all pairwise comparison
was used. All statistical analyses were performed using SPSS 16.0
statistical software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of LPS on A549 cell
viability
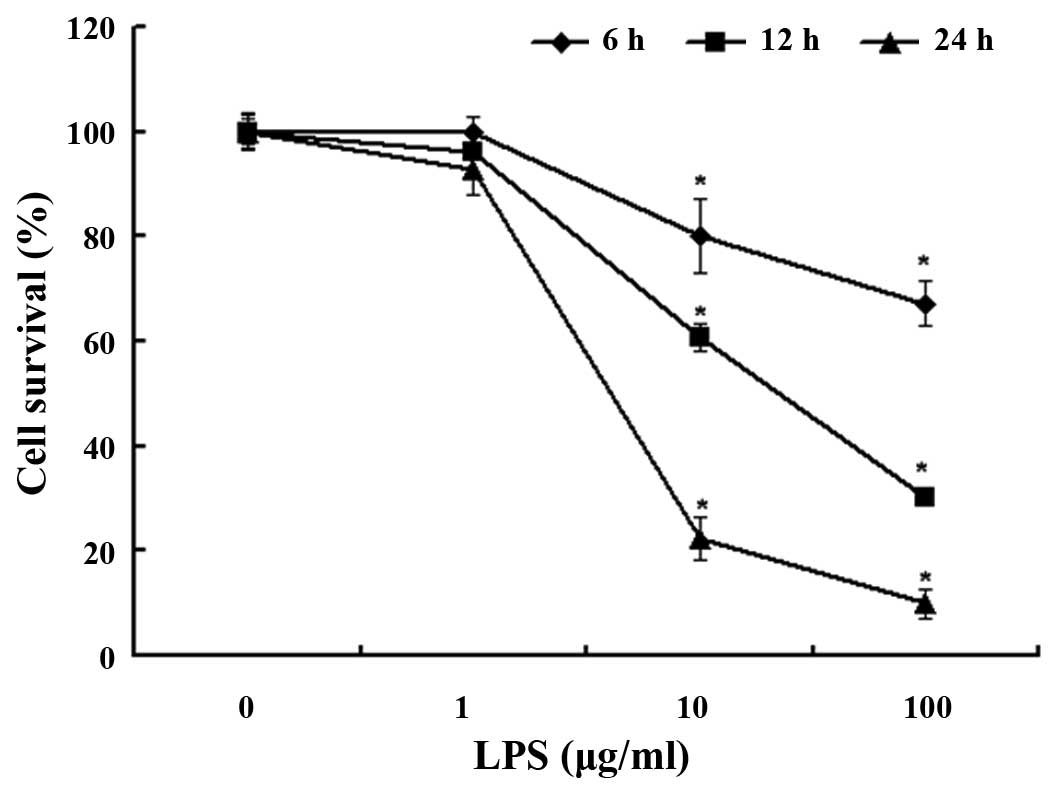
To investigate the effect of LPS on the A549 cells,
the cells were treated with 1, 10 and 100 µg/ml LPS at 6, 12 and 24
h. Cell viability was observed using an MTT assay. The survival
rate was ~80% after 6 h, 60% after 12 h and 22% after 24 h with LPS
10 µg/ml treatment (Fig. 1). The cell
viability of the LPS-treated group was significantly decreased when
compared with the untreated cells (P<0.05). These results
suggest that the A549 cells were damaged by LPS in a dose- and
time-dependent manner.
Effect of IA on A549 cell
viability
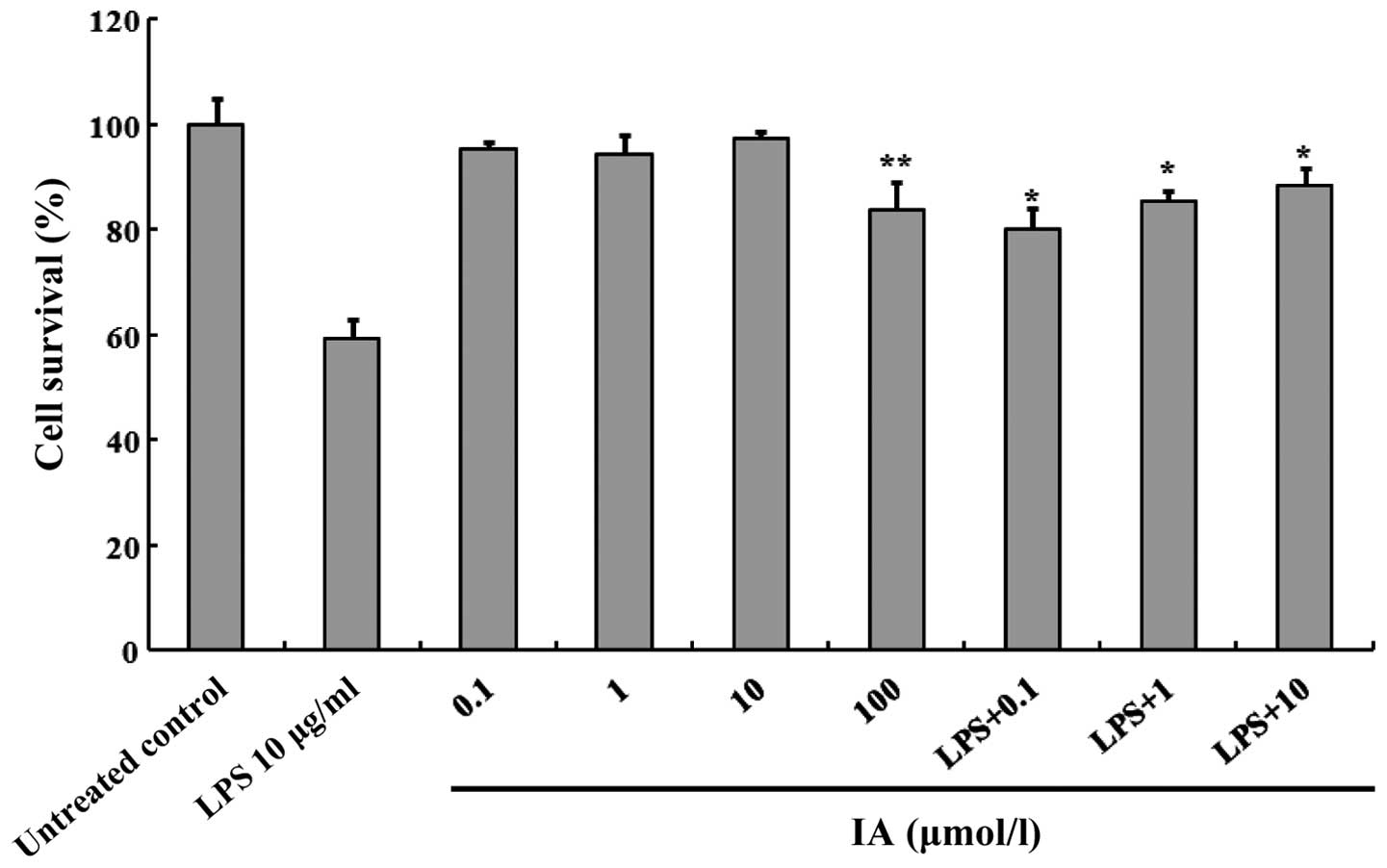
The effect of IA on the viability of the A549 cells
was subsequently investigated. The cells were incubated with
varying IA concentrations for a period of 12 h in a serum-free
medium. Cell viability was determined using an MTT reduction assay.
As presented in Fig. 2, a
concentration of 100 µmol/l IA was observed to significantly
decrease cellular MTT reduction (P<0.05), but did not exhibit
any toxicity at ≤10 µmol/l. Additionally, IA was tested at 0.1–10
µmol/l to determine whether it improved LPS-induced cell
inhibition. For this experiment, the A549 cells were treated with
10 µg/ml LPS in the presence or absence of IA at a concentration of
0.1–10 µmol/l. IA significantly increased cell growth in the
presence of 10 µg/ml LPS (P<0.05). The results suggested that IA
possesses the ability to attenuate the cell injury induced by
LPS.
Effect of IA on LPS-induced IL-8 and
PGE2 production in A549 cells
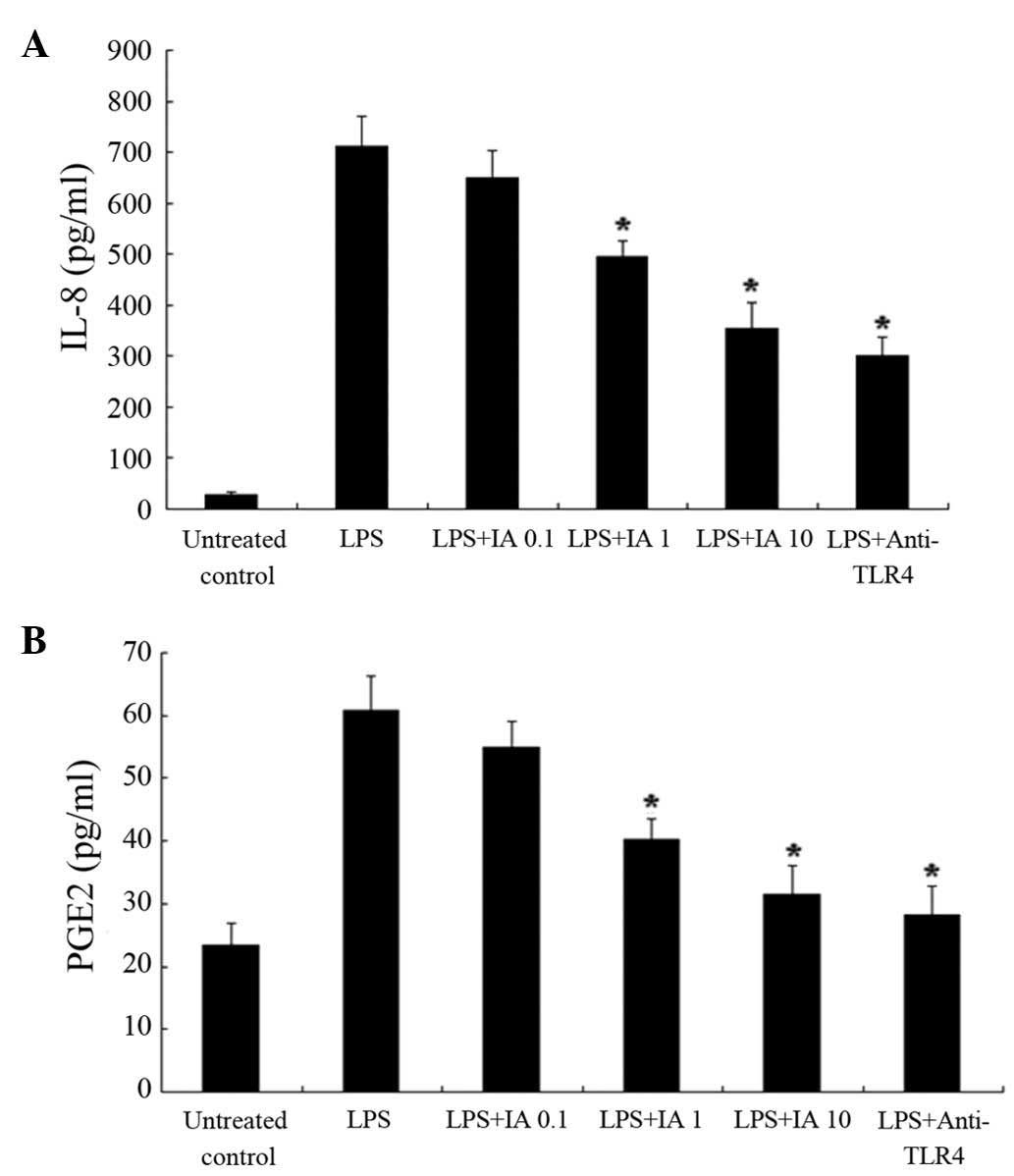
LPS was used as an inflammatory agent in the
subsequent experiment. To investigate the effect of IA on the
production of LPS-induced IL-8 and PGE2 (a major COX product in
A549 cells), 0.1–10 µmol/l IA was added concomitantly with 10 µg/ml
LPS. The level of IL-8 and PGE2 in the medium was quantitatively
measured using an ELISA. As presented in Fig. 3, LPS significantly increased the
amount of IL-8 and PGE2. The LPS-induced IL-8 production was
significantly reduced by nearly 31 and 50% by 1 and 10 µmol/l IA,
respectively (P<0.05). PGE2 production was reduced significantly
by nearly 34 and 48% by 1 and 10 µmol/l IA (P<0.05),
respectively. In addition, TLR4, the primary receptor for the
recognition of LPS on the cell surface, was used as a positive
control. LPS-induced IL-8 and PGE2 production in the A549 cells was
abolished by the presence of a neutralizing antibody against TLR4
(20 µg/ml).
Effect of IA on COX2 expression
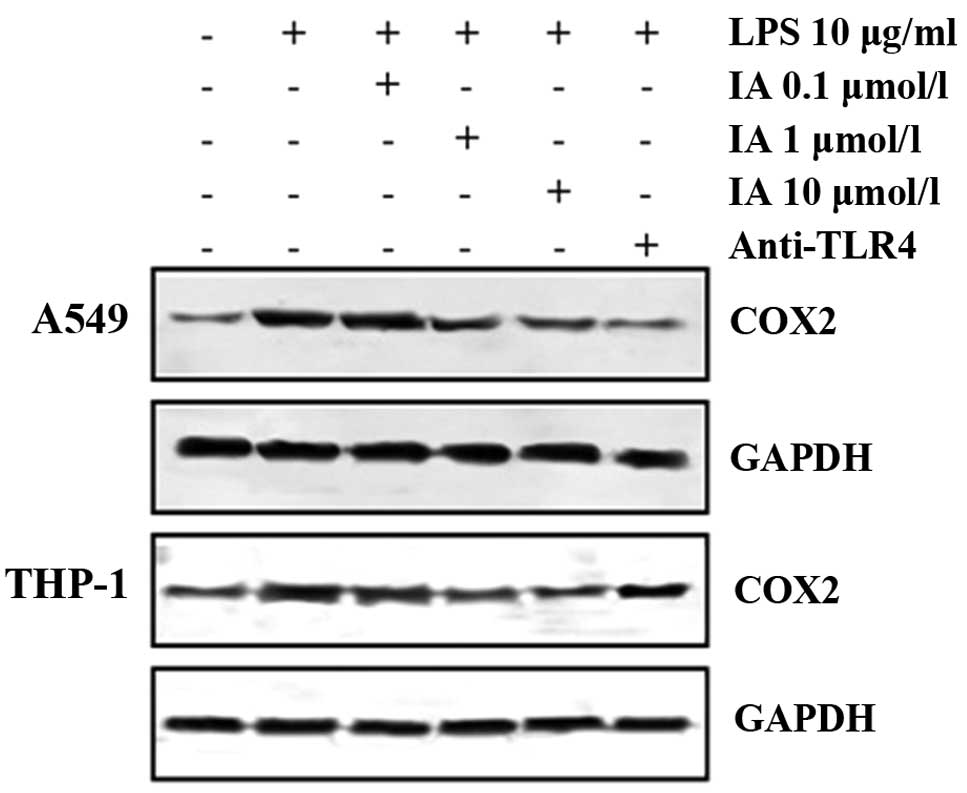
To examine the sensitivity of A549 cells to LPS, the
cells were incubated with LPS at a concentration of 10 µg/ml for 12
h. COX2 expression was analyzed using western blot analysis with
anti-COX2 antibodies. As presented in Fig. 4, the expression of COX2 was
significantly increased by treatment with LPS. The expression of
COX2, induced by LPS, in the A549 cells was suppressed by the
presence of a neutralizing antibody against TLR4, indicating that
the response is mediated by TLR4 activation. Furthermore,
LPS-induced COX2 expression was significantly inhibited by the
presence of 10 µmol/l IA (P<0.05) and was inhibited overall by
IA in a concentration-dependent manner. To observe whether the
aforementioned effect of IA was specific to the A549 cells, the
human monocytic THP-1 cell line was also analyzed. As presented in
Fig. 4, the expression of COX2 in the
THP-1 cells was substantially increased by treatment with LPS. The
LPS-induced COX2 expression in the THP-1 cells was also inhibited
by 1 and 10 µmol/l IA.
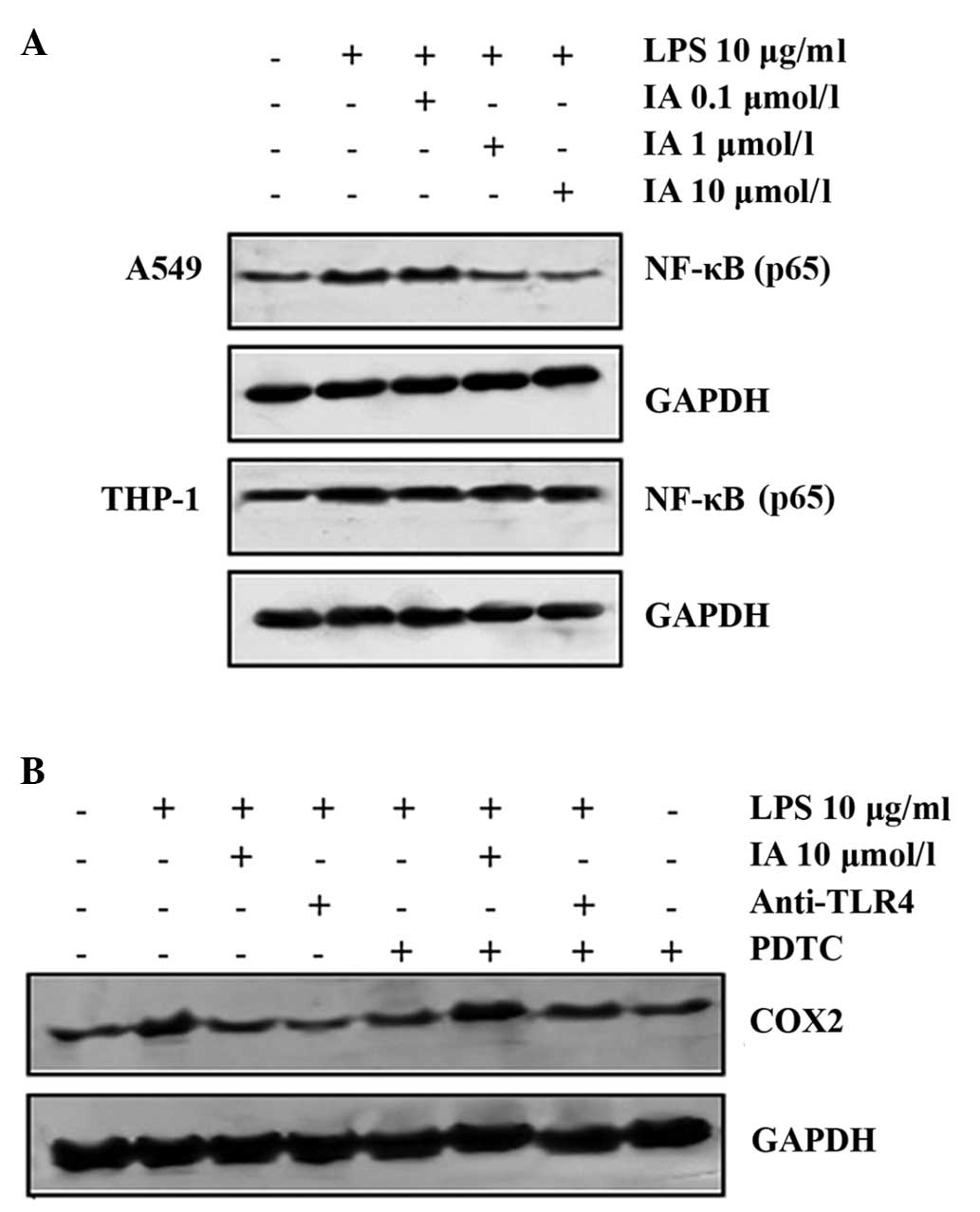
IA inhibits COX2 production through
the NF-κB pathway
Expanding on previous studies that have reported
NF-κB activation as a key signaling mechanism of LPS-induced COX2
expression in A549 and RAW 264.7 cells (17), the molecular basis for IA inhibition
of NF-κB-mediated inflammatory cytokine production was
investigated. Upon IA treatment, the LPS-induced NF-κB was
suppressed in the A549 cells (P<0.05), but not in the THP-1
cells (Fig. 5A). To confirm the
inhibition of COX2 production by the NF-κB pathway, the effect of
IA treatment on LPS-induced COX2 in the A549 cells in the presence
or absence of PDTC (a specific inhibitor of NF-κB) was assessed.
Fig. 5B demonstrates that IA
inhibited LPS-induced COX2 production. Furthermore, PDTC (50
µmol/l) reversed IA-suppressed COX2 production in the A549 cells.
Therefore, it can be concluded that IA suppresses COX2 production
via the NF-κB pathway.
Discussion
IA was isolated and characterized from the whole
plant of Picria fel-terrae Lour (18). However, the mechanism of IA is not
well understood. The present study is the first of its type to
investigate the immunoregulatory functions of IA on LPS-induced
COX2, IL-8 and PGE2 responses in lung epithelial cells.
Additionally, the study also established that the immunoregulatory
effects of IA on LPS-induced epithelial cell responses are mediated
partially by NF-κB inhibition.
The key finding of the current study was that IA
significantly inhibited LPS-induced COX-2 expression, and IL-8 and
PGE2 production in human pulmonary adenocarcinoma epithelial A549
cells. As the increased expression of COX2, and the production of
IL-8 and PGE2 are indicated to serve an important role in airway
inflammatory mechanisms, the inhibitory effect of IA may therefore
facilitate the treatment of respiratory diseases.
In the present study, LPS was employed to induce
cytokine release in the A549 cells. The primary receptor for LPS is
TLR4 (19), with its signaling
requiring numerous accessory proteins, including cluster of
differentiation (CD)14 and lymphocyte antigen 96 (20,21). It
has been previously reported that A549 cells respond to LPS through
TLR4 in the presence of 1% serum, but that in the absence of serum
they are unresponsive to LPS due to the low expression of CD14 and
TLR4 (22). In the present study, the
A549 cells responded to LPS and the response was subsequently
blocked by an anti-TLR4 neutralizing antibody, therefore indicating
that the A549 cell response is mediated by TLR4. The A549 cell
cultures were moved to a serum-free medium 24 h prior to assays,
however, serum influence may have remained in the cultures.
IL-8 is a chemokine that possesses a wide spectrum
of responsibilities, including chemotaxis, cell adhesion, reactive
oxygen species generation, angiogenesis promotion and azurophil
granule release (23). Despite IL-8
being commonly recognized as a neutrophil-specific chemokine, it
has also been indicated to modulate the function of a number of
inflammatory cells, including T and B lymphocytes, natural killer
cells, basophils, monocytes and eosinophils. The proinflammatory
and regulatory cytokine IL-8 can be identified in lung inflammation
(24). In the present study, IA
decreased LPS-induced IL-8 production in the A549 cells (Fig. 3A). PGE2 has been previously reported
to activate IL-8 gene expression in certain cell types.
Prostaglandins, particularly PGE2 are produced in large quantities
in osteoarthritic cartilage, and function as modulators of
inflammation, inflammatory pain and tissue destruction (25). Prostaglandins are synthesized from
arachidonic acid through the production of COX enzymes and
prostaglandin synthases (26). The
present study observed that IA also suppressed PGE2 production
(Fig. 3B) and COX2 expression
(Fig. 4). These results suggest that
IA has possible anti-inflammatory effects.
A previous study (27)
indicated that IA extracts suppressed the expression of COX2, the
production of NO and PGE2, and the secretion of the proinflammatory
cytokines IL-6, IL-8 and tumor necrosis factor-α in LPS-mediated
RAW264.7 cells. Furthermore, these effects were demonstrated to be
closely associated with the inhibition of the NF-κB p50/p65
subunits (27). Therefore, in the
present study, the effect on LPS-induced COX2 expression in THP-1
cells in the presence of IA was observed (Fig. 4). It has been reported that NF-κB is a
key signaling mechanism in LPS-induced COX2 expression (28). IA is likely to act on these common
mechanisms, thus inducing COX2 expression in the A549 and THP-1
cells (Fig. 4).
The present study has demonstrated that IA can
downregulate IL-8 and PGE2 production and COX2 expression via the
pulmonary epithelial cell line in response to LPS. The results
indicated that the mechanism of this modulation was partially
initiated by NF-κB inhibition. Further understanding of the
molecular mechanisms by which inflammation is regulated in the
lungs is required to aid the production of effective therapeutics
that may eventually treat inflammatory lung diseases. Having
demonstrated the possible advantages of the anti-inflammatory
effects of IA for the treatment of respiratory disorders in the
present study, future studies may decide to focus on the effects of
IA using animal models of airway inflammation in vivo.
Acknowledgements
This study was financially supported by the Shanghai
Natural Science Foundation (grant no. 15ZR1442000), the Budgetary
Research Projects of the Shanghai Municipal Education Commission
(grant no. 2014YSN41) and the Doctoral Fund of the Ministry of
Education of China (grant no. 20123107120010).
References
|
1
|
Wen L, Wei Q, Chen G, Liu F, Zhang S and
You T: Bioassay and liquid chromatography/mass spectrometry-guided
acetylcholinesterase inhibitors from Picriafel-terrae. Pharmacogn
Mag. 9((Suppl 1)): S25–S31. 2013.PubMed/NCBI
|
|
2
|
Ueki T, Akaishi T, Okumura H and Abe K:
Extract from Nandina domestica inhibits
lipopolysaccharide-induced cyclooxygenase-2 expression in human
pulmonary epithelial A549 cells. Biol Pharm Bull. 35:1041–1047.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bals R and Hiemstra PS: Innate immunity in
the lung: How epithelial cells fight against respiratory pathogens.
Eur Respir J. 23:327–333. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Skerrett SJ, Liggitt HD, Hajjar AM, et al:
Respiratory epithelial cells regulate lung inflammation in response
to inhaled endotoxin. Am J Physiol Lung Cell Mol Physiol.
287:L143–L152. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cornell TT, Hinkovska-Galcheva V, Sun L,
et al: Ceramide-dependent PP2A regulation of TNFalpha-induced IL-8
production in respiratory epithelial cells. Am J Physiol Lung Cell
Mol Physiol. 296:L849–L856. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abate W, Alghaithy AA, Parton J, Jones KP
and Jackson SK: Surfactant lipids regulate LPS-induced
interleukin-8 production in A549 lung epithelial cells by
inhibiting translocation of TLR4 into lipid raft domains. J Lipid
Res. 51:334–344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen H, Ma F, Hu X, Jin T, Xiong C and
Teng X: Elevated COX2 expression and PGE2 production by
downregulation of RXRα in senescent macrophages. Biochem Biophys
Res Commun. 440:157–162. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu S, Duan S, Zhao S, et al: Atorvastatin
reduces lipopolysaccharide-induced expression of cyclooxygenase-2
in human pulmonary epithelial cells. Respir Res. 6:272005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin CH, Nai PL, Bien MY, Yu CC and Chen
BC: Thrombin-induced CCAAT/enhancer-binding protein β activation
and IL-8/CXCL8 expression via MEKK1, ERK, and p90 ribosomal S6
kinase 1 in lung epithelial cells. J Immunol. 192:338–348. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin CH, Yu MC, Chiang CC, et al:
Thrombin-induced NF-κB activation and IL-8/CXCL8 release is
mediated by c-Src-dependent Shc, Raf-1, and ERK pathways in lung
epithelial cells. Cell Signal. 25:1166–1175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maza PK, Oliveira P, Toledo MS, et al:
Paracoccidioides brasiliensis induces secretion of IL-6 and
IL-8 by lung epithelial cells. Modulation of host cytokine levels
by fungal proteases. Microbes Infect. 14:1077–1085. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hildebrand F, Stuhrmann M, van Griensven
M, et al: Association of IL-8-251A/T polymorphism with incidence of
Acute Respiratory Distress Syndrome (ARDS) and IL-8 synthesis after
multiple trauma. Cytokine. 37:192–199. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bao Z, Ye Q, Gong W, Xiang Y and Wan H:
Humanized monoclonal antibody against the chemokine CXCL-8 (IL-8)
effectively prevents acute lung injury. Int Immunopharmacol.
10:259–263. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Choi KC, Lee YH, Jung MG, et al: Gallic
acid suppresses lipopolysaccharide-induced nuclear factor-kappaB
signaling by preventing RelA acetylation in A549 lung cancer cells.
Mol Cancer Res. 7:2011–2021. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu L, Gu H, Liu H, et al: Protective
effect of resveratrol against IL-1β-induced inflammatory response
on human osteoarthritic chondrocytes partly via the
TLR4/MyD88/NF-κB signaling pathway: An ‘in vitro study’. Int J Mol
Sci. 15:6925–6940. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou QM, Wang XF, Liu XJ, et al: Curcumin
improves MMC-based chemotherapy by simultaneously sensitising
cancer cells to MMC and reducing MMC-associated side-effects. Eur J
Cancer. 47:2240–2247. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Du X, He S, Jiang Y, Wei L and Hu W:
Adiponectin prevents islet ischemia-reperfusion injury through the
COX2-TNFα-NF-κB-dependent signal transduction pathway in mice. J
Endocrinol. 218:75–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zou JM, Wang LS, Ma XM, Guo YJ and Shi RB:
A new cucurbitacin from Picria fel-terrae. J Asian Nat Prod Res.
8:367–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Medzhitov R, Preston-Hurlburt P and
Janeway CA Jr: A human homologue of the Drosophila Toll
protein signals activation of adaptive immunity. Nature.
388:394–397. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Frey EA, Miller DS, Jahr TG, et al:
Soluble CD14 participates in the response of cells to
lipopolysaccharide. J Exp Med. 176:1665–1671. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shimazu R, Akashi S, Ogata H, et al: MD-2,
a molecule that confers lipopolysaccharide responsiveness on
Toll-like receptor 4. J Exp Med. 189:1777–1782. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thorley AJ, Grandolfo D, Lim E, et al:
Innate immune responses to bacterial ligands in the peripheral
human lung - role of alveolar epithelial TLR expression and
signalling. PLoS One. 6:e218272011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hay DW and Sarau HM: Interleukin-8
receptor antagonists in pulmonary diseases. Curr Opin Pharmacol.
1:242–247. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mukaida N: Pathophysiological roles of
interleukin-8/CXCL8 in pulmonary diseases. Am J Physiol Lung Cell
Mol Physiol. 284:L566–L577. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vuolteenaho K, Koskinen A, Kukkonen M, et
al: Leptin enhances synthesis of proinflammatory mediators in human
osteoarthritic cartilage - mediator role of NO in leptin-induced
PGE2, IL-6, and IL-8 production. Mediators Inflamm.
2009:3458382009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Higaki T, Okano M, Fujiwara T, et al:
COX/PGE(2) axis critically regulates effects of LPS on
eosinophilia-associated cytokine production in nasal polyps. Clin
Exp Allergy. 42:1217–1226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang DJ, Chang YY, Lin HW, Chen YC, Hsu SH
and Lin JT: Inhibitory effect of litchi (Litchi chinensis
Sonn.) flower on lipopolysaccharide-induced expression of
proinflammatory mediators in RAW264.7 cells through NF-κB, ERK, and
JAK2/STAT3 inactivation. J Agric Food Chem. 62:3458–3465. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee KH, Yeh MH, Kao ST, et al: Xia-bai-san
inhibits lipopolysaccharide-induced activation of intercellular
adhesion molecule-1 and nuclear factor-kappa B in human lung cells.
J Ethnopharmacol. 124:530–538. 2009. View Article : Google Scholar : PubMed/NCBI
|