Introduction
Cancer cells differ from untransformed cells by a
large number of bioenergetic and metabolic changes, which sustain
their high rate of growth and proliferation (1–3). According
to the Warburg effect, the bioenergetic switch from a high-energy
yielding process (TCA cycle) to a low energy yielding process
(glycolysis) is a hallmark of most cancer cells. Therefore,
targeting of metabolic adaptations that inhibit glycolysis has been
widely studied, as part of the search for novel therapies for
cancer cells (4,5). Currently, inhibitors of glycolysis and
agents that impair mitochondrial oxidative phosphorylation (OXPHOS)
have been exploited to target malignant cancer cells by inducing
apoptosis or autophagic cell death (4,5).
ErbB2 (Her2/neu) is an oncogene that is
overexpressed in ~30% of breast cancers, and its expression is
correlated with a poor prognosis (6).
The overexpression of ErbB2 increases the transformation and/or
metastatic potential of breast cancer cells (7). Additionally, it has been reported that
overexpression of ErbB2 leads to increased glucose uptake, lactate
production and decreased oxygen consumption in multiple human
breast cancer cell lines (8). In a
recent study, a novel mitochondrial localization of ErbB2 that
negatively regulates mitochondrial respiration in breast cancer
cells was reported (9). Furthermore,
breast cancer cells with high levels of mitochondrial ErbB2 were
more resistant to the ErbB2-targeting antibody trastuzumab
(9).
Hexokinase (HK) is a key enzyme that catalyzes the
first step in the glycolysis pathway. This enzyme transfers a
phosphate group from ATP to glucose, forming glucose-6-phosphate.
In human cells, there are four isoforms of HK (I–IV). Regulation of
HK II is involved in glucose or lactate metabolism, and blocking
its activity is an effective way of inhibiting glycolysis (10). Glycolysis of cancer cell metabolism is
associated with the binding of HK II to the outer mitochondrial
membrane protein voltage-dependent anion channel, and this
association appears important for mitochondrial homeostasis
(10,11). HK II association with the
mitochondrial outer membrane is involved in mitochondria-mediated
apoptosis and increased resistance of cancer cells to
chemotherapeutic drugs (11,12). 3-Bromopyruvate (3-BrPA) is a
halogenated and alkylating analog of pyruvic acid, and is able to
inhibit glycolysis through the dissociation of HK II from
mitochondria (13). Previous studies
demonstrated that 3-BrPA can induce cell death through the
mitochondria-mediated intrinsic apoptotic pathway, which is coupled
with the release of cytochrome c from mitochondria to
cytoplasm leading to activation of the caspase cascade (14). The aim of this study was to examine
whether inhibition of glucose metabolism pathway may specifically
inhibit the ErbB2-overexpressing cells. Our study may provide novel
perspectives for clinical applications of cancer cell treatment by
targeting on the ErbB2-promoted glycolysis.
Materials and methods
Cell lines and culture conditions
The MCF7, MDA-MB-231 and BT474 human breast cancer
cell lines were purchased from the American Type Culture Collection
(Manassas, VA, USA). The cells were cultured in Dulbecco's modified
Eagle's medium (DMEM)/F-12 (Mediatech Inc., Manassas, VA, USA) with
10% fetal bovine serum (FBS) at 37°C, in a 5% CO2
humidified incubator.
Western blotting and antibodies
The cells were harvested and lysed in a buffer
containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1%
Triton, 1 mM PMSF and Protease Inhibitor Cocktail (Sigma, St.
Louis, MO, USA) for 20 min on ice. Lysates were cleared by
centrifugation at 14,000 rpm at 4°C for 10 min. Supernatants were
collected and protein concentrations were determined by the
Bradford assay (Bio-Rad, Hercules, CA, USA). The proteins were then
separated with a SDS/polyacrylamide gel and transferred to a
nitrocellulose membrane (Bio-Rad). After blocking in
phosphate-buffered saline (PBS) with 5% non-fat dry milk for 1 h,
the membranes were incubated overnight at 4–8°C with the primary
antibodies in PBS with 5% non-fat dry milk. The following
antibodies were utilized: anti-ErbB2 mouse antibody (1:1000, OP15,
Calbiochem, National Harbor, MD, USA), anti-HK II mouse antibody
(1:1000, no. 8169, Cell Signaling Technology Inc., Beverly, MA,
USA), anti-prohibitin rabbit antibody (1:1000, no. 2426, Cell
Signaling Technology Inc.), anti-α-Tubulin rabbit antibody (1:1000,
no. 2125, Cell Signaling Technology Inc.), anti-mtHSP70 mouse
antibody (1:1000, MA3-028, Thermo Scientific, Carlsbad, CA, USA),
anti-cytochrome C rabbit antibody (1:1000, no. 11940, Cell
Signaling Technology Inc.), anti-PARP rabbit antibody (1:1000, no.
9542, Cell Signaling Technology Inc.), anti-AIF rabbit antibody
(1:1000, no. 4642, Cell Signaling Technology Inc.) and anti-β-actin
rabbit antibody (1:2000, no. 4970, Cell Signaling Technology Inc.).
Membranes were extensively washed with PBS and incubated with
horseradish peroxidase-conjugated secondary anti-mouse antibody or
anti-rabbit antibody (1:2,000, Bio-Rad). After additional washes
with PBS, antigen-antibody complexes were visualized with the
enhanced chemiluminescence kit (Pierce, Rockford, IL, USA).
Cell viability assay
The cancer cells were treated with 3-BrPA (Sigma
Aldrich, St. Louis, MO, USA) with the indicated concentrations for
24 h. The cells were seeded in a 96-well plate, at a density of
3×103 cells/well in 0.2 ml DMEM containing 10% FBS.
After overnight incubation under the same cultivating conditions,
each well was refreshed with 0.2 ml serum-free medium (SFM) for
another day. The cells were then treated with 0.2 ml SFM containing
various concentrations of 3-BrPA. The drug-containing SFM was
refreshed after 2 days, and incubated under the same conditions for
another 2 days. Cell viability was accessed with an MTT reagent
(Sigma Diagnostics, Inc., St. Louis, MO, USA), and by measuring the
absorbance at 590 nm using a SpectraMax microplate reader
(340PC384; Molecular Devices, Sunnyvale, CA, USA). Relative
viability was obtained from the absorbance at 590 nm (A590 nm) of
drug-treated cells/A590 nm of untreated cells. The experiment was
repeated three times.
Small interfering (si)RNA and plasmid
DNA transfection
Specific siRNA for the knockdown of ErbB2 was
purchased from Sigma-Aldrich (MISSION® siRNA SIHK0723) and a
plasmid vector containing wild-type Myc-DDK-tagged ErbB2 (cat no.
RC212583) was purchased from OriGene Technologies, Inc., Rockville,
MD, USA. Transfection was performed using the Oligofectamine™
transfection reagent (Invitrogen Life Technologies, Carlsbad, CA,
USA) according to the manufacturer's instructions. Forty-eight
hours after transfection, whole-cell lysates were prepared for
further analysis by Western blot and cytotoxicity assay.
Isolation of mitochondria
High-purity, intact mitochondria were isolated using
the Qproteome® Mitochondria Isolation kit (cat no. 37612, Qiagen
GmbH, Hilden, Germany) according to the manufacturer's
instructions.
Detection of mitochondrial membrane
potential
Mitochondrial membrane potential was detected using
a kit from BD™ MitoScreen (JC-1; BD Pharmingen, San Diego, CA, USA)
according to the manufacturer's instructions.
HK activity
HK activity was measured using the Hexokinase
colorimetric assay kit (Sigma-Aldrich) according to the
manufacturer's instructions. Absorbance was measured at 563 nm
using a SpectraMax M5 plate reader (Molecular Devices). One unit of
HK is the amount of enzyme that will generate 1.0 mM of NADH per
min at pH 8.0 and room temperature. The results were normalized to
the amount of total protein compared to the control cells.
Xenograft experiments
Female athymic nude mice were used in the present
study. Nude mice were subcutaneously injected into the right flank
with 2×106 MDA-MB-231 empty vector cells (231V) or
MDA-MB-231 ErbB2 cells (231 ErbB2). When the tumors reached >150
mm3 in size, the mice were randomly divided into four
groups (8 mice per group) as follows: i) 231V cells treated with
PBS (control) and 3-BrPA (10 mg/kg intraperitoneal, twice/week for
3 weeks); ii) 231 ErbB2 cells treated with PBS-treated control and
3-BrPA. Mice were weighed weekly and tumor diameters were measured
with calipers twice per week for >5 weeks. The tumor volumes
were calculated using the formula: volume (mm3) = (W ×
L)/2, where W and L are the minor and major diameters (in mm),
respectively. All of the experiments involving mouse models
complied with Chinese laws and the guidelines of the Ethics
Committee of Jilin University, Changchun, China.
Statistical analysis
The unpaired Student's t-test was used for data
analysis. Data were shown as mean ± standard error (SE). P<0.05
was considered to represent a statistically significant
difference.
Results
ErbB2-positive breast cancer cells are
more sensitive to glucose starvation
A previous study demonstrated that ErbB2 promotes
glycolysis through the activation of LDHA in breast cancer cells
(8). To evaluate the ability of ErbB2
to modulate sensitivity of breast cancer cells to glucose
starvation, an overexpressed vector containing wild-type ErbB2 was
transfected into two ErbB2-negative cell lines, MDA-MB-231 and
MCF7. Additionally, ErbB2 expression was knocked down by siRNA in
BT474 cells, which normally express high levels of ErbB2. The
enforced expression and knockdown of ErbB2 protein expression was
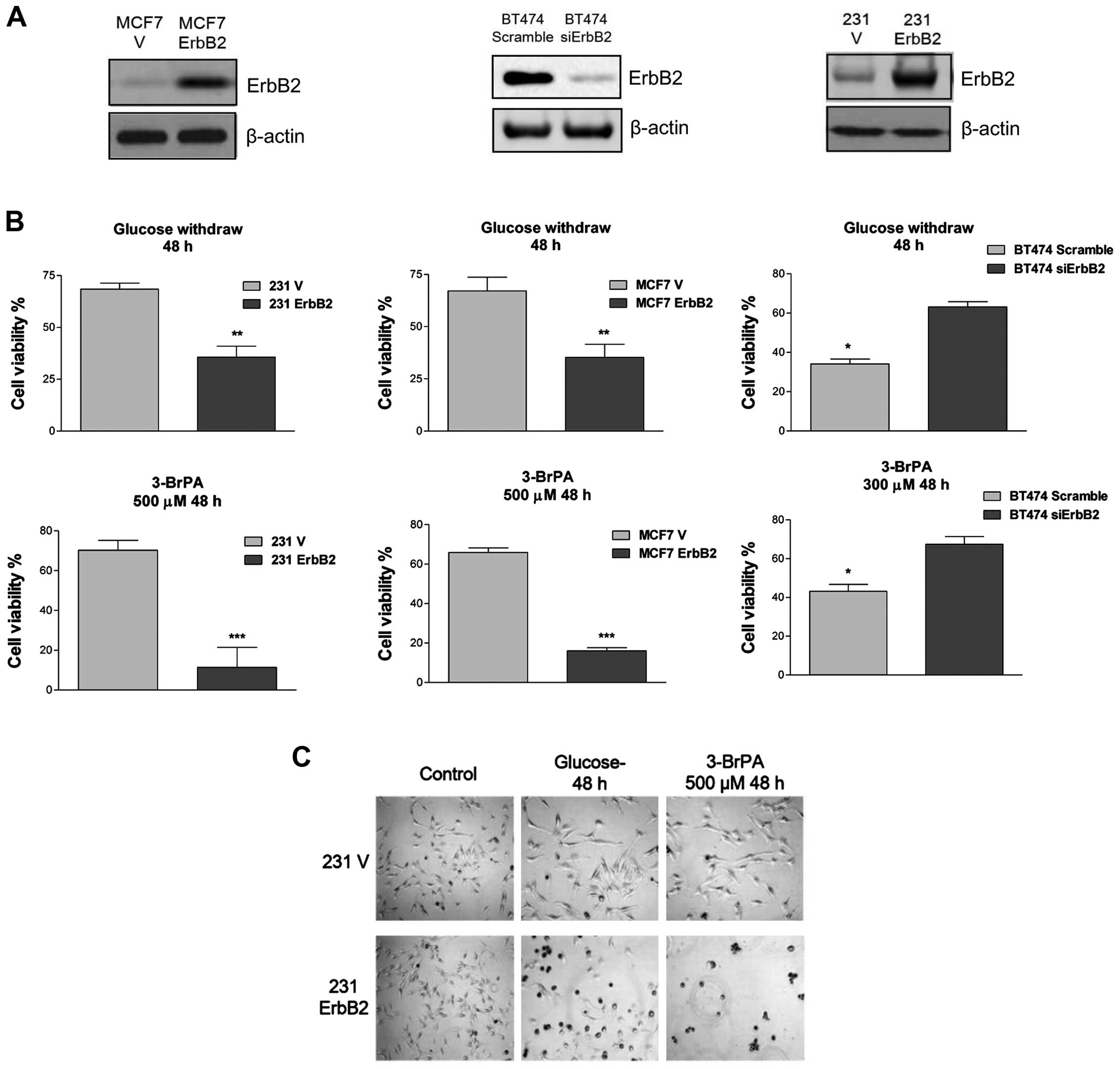
confirmed by western blot analysis (Fig.
1A). To investigate whether ErbB2 signaling is involved in
rapid cell death following glucose withdrawal, the present study
examined the response of these cell lines to the withdrawal of
glucose and the glycolytic product pyruvate, an alternate substrate
for the TCA cycle. Within 48 h of glucose and pyruvate withdrawal,
cells with a high ErbB2 expression (231ErbB2, MCF7ErbB2 and BT474)
exhibited rapid cell death, whereas cells that expressed low levels
of ErbB2 exhibited only a minor (25%) loss of viability (Fig. 1B). These results indicated that
overexpression of ErbB2 in breast cancer cells enhanced their
sensitivity to nutrition depletion. Following this, the viability
of these cells in response to treatment with 3-BrPA, a glycolysis
inhibitor that specifically targets HK II, was determined. The
231ErbB2 and MCF7ErbB2 cells exhibited a large decrease in the
viability ratio compared with control cells, and knockdown of ErbB2
in BT474 cells revealed resistance to 3-BrPA treatment (Fig. 1C). This indicated that breast cancer
cells overexpressing ErbB2 were more sensitive to the glycolysis
inhibitor, the latter being a potential therapeutic target for the
treatment of ErbB2-positive breast cancer patients.
ErbB2 enhances mitochondrial HK II
distribution and increases HK activity
The putative mechanisms underlying ErbB2-mediated
sensitivity to glucose starvation were investigated. Since the
major function of the 3-BrPA glycolysis inhibitor is to dissociate
HK II from mitochondrial outer membrane, the expression of HK II in
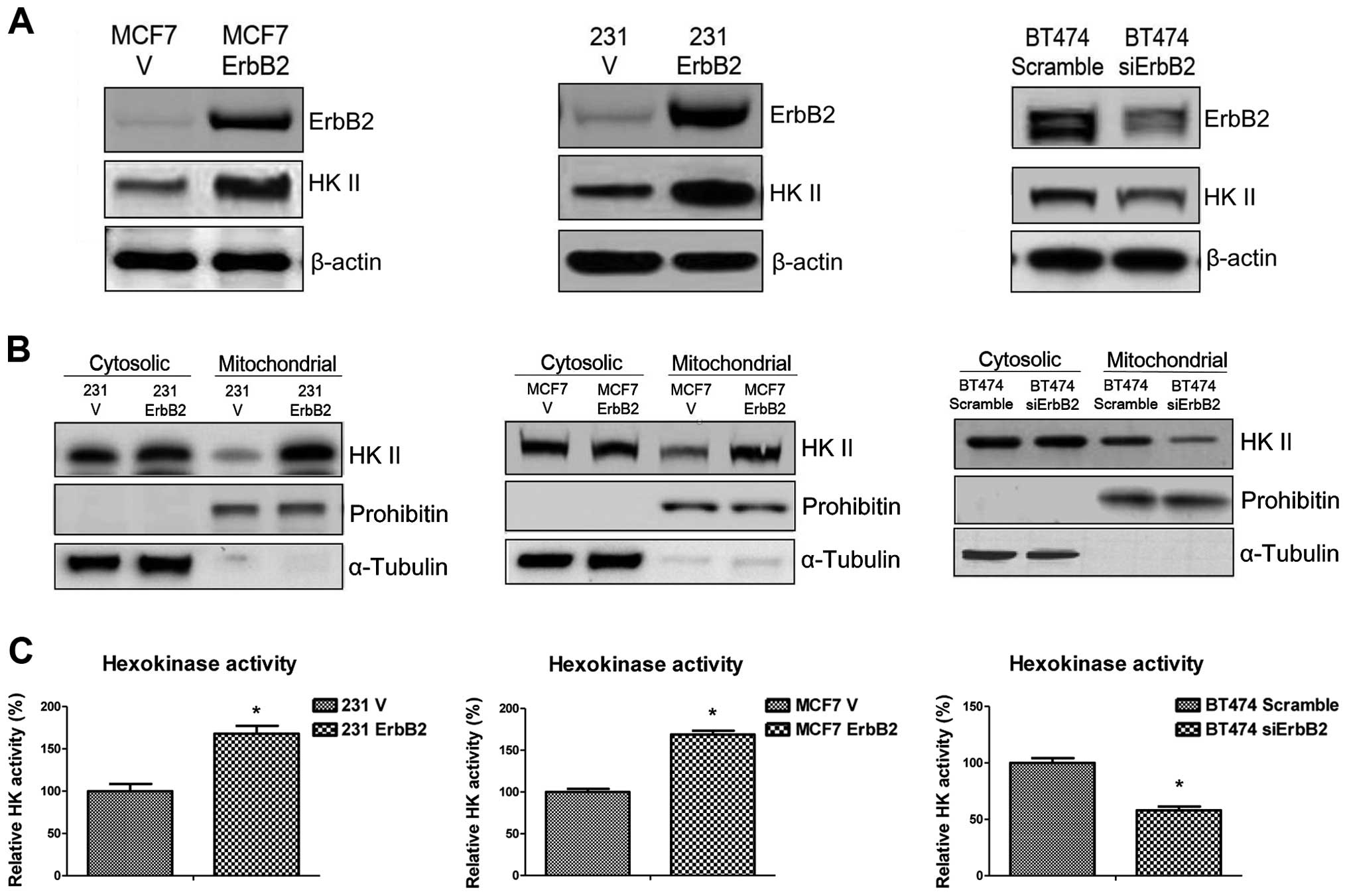
ErbB2-overexpressing cells was examined. Western blot analysis
revealed that ErbB2 expression upregulated HK II expression in
231ErbB2 and MCF7ErbB2 cells, and HK II expression was
significantly decreased when ErbB2 was knocked down in BT474 cells
(Fig. 2A). HK II associates with the
outer surface of the external mitochondrial membrane through
specific binding to voltage-dependent anion channels, where it
catalyzes the first step of glycolysis through the phosphorylation
of glucose. Therefore, we hypothesized that ErbB2 may regulate the
association of HK II with the mitochondrial outer membrane. As
demonstrated in Fig. 2B, the
overexpression of ErbB2 increased mitochondrial HK II distribution,
but did not regulate cytosolic HK II in 231ErbB2 and MCF7ErbB2
cells. Similar results were obtained in cells with ErbB2 knockdown
(Fig. 2B). HK activity was then
measured in the MCF7, 231 and BT474 cells. In concordance with the
earlier results, ErbB2 increased the activity of HK (Fig. 2C). Thus, the results demonstrated that
ErbB2 promotes mitochondrial HK II expression and activity in the
three breast cancer cell lines.
ErbB2 promotes the dissociation of
HKII from mitochondria in response to 3-BrPA treatment
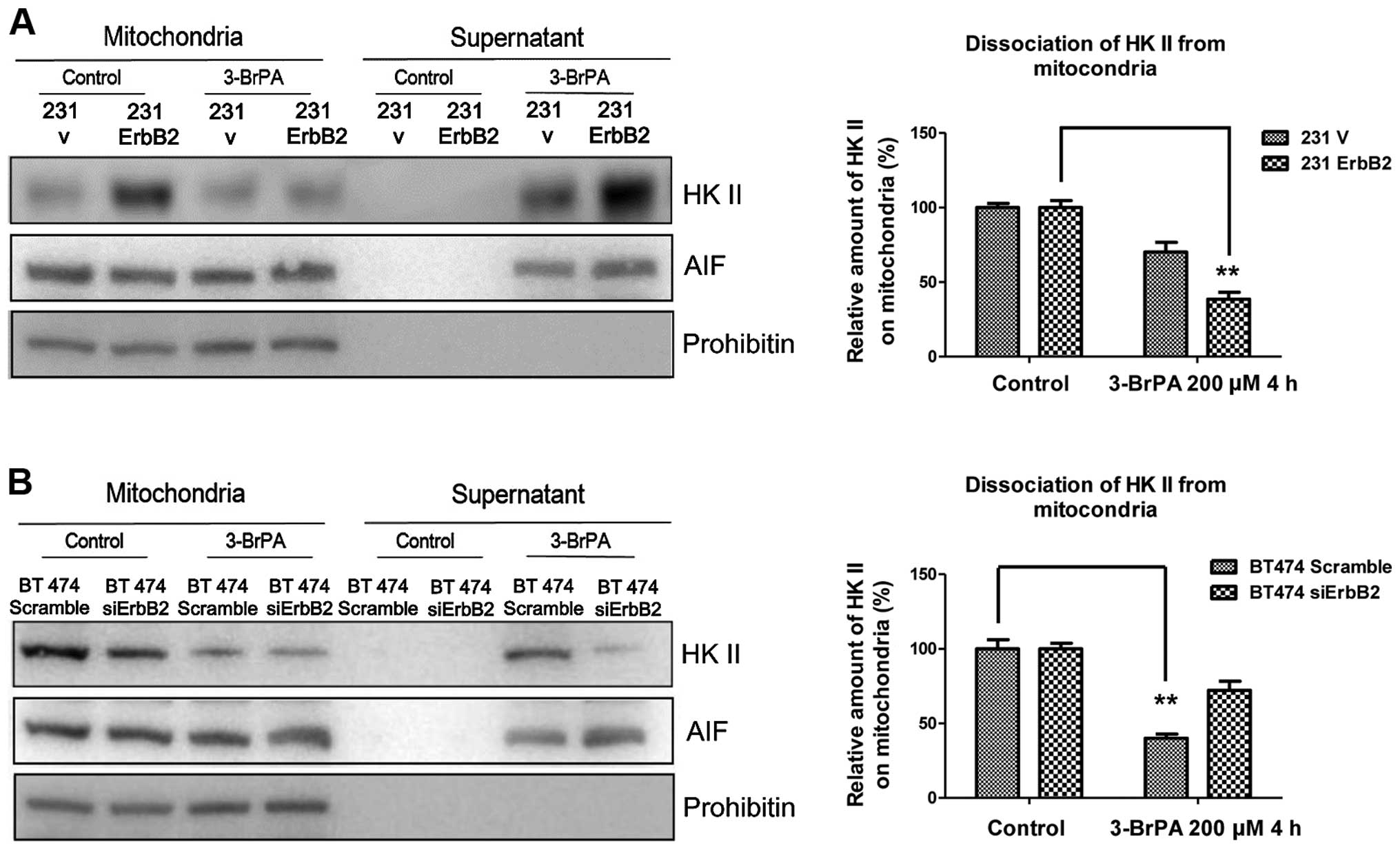
A previous study has demonstrated that
mitochondria-associated HK II is involved in the regulation of
apoptosis (12). To investigate the
putative mechanism, the detachment of HK II from mitochondria under
the 3-BrPA treatment was investigated. The rationale of these
experiments was that proteins released from the isolated
mitochondria would be present in the supernatant and could
therefore be separated from the mitochondrial-associated proteins
by centrifugation. The mitochondrial fractions were isolated from
cells and treated with 3-BrPA for 2 h. The supernatant and
mitochondria pellets were separated by centrifugation and
immunoblotted for HK II and control markers. As expected, the
overexpression of ErbB2 significantly promoted the dissociation of
HK II from mitochondria following 3-BrPA treatment (Fig. 3A). By contrast, BT474 ErbB2 knockdown
cells had less HK II dissociation (Fig.
3B). The results indicate that the high ErbB2-expressing cells
are more susceptible to losing the association between HK II and
the mitochondrial outer membrane when treated with the specific HK
II inhibitor, 3-BrPA.
The dissociation of HK II from
mitochondria leads to cytochrome c release from mitochondria to the
cytoplasm
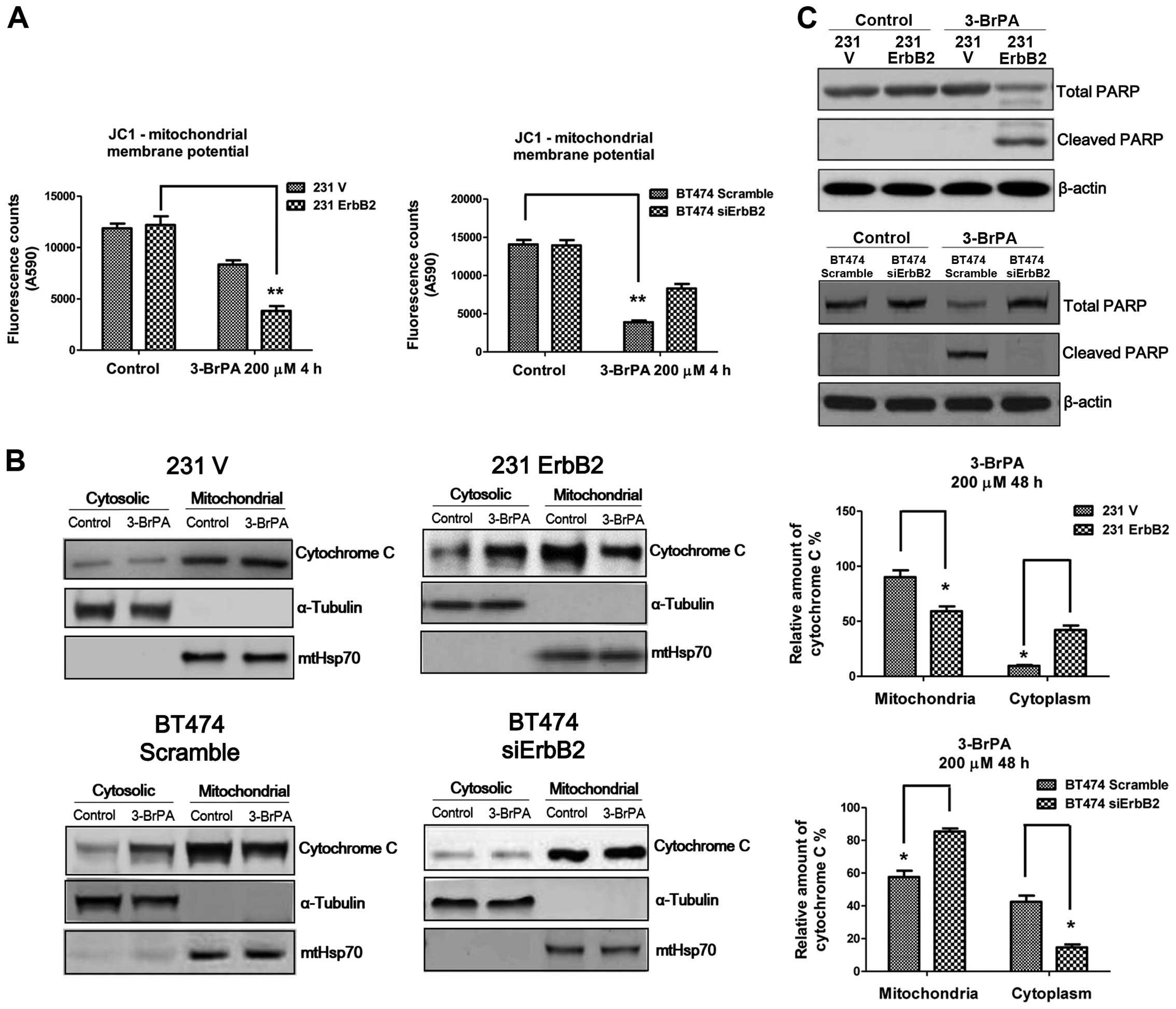
Previous studies demonstrated that 3-BrPA can induce
cell death through the mitochondria-mediated intrinsic apoptotic
pathway, which is coupled to the release of cytochrome c
from mitochondria to the cytosol, activating the caspase cascade
(12). The role of ErbB2 in altering
mitochondrial membrane potential in response to 3-BrPA treatment
was investigated using the JC1 assay. Fig. 4A demonstrates significant changes in
mitochondrial membrane potential in the ErbB2-overexpressing and
knockdown cells following 3-BrPA treatment, compared to the control
cells. The 231ErbB2 cells exhibited increased cytochrome c
release from mitochondria to the cytoplasm compared to the control
cells, and knockdown of ErbB2 in BT474 cells reduced the release of
cytochrome c into the cytoplasm (Fig. 4B). Levels of cleaved PARP in
ErbB2-overexpressing cells under 3-BrPA treatments were then
compared. Consistently, the results demonstrated increased levels
of cleaved PARP in the high ErbB2-expressing cells, following
3-BrPA treatment (Fig. 4C). In
general, breast cancer cells with overexpression of ErbB2 were more
sensitive to 3-BrPA through the dissociation of HK II from
mitochondria, leading to the release of cytochrome c from
mitochondria to the cytosol and activation of the mitochondrial
apoptosis pathway.
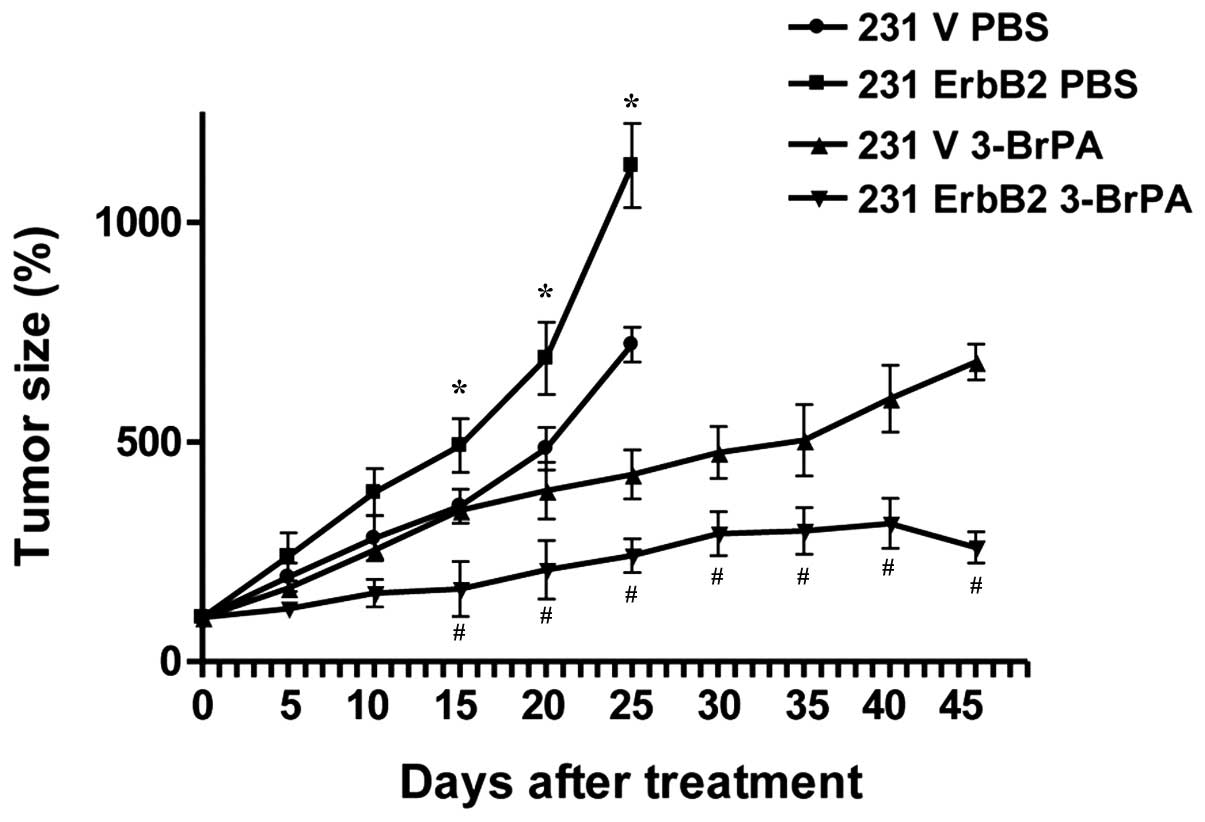
3-BrPA exhibits higher therapeutic
efficiency in ErbB2-overexpressing cancer cells in vivo
To confirm our in vitro results in
vivo, a xenograft nude mouse model was used to further
investigate 3-BrPA treatment. The 231V, 231ErbB2, MCF7V and
MCF7ErbB2 cells were inoculated into the mammary fat pads of
6-week-old nude mice. Following tumor formation, tumor xenografts
were treated with 3-BrPA. Forty-five days following injection, mice
were sacrificed by exsanguination under general anesthesia. As
expected, 3-BrPA significantly inhibited 231ErbB2-derived tumor
growth compared with 231V-derived tumors (Fig. 5; P<0.05). These in vivo data
support our in vitro results demonstrating that ErbB2
promoted the dissociation of HKII from mitochondria, resulting in
increased sensitivity to 3-BrPA treatment.
Discussion
In the 1920s, Warburg hypothesized that cancer cells
possess a unique energy metabolism, and suggested this was due to a
shift in energy production from mitochondrial OXPHOS to aerobic
glycolysis (1). Studies of the
changes of cancer cell metabolism predicted by Warburg, may provide
a rationale and an insight for anticancer therapies. Thus,
regulation of the enzymes involved in glucose or lactate
production, such as HK II, glucose transporter 1 and lactate
dehydrogenase A (LDHA), may be an effective means of inhibiting
cancer cells (15). Multiple
oncogenes and tumor suppressors have been reported to reprogram
glucose metabolism in cancer cells, including Ras (16), ErbB2 (9), epidermal growth factor receptor
(17), protein kinase B (also known
as Akt) (18), Src (19), phosphatase and tensin homolog
(20) and p53 (21). For example, oncogenic signaling
through the PI3K/Akt pathway, commonly upregulated in cancer,
promotes glycolysis through multiple mechanisms. Akt signaling
increases the expression and membrane localization of glucose
transporters and increases the activity of glycolytic enzymes, such
as phosphofructokinase and HK II (18). Furthermore, the p53 tumor suppressor
negatively regulates the expression of the glycolytic protein
phosphoglycerate mutase-2 (21).
Thus, oncogene signaling, not only activates cancer cell mitogenic
pathways that drive unchecked growth of cancer, but also promotes a
coordinated metabolic transformation of cancer cells by activating
metabolic pathways and transcriptionally regulating metabolic
enzymes. As a well-studied oncogene, ErbB2 promotes cancer cell
glycolysis and is important in the regulation of the anti-apoptosis
pathway during cell stress. In breast cancer cells, the ErbB2
oncogene activates signaling pathways that regulates the
proliferation, metastasis, invasion and resistance to
chemotherapeutic drugs of cancer cells. Additionally, ErbB2
signaling has been demonstrated to transcriptionally upregulate the
glycolytic enzyme LDHA (8). To the
best of our knowledge, the present study, demonstrates for the
first time that increased mitochondrial localization of HK II is
regulated by ErbB2. Overexpression of ErbB2 in breast cancer cells
upregulated HK II expression and promoted the localization of HK II
on the mitochondrial outer membrane. The results of the present
study demonstrate a novel mechanism for ErbB2-stimulated glycolysis
via the activation of HK II.
Cancer cells expressing higher levels of ErbB2 are
highly dependent on glucose supply, therefore, we investigated
whether cancer cells expressing high levels of ErbB2 were more
sensitive to the depletion of glucose than ErbB2 negative cells.
The present study demonstrates that ErbB2-overexpressing breast
cancer cells exhibit a higher apoptosis tendency under glucose
withdrawal conditions, compared to ErbB2-negative breast cancer
cells. This result indicates a dual role of ErbB2 in the regulation
of cancer cell apoptosis. It has been reported previously that
c-Myc enhances glycolysis by increasing glucose uptake,
upregulating the expression of LDH, and favoring the production of
the M2 isoform of pyruvate kinase 2 a key regulator of glycolytic
flux (22). An additional study
demonstrated that glucose deprivation induces the apoptosis of
c-Myc-transformed cancer cells. Furthermore, exogenously enhanced
glycolysis by overexpression of LDHA sensitizes cells to glucose
deprivation-induced apoptosis, suggesting that cells with
oncogene-activated upregulation of glycolysis are more susceptible
to glucose depletion. Similar results were observed in the present
study.
ErbB2-overexpressing cancer cells are more
susceptible to apoptosis when treated with other glycolysis
inhibitors such as 2-DG and Oxamate (8). In the present study, cancer cells
expressing higher levels of ErbB2 underwent more apoptosis in
glucose starvation conditions, thus, we investigated whether
inhibition of glucose metabolism pathway could specifically inhibit
ErbB2-overexpressing cells. 3-BrPA is a specific inhibitor of HK
II, through the induction of apoptosis. ErbB2-overexpressing breast
cancer cells were observed to be more sensitive to 3-BrPA treatment
through the dissociation of HK II from the mitochondrial outer
membrane, suggesting 3-BrPA may be a therapeutic anti-cancer drug
for the specific inhibition of ErbB2 high-expressing cancer cells.
The increased dissociation of HK II from the mitochondrial membrane
induced mitochondria-mediated apoptosis. However, the detailed
mechanisms for the ErbB2-induced dissociation of HK II remain
unclear. The anti-tumor efficacy of 3-BrPA in vivo was
evaluated by measuring tumor volumes. The xenograft model
experiment in the present study demonstrated 3-BrPA treatments were
more effect in ErbB2-overexpressing breast cancer cells, supporting
our in vitro results.
In conclusion, we suggest a novel mechanism for
ErbB2-activated glycolysis in breast cancer cells in vitro
and in vivo, and this may contribute to the development of
clinical therapies for the treatment of breast cancer patients.
Acknowledgements
The present authors thank Dr Shengnan Ren for
critically reviewing and revising the manuscript, and Mr. Zhuo Liu
for assistance with data analysis. We also thank Dr Xuebo Chen for
help with animal experiments. This study was supported by a
Pre-doctoral Fellowship (grant no: 201201037), awarded to Ms. Sujie
Gao by Jilin Provincial Science & Technology Department, Jilin
University.
References
|
1
|
Warburg O: On respiratory impairment in
cancer cells. Science. 124:269–270. 1956.PubMed/NCBI
|
|
2
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect, The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferreira LM: Cancer metabolism: the
Warburg effect today. Exp Mol Pathol. 89:372–380. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao Y, Butler EB and Tan M: Targeting
cellular metabolism to improve cancer therapeutics. Cell Death Dis.
4:e5322013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vander Heiden: MG:T argeting cancer
metabolism: A therapeutic window opens. Nat Rev Drug Discov.
10:671–684. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gutierrez C and Schiff R: HER2: Biology
detection, and clinical implications. Arch Pathol Lab Med.
135:55–62. 2011.PubMed/NCBI
|
|
7
|
Angelini PD, Fluck Zacarias MF, Pedersen
K, Parra-Palau JL, Guiu M, et al: Constitutive HER2 signaling
promotes breast cancer metastasis through cellular senescence.
Cancer Res. 73:450–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao YH, Zhou M, Liu H, Ding Y, Khong HT,
Yu D, Fodstad O and Tan M: Upregulation of lactate dehydrogenase A
by ErbB2 through heat shock factor 1 promotes breast cancer cell
glycolysis and growth. Oncogene. 28:3689–3701. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Patel NI: Barrientos A and Landgraf R: The
growth factor receptor ERBB2 regulates mitochondrial activity on a
signaling time scale. J Biol Chem. 288:35253–35265. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wolf A, Agnihotri S, Micallef J, Mukherjee
J, Sabha N, Cairns R, Hawkins C and Guha A: Hexokinase 2 is a key
mediator of aerobic glycolysis and promotes tumor growth in human
glioblastoma multiforme. J Exp Med. 208:313–326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gershon TR, Crowther AJ, Tikunov A, Garcia
I, Annis R, Yuan H, Miller CR, Macdonald J, Olson J and Deshmukh M:
Hexokinase-2-mediated aerobic glycolysis is integral to cerebellar
neurogenesis and pathogenesis of medulloblastoma. Cancer Metab.
1:22013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shulga N, Wilson-Smith R and Pastorino JG:
Hexokinase II detachment from the mitochondria potentiates
cisplatin induced cytotoxicity through a caspase-2 dependent
mechanism. Cell Cycle. 8:3355–3364. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ganapathy-Kanniappan S, Geschwind JF,
Kunjithapatham R, Buijs M, et al: 3-Bromopyruvate induces
endoplasmic reticulum stress, overcomes autophagy and causes
apoptosis in human HCC cell lines. Anticancer Res. 30:923–935.
2010.PubMed/NCBI
|
|
14
|
Chen Z, Zhang H, Lu W and Huang P: Role of
mitochondria-associated hexokinase II in cancer cell death induced
by 3-bromopyruvate. Biochim Biophys Acta. 87:553–560. 2009.
View Article : Google Scholar
|
|
15
|
Teicher BA, Linehan WM and Helman LJ:
Targeting cancer metabolism. Clin Cancer Res. 18:5537–5545. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gaglio D, Metallo CM, Gameiro PA, Hiller
K, et al: Oncogenic K-Ras decouples glucose and glutamine
metabolism to support cancer cell growth. Mol Syst Biol. 7:5232011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huber SM, Misovic M, Mayer C, Rodemann HP
and Dittmann K: EGFR-mediated stimulation of sodium/glucose
cotransport promotes survival of irradiated human A549 lung
adenocarcinoma cells. Radiother Oncol. 103:373–379. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Elstrom RL, Bauer DE, Buzzai M, Karnauskas
R, Harris MH, Plas DR, et al: Akt stimulates aerobic glycolysis in
cancer cells. Cancer Res. 64:3892–3899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Valle-Casuso JC, González-Sánchez A,
Medina JM and Tabernero A: HIF-1 and c-Src mediate increased
glucose uptake induced by endothelin-1 and connexin43 in
astrocytes. PLoS One. 7:e324482012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Blouin MJ, Zhao Y, Zakikhani M, Algire C,
Piura E and Pollak M: Loss of function of PTEN alters the
relationship between glucose concentration and cell proliferation,
increases glycolysis, and sensitizes cells to 2-deoxyglucose.
Cancer Lett. 289:246–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheung EC and Vousden KH: The role of p53
in glucose metabolism. Curr Opin Cell Biol. 2:186–191. 2010.
View Article : Google Scholar
|
|
22
|
David CJ, Chen M, Assanah M, Canoll P and
Manley JL: HnRNP proteins controlled by c-Myc deregulate pyruvate
kinase mRNA splicing in cancer. Nature. 7279:364–368. 2010.
View Article : Google Scholar
|