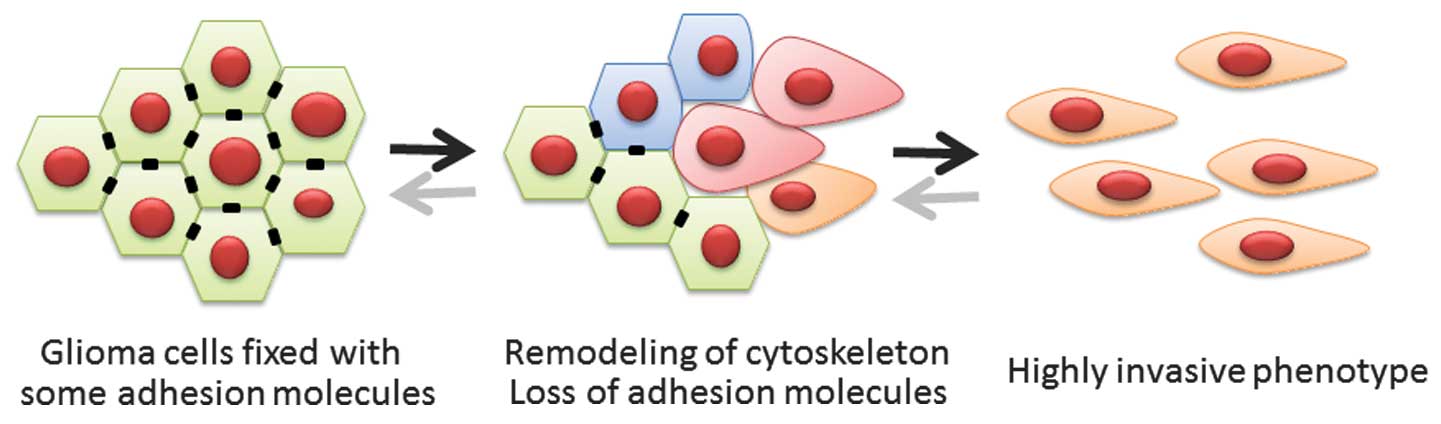
Epithelial-mesenchymal transition (EMT) is a
biological process in which polarized epithelial cells are induced
to undergo numerous biochemical changes; this results in a
mesenchymal phenotype, defined by an enhanced migratory capacity
and elevated resistance to genotoxic agents (1,2). EMT is
indispensable for wound healing, embryonic development and tissue
remodeling. As a pathological process, EMT also induces migratory
and invasive capabilities in epithelial tumor cells without a loss
in viability (1,2). The process of EMT includes the
detachment of tumor cells from the basement membrane. Although the
central nervous system (CNS) lacks this critical tissue component,
key invasive mechanisms overlap between cancers of the CNS and
other cancer types (3). The factors
that induce EMT in other cancers may also activate mesenchymal
features in gliomas (Fig. 1).
Furthermore, EMT is an important inducer of the cancer stem cell
phenotype (4). The mesenchymal
subtype of glioblastoma (GBM) typically expresses neural stem cell
markers and is associated with an aggressive phenotype (5–7). Glioma
cells that express stem cell markers are highly invasive and
resistant to chemotherapy and radiotherapy in vitro
(8–10)
and in the clinical setting (11).
Gliomas are classified according to their
histopathological features; these features allow clinicians to
distinguish between two cellular lineages (astrocytic and
oligodendrocytic) and four grades of malignancy (grades I to IV)
(12). The most malignant form of
grade IV is GBM, which originates from progenitor or stem cells in
the astrocytic lineage. Recent genotyping and expression profiling
analyses have demonstrated that GBMs may be categorized into four
subclasses dependent on their neural differentiation (5,6). The
proneural subtype is associated with a positive prognosis, whilst
the mesenchymal subtype is characterized by higher percentages of
cycling cells and neoangiogenesis, with a highly invasive nature
and poor prognosis (5,6). Furthermore, non-mesenchymal subtypes of
tumors typically acquire mesenchymal features at recurrence
(6). A shift towards the mesenchymal
subtype appears to be a common pattern in disease progression,
similar to cancer cells undergoing EMT in order to acquire a more
aggressive nature (13).
Paradoxically, migrating tumor cells are required to
lose the mesenchymal phenotype to establish a secondary tumor at
distant sites (1,2). This suggests that EMT is a reversible
process, and is most likely to be mediated by epigenetic
alterations that are induced by microenvironmental stimuli, rather
than as a result of genetic alterations (14,15).
Differentiated tumor cells change their phenotype through a dynamic
reprogramming process that is affected by a repair-associated
process or pathological stresses, such as hypoxic insults (16–20).
Acquisition of the stem cell phenotype may be closely associated
with epigenetic alterations that are induced by EMT. Although EMT
may be a common pattern in glioma progression, numerous therapeutic
interventions affect the occurrence and magnitude of EMT during the
clinical course of GBM (21–23). The present review discusses the
participation of EMT in GBM progression, and the resulting
acquisition of the stem-cell phenotype.
EMT type 2 occurs in adults and is associated with
tissue regeneration, wound healing and organ fibrosis, in which
fibroblasts are formed in injured tissues. Organ fibrosis is
mediated by fibroblasts and inflammatory cells that secrete a
number of inflammatory signals alongside the components of a
complex extracellular matrix, consisting of elastin, collagens,
tenacin and laminins. The transition of epithelial cells into
fibroblasts occurring over a few days in culture is one line of
evidence for this type of EMT, and an active diversion to MET
occurs in the presence of bone morphogenic protein-7 (25). Cancer-associated fibroblasts in
primary epithelial tumor nodules have recently been demonstrated to
share certain genetic mutations with tumor cells, suggesting that
type 2 EMT emerges prior to the full onset of tumorigenesis
(26).
EMT type 3 is observed in subsets of cancer cells
undergoing a phenotypic conversion to increase migration, invasion
and metastasis. Certain studies have noted that transforming growth
factor (TGF)-β can induce EMT in epithelial cancer cells through
Smad or p38 mitogen-activated protein kinase/Ras homolog family
member A pathways (27–29). Activation of EMT programs, through
tumor microenvironment stimuli, has been proposed as the critical
mechanism for the acquisition of highly malignant phenotypes of
cancer cells (30). In type 3 EMT,
certain cancer cells with a transitioning mesenchymal phenotype
undergo MET to form metastatic tumor nodules at distant sites
(1,2).
The genetic and epigenetic alterations that cancer
cells undergo render them sensitive to EMT-inducing signals. Highly
motile mesenchyme-like cancer cells are typically observed at the
invasive front, suggesting that dedifferentiating signals usually
originate from the tumor microenvironment (30). As aforementioned, the reversibility of
EMT suggests that epigenetic alterations, as a result of
environmental signals, generate highly aggressive tumor phenotypes
(31–34).
A hypoxic microenvironment is generally regarded as
a potent inducer of EMT in various types of epithelial cancer
(30,35). In gliomas, inflammatory processes, or
a hypoxic microenvironment within the tumor or neighboring normal
tissues, may result in the recruitment of circulating or
residential myeloid cells (including macrophages or microglia) into
the tumor stroma (34). These cells
release a number of growth factors, including TGF-β, epidermal
growth factor, platelet-derived growth factor and fibroblast growth
factor-2, which trigger alterations in the levels of transcription
factors required for the initiation of EMT, and also in numerous
proteases that increase invasiveness into the surrounding normal
brain (17,21,34,36). Thus,
glioma cells that are affected by the bystander myeloid cells and
such signaling molecules may undergo EMT in a hypoxic
microenvironment.
Twist is a protein with a basic helix-loop-helix
structure and is transcriptionally active during cell
differentiation and lineage determination (37,38).
During the establishment of cancer metastases by EMT, Twist acts
independently of Snail to suppress E-cadherin and to upregulate
N-cadherin and fibronectin (38).
Using a brain slice culture and an orthotopic model of
xenotransplantation, it has been reported that Twist is upregulated
in malignant gliomas, and promotes glioma cell invasion through the
mesenchymal target gene Slug and the fibroblast activation protein,
independent of the cadherin switch (39,40). It
has also been demonstrated that the inhibition of Twist expression
results in a significant reduction in GBM stem cell sphere growth
and formation. Nagaishi et al (41) observed that the expression of Twist is
characteristic of mesenchymal areas of gliosarcomas, indicating
that EMT is involved in the formation of biphasic tumor
gliosarcoma.
Snail is a member of the SNAIL family of
transcriptional activators and is a primary suppressor of
E-cadherin expression (1,2,42). Snail
regulates a range of other EMT phenotypes, including the decreased
expression of various epithelial markers (occludins, claudins and
cytokeratin) and the increased expression of mesenchymal markers
(vitronectin and fibronectin) (43).
The transcriptional activity of Snail is predominantly regulated by
its subcellular localization. Phosphorylation of Snail results in
its exportation from the nucleus to the cytoplasm, leading to
inactivation of the protein as a transcription factor (42). TGF-β is secreted from mesenchymal
cells following irradiation and induces the nuclear localization of
Snail via Smad2/3 pathways (22).
Slug is another member of the SNAIL family of
transcriptional activators and serves an important role in
suppressing the epithelial phenotype in numerous cancer cells
(1,2,44). Slug is
closely associated with the increased migration and invasion of
malignant gliomas (45). A
multi-cancer mesenchymal transition signature of mRNA expression
levels from The Cancer Genome Atlas (TCGA) data has been
highlighted by strong expression of Slug and cluster of
differentiation (CD)44 (5,6).
The zinc finger E-box-binding homeobox (ZEB)
proteins, ZEB1 and smad1-interacting protein-1 (also known as
ZEB2), are another family of noteworthy transcription factors that
are responsible for the mediation of EMT in numerous types of
cancer and glioma (1,2,46). ZEB
proteins bind to the promoter region of E-cadherin and suppress its
expression, resulting in the loss of cell-cell contact and
increased motility (47,48). Wang et al (46) observed that patients with GBM
containing high levels of ZEB2 demonstrated significantly earlier
recurrence with malignant transformation compared to those with low
levels of ZEB2. Connective tissue growth factor also renders glioma
cells highly invasive through the activation of nuclear factor-κB,
which subsequently initiates ZEB1 expression (49).
In multiple types of cancer, β-catenin is
sequestered in the cytoplasm by E-cadherin, with the translocation
of β-catenin into the nucleus following the downregulation of
E-cadherin being directly correlated with acquisition of the
mesenchymal phenotype (1,50,51).
Although the majority of GBMs do not express E-cadherin, nuclear
localization of β-catenin is primarily observed at the invasive
front of the tumor (52).
Furthermore, GBMs that express high levels of WNT/β-catenin are
correlated with significantly shorter patient survival times
(53). The WNT/β-catenin pathway is
an important stem cell maintenance pathway and is involved in
therapy resistance (54). GBM cells,
in which the WNT/β-catenin pathway is activated, trigger the
expression of a set of EMT activators, including Twist1, ZEB1,
Snail and Slug (55). Furthermore,
high expression levels of the WNT/β-catenin receptor, Frizzled-4,
promotes the expression of Snail and the acquirement of a
mesenchymal phenotype in GBM (56).
NOTCH is a cell surface receptor that serves an
important function in the development of numerous types of cells
and tissues (1). NOTCH signaling is a
primary inducer of EMT in a number of epithelial cancers, including
cancer of the lung, breast and pancreas (57). Fan et al (58) reported that inhibition of this
signaling pathway by γ-secretase inhibitors reduces CD133-positive
stem-like cells in GBMs. In addition to WNT/β-catenin, NOTCH is a
major regulator of glioma stem cells within their
microenvironments. NOTCH is also directly correlated with
phosphoinositide-3 kinase/Akt pathway activation (59–61).
CD44 is a hyaluronic acid receptor that interacts
with ligands such as collagens, osteopontin and matrix
metalloproteases (62,63). In addition to the standard isoform of
CD44 (CD44s), alternative splicing results in 11 other isoforms of
CD44 variants (CD44v2-v12) (64).
CD44s is a primary inducer of EMT in breast and colorectal cancer.
TCGA data indicates that GBMs with high levels of mRNA expression
of EMT-inducing signature molecules, including Slug and CD44, are
correlated with increased resistance to therapies and tumor
invasion (65). However, functional
data for CD44-mediated EMT in GBM have not been fully elucidated
(66).
miRs are small, 20–23-nucleotide non-coding RNAs
that serve as epigenetic regulators of gene expression through the
downregulation of target genes; this occurs through the binding of
miRs to regions of partial complementarity in the target gene
3′-untranslated regions (67). Each
miR has hundreds of target genes, and numerous genes are targeted
by multiple miRs, creating a highly complex gene expression
regulatory network (68). Control of
gene expression by miRs is one of the most important modulating
processes in cellular differentiation during normal embryogenesis
(69,70). A number of studies have demonstrated
that miRs may function as negative regulators of gene expression in
normal tissues and as tumor suppressors or oncogenes in various
tumors (67,70,71). In
several types of cancer, epigenetic regulation (involving miRs) is
a core mechanism of EMT modulation, and thus, reversible modulation
of the genes that mediate EMT is possible (72). The majority of miRs are negatively
correlated with tumorigenesis, tumor invasion and mesenchymal
changes in gliomas. Notably, the expression of miR-21, −34a, −128a,
−124 and −184 is correlated with the downregulation of mesenchymal
markers and decreased invasiveness. By contrast, a relatively small
number of miRs are oncogenic and may function as therapeutic
targets. The inhibition of a Dicer enzyme for a specific oncogenic
miR was recently indicated to block maturation of the miR and
suppress tumor invasion (73).
Furthermore, evidence is growing concerning the effect of miRs on
the progression and maintenance of glioma stem cells (67,71).
Radiation therapy is a major modality of cancer
therapy and also serves a key role in the multimodal treatment of
GBM. However, irradiation that is sublethal to malignant glioma
cells consequently promotes cell migration and invasion through the
expression of TGF-β, epidermal growth factor, vascular endothelial
growth factor (VEGF) and the hepatocyte growth factor pathway
(74–76). Glioma cells that are resistant to
irradiation have a gene expression signature that is enriched in
the EMT pathway, leading to highly invasive recurrence patterns
(22,23,77,78). TCGA
data indicates a shift from a proneural to mesenchymal phenotype at
the time of tumor recurrence. Recently, Mahabir et al
(22) observed that two different
pathways are involved in the radiation-associated EMT induction in
malignant gliomas; TGF-β, derived from the mesenchymal cells in the
tumor environment, evokes the activation of Smad2/3, whilst
reactive oxygen species activate extracellular signal-regulated
kinase1/2, with each pathway leading to the nuclear localization of
Snail. Such data suggests that EMT serves a crucial role in the
acquisition of radiation resistance. Furthermore, emerging evidence
suggests that such a role for EMT in the generation of refractory
cancer cells is associated with an accumulation of stem cell
markers. The NOTCH pathway and WNT/β-catenin signaling are
important for stem cell maintenance and are associated with the
radiation resistance of GBM (78).
A further important aspect of the biological effects
of radiation therapy on GBM is the induction of hypoxia or
necrosis. Tissue hypoxia directly induces EMT and recruits myeloid
cells into tumor tissues (15–20).
Glioma cells, under a hypoxic microenvironment, and recruited
myeloid cells each secrete TGF-β, leading to the induction of
hypoxia-inducible factor-1α (HIF-1α), which subsequently promotes
the malignant progression of glioma cells (21,79).
VEGF is one of the most important factors
facilitating angiogenesis and resultant tumor growth in GBM.
Inhibiting the VEGF-VEGF receptor (VEGFR) signal transduction
pathway with anti-VEGF therapy (including the use of bevacizumab)
and VEGFR inhibitors (including sunitinib) is a promising strategy
in cancer therapy (80). Bevacizumab
is effective in prolonging progression-free survival (PFS) in newly
diagnosed GBM patients, but is not effective in prolonging overall
survival (81,82). During the early phases of bevacizumab
therapy, tumor oxygenation improves through the process of vascular
normalization (83). However, with
prolonged treatment with bevacizumab, in a similar manner to
radiation therapy, the tumor develops progressive hypoxia that
directly or indirectly promotes the mesenchymal phenotype (21,79,83).
Furthermore, hypoxia induces the release of HIF-1α from glioma
cells and subsequently attracts myeloid cells, including
macrophages and granulocytes, from bone marrow into the glioma
tissues (21,23). The recruited myeloid cells release
TGF-β, which then directly induces EMT in the glioma cells. Myeloid
cells also secrete multiple growth factors, including interleukin
(IL)-6, IL-10 and matrix metalloproteinases (15–19).
TGF-β, alongside VEGF, also recruits mesenchymal stem cells into
the glioma tissues, which contributes to the further malignant
progression of GBMs (83). By
contrast, VEGFR inhibitors lack the efficacy in PFS prolongation,
due to the induction of hypoxia in the early phase, without the
vascular normalization phase, or due to dose-limiting adverse
events. Similarly, anti-VEGF therapy is more effective than VEGFR
inhibitors in decreasing myeloid cell infiltration, which may
contribute to the efficacy of bevacizumab observed during early
phases.
Glioma cells undergoing EMT acquire the potential to
initiate metastasis and invasion. This process is highly affected
by the tumor microenvironment, particularly a hypoxic environment
or one involving the release of proinflammatory molecules from
recruited myeloid or mesenchymal stem cells. The evidence that type
I and II EMT occur during the normal physiological processes of
embryogenesis and wound healing in a relatively short time suggests
that epigenetic mechanisms are more crucial than genetic changes.
This notion is also supported by the evidence that migrating tumor
cells that have undergone EMT may also undergo MET to establish
metastatic tumor nodules. Therefore, tumor microenvironments are
emerging as a therapeutic target, particularly when in a hypoxic
state, which controls epigenetic alterations in tumor cells. The
microenvironmental modifier, bevacizumab, has recently been
developed; however, future clinical trials to maximize the efficacy
of anti-VEGF therapy are required, with the aim that such treatment
will normalize oxygen concentration and suppress the excessive
recruitment of myeloid and mesenchymal stem cells.
|
1
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kahlert UD, Nikkhah G and Maciaczyk J:
Epithelial-to-mesenchymal (−like) transition as a relevant
molecular event in malignant gliomas. Cancer Lett. 331:131–138.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Phillips HS, Kharbanda S, Chen R, Forrest
WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et
al: Molecular subclasses of high-grade glioma predict prognosis,
delineate a pattern of disease progression, and resemble stages in
neurogenesis. Cancer Cell. 9:157–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Verhaak RGW, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al:
Cancer Genome Atlas Research Network: Integrated genomic analysis
identifies clinically relevant subtypes of glioblastoma
characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1.
Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zarkoob H, Taube JH, Singh SK, Mani SA and
Kohandel M: Investigating the link between molecular subtypes of
glioblastoma, epithelial-mesenchymal transition, and CD133 cell
surface protein. PLoS One. 8:e641692013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bhat KPL, Balasubramaniyan V, Vaillant B,
Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L,
James JD, Goodman LD, et al: Mesenchymal differentiation mediated
by NF-κB promotes radiation resistance in glioblastoma. Cancer
Cell. 24:331–346. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang X, Zhang W, Mao XG, Zhen HN, Cao WD
and Hu SJ: Targeting role of glioma stem cells for glioblastoma
multiforme. Curr Med Chem. 20:1974–1984. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murat A, Migliavacca E, Gorlia T, Lambiv
WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven
MC, et al: Stem cell-related self-renewal signature and high
epidermal growth factor receptor expression associated with
resistance to concomitant chemoradiotherapy in glioblastoma. J Clin
Oncol. 26:3015–3024. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kleihues P and Cavenee WK: Pathology and
genetics of tumors of the nervous system. In: World Health
Organization Classification of Tumours. Pathology and Genetics of
Head and Neck Tumours (Lyon, France). IARC Press. 9–15. 2000.
|
|
13
|
Kahlert UD, Maciaczyk D, Doostkam S, Orr
BA, Simons B, Bogiel T, Reithmeier T, Prinz M, Schubert J,
Niedermann G, et al: Activation of canonical WNT/β-catenin
signaling enhances in vitro motility of glioblastoma cells by
activation of ZEB1 and other activators of
epithelial-to-mesenchymal transition. Cancer Lett. 325:42–53. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brabletz T: To differentiate or not -
routes towards metastasis. Nat Rev Cancer. 12:425–436. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baysan M, Woolard K, Bozdag S, Riddick G,
Kotliarova S, Cam MC, Belova GI, Ahn S, Zhang W, Song H, et al:
Micro-environment causes reversible changes in DNA methylation and
mRNA expression profiles in patient-derived glioma stem cells. PLoS
One. 9:e940452014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Heddleston JM, Li Z, McLendon RE,
Hjelmeland AB and Rich JN: The hypoxic microenvironment maintains
glioblastoma stem cells and promotes reprogramming towards a cancer
stem cell phenotype. Cell Cycle. 8:3274–3284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cooper LA, Gutman DA, Chisolm C, Appin C,
Kong J, Rong Y, Kurc T, Van Meir EG, Saltz JH, Moreno CS and Brat
DJ: The tumor microenvironment strongly impacts master
transcriptional regulators and gene expression class of
glioblastoma. Am J Pathol. 180:2108–2119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bar EE, Lin A, Mahairaki V, Matsui W and
Eberhart CG: Hypoxia increases the expression of stem-cell markers
and promotes clonogenicity in glioblastoma neurospheres. Am J
Pathol. 177:1491–1502. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Evans SM, Judy KD, Dunphy I, Jenkins WT,
Hwang WT, Nelson PT, Lustig RA, Jenkins K, Magarelli DP, Hahn SM,
et al: Hypoxia is important in the biology and aggression of human
glial brain tumors. Clin Cancer Res. 10:8177–8184. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schonberg DL, Lubelski D, Miller TE and
Rich JN: Brain tumor stem cells: Molecular characteristics and
their impact on therapy. Mol Aspects Med. 39:82–101. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Piao Y, Liang J, Holmes L, Zurita AJ,
Henry V, Heymach JV and de Groot JF: Glioblastoma resistance to
anti-VEGF therapy is associated with myeloid cell infiltration,
stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol.
14:1379–1392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mahabir R, Tanino M, Elmansuri A, Wang L,
Kimura T, Itoh T, Ohba Y, Nishihara H, Shirato H, Tsuda M and
Tanaka S: Sustained elevation of Snail promotes glial-mesenchymal
transition after irradiation in malignant glioma. Neuro Oncol.
16:671–685. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim YH, Yoo KC, Cui YH, Uddin N, Lim EJ,
Kim MJ, Nam SY, Kim IG, Suh Y and Lee SJ: Radiation promotes
malignant progression of glioma cells through HIF-1alpha
stabilization. Cancer Lett. 354:132–141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Knecht AK and Bronner-Fraser M: Induction
of the neural crest: A multigene process. Nat Rev Genet. 3:453–461.
2002.PubMed/NCBI
|
|
25
|
Zeisberg M, Hanai J, Sugimoto H, Uddin N,
Lim EJ, Kim MJ, Nam SY, Kim IG, Suh Y and Lee SJ: BMP-7 counteracts
TGF-beta-1-induced epithelial-to mesenchymal transition and
reverses chronic renal injury. Nat Med. 9:964–968. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ and
Feng YM: Cancer-associated fibroblasts induce
epithelial-mesenchymal transition of breast cancer cells through
paracrine TGF-β signaling. Br J Cancer. 110:724–732. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bierie B and Moses HL: Tumour
microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer.
Nat Rev Cancer. 6:506–520. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Song J: EMT or apoptosis: A decision for
TGF-beta. Cell Res. 17:289–290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhowmick NA, Zent R, Ghiassi M, McDonnell
M and Moses HL: Integrin beta 1 signaling is necessary for
transforming growth factor-beta activation of p38MAPK and
epithelial plasticity. J Biol Chem. 276:46707–46713. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Charles NA, Holland EC, Gilbertson R,
Glass R and Kettenmann H: The brain tumor microenvironment. Glia.
59:1169–1180. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dumont N, Wilson MB, Crawford YG, Reynolds
PA, Sigaroudinia M and Tlsty TD: Sustained induction of epithelial
to mesenchymal transition activates DNA methylation of genes
silenced in basal-like breast cancers. Proc Natl Acad Sci USA.
105:14867–14872. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hirohashi S: Inactivation of the
E-cadherin-mediated cell adhesion system in human cancers. Am J
Pathol. 153:333–339. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Birchmeier W and Behrens J: Cadherin
expression in carcinomas: Role in the formation of cell junctions
and the prevention of invasiveness. Biochim Biophys Acta.
1198:11–26. 1994.PubMed/NCBI
|
|
34
|
Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen
L, Xiao HL, Wang B, Yi L, Wang QL, et al: Tumor-associated
microglia/macrophages enhance the invasion of glioma stem-like
cells via TGF-β1 signaling pathway. J Immunol. 189:444–453. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jensen RL: Brain tumor hypoxia:
Tumorigenesis, angiogenesis, imaging, pseudoprogression, and as a
therapeutic target. J Neurooncol. 92:317–335. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Iwadate Y, Sakaida T, Hiwasa T, Nagai Y,
Ishikura H, Takiguchi M and Yamaura A: Molecular classification and
survival prediction in human gliomas based on proteome analysis.
Cancer Res. 64:2496–2501. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Castanon L and Baylies MK: A Twist in
fate: Evolutionary comparison of Twist structure and function.
Gene. 287:11–22. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang MH, Wu MZ, Chiou SH, Chen PM, Chang
SY, Liu CJ, Teng SC and Wu KJ: Direct regulation of TWIST by
HIF-1alpha promotes metastasis. Nat Cell Biol. 10:295–305. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Elias MC, Tozer KR, Silber JR, Mikheeva S,
Deng M, Morrison RS, Manning TC, Silbergeld DL, Glackin CA, Reh TA
and Rostomily RC: Twist is expressed in human gliomas and promotes
invasion. Neoplasia. 7:824–837. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mikheeva SA, Mikheev AM, Petit A, Beyer R,
Oxford RG, Khorasani L, Maxwell JP, Glackin CA, Wakimoto H,
González-Herrero I, et al: TWIST1 promotes invasion through
mesenchymal change in human glioblastoma. Mol Cancer. 9:1942010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nagaishi M, Paulus W, Brokinkel B, Vital
A, Tanaka Y, Nakazato Y, Giangaspero F and Ohgaki H:
Transcriptional factors for epithelial-mesenchymal transition are
associated with mesenchymal differentiation in gliosarcoma. Brain
Pathol. 22:670–676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Boutet A, De Frutos CA, Maxwell PH, Mayol
MJ, Romero J and Nieto MA: Snail activation disrupts tissue
homeostasis and induces fibrosis in the adult kidney. EMBO J.
25:5603–5613. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cheng WY, Kandel JJ, Yamashiro DJ, Canoll
P and Anastassiou D: A multi-cancer mesenchymal transition gene
expression signature is associated with prolonged time to
recurrence in glioblastoma. PLoS One. 7:e347052012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang HW, Menon LG, Black PM, Carroll RS
and Johnson MD: SNAI2/Slug promotes growth and invasion in human
gliomas. BMC Cancer. 10:3012010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xie YK, Huo SF, Zhang G, Zhang F, Lian ZP,
Tang XL and Jin C: CDA-2 induces cell differentiation through
suppressing Twist/SLUG signaling via miR-124 in glioma. J
Neurooncol. 110:179–186. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang Q, Li X, Zhu Y and Yang P:
MicroRNA-16 suppresses epithelial-mesenchymal transition-related
gene expression in human glioma. Mol Med Rep. 10:3310–3314.
2014.PubMed/NCBI
|
|
47
|
Sánchez-Tilló E, Liu Y, de Barrios O,
Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A
and Postigo A: EMT-activating transcription factors in cancer:
Beyond EMT and tumor invasiveness. Cell Mol Life Sci. 69:3429–3456.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Qi S, Song Y, Peng Y, Wang H, Long H, Yu
X, Li Z, Fang L, Wu A, Luo W, et al: ZEB2 mediates multiple
pathways regulating cell proliferation, migration, invasion, and
apoptosis in glioma. PLoS One. 7:e388422012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Edwards LA, Woolard K, Son MJ, Li A, Lee
J, Ene C, Mantey SA, Maric D, Song H, Belova G, et al: Effect of
brain- and tumor-derived connective tissue growth factor on glioma
invasion. J Natl Cancer Inst. 103:1162–1178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim K, Lu Z and Hay ED: Direct evidence
for a role of beta-catenin/LEF-1 signaling pathway in induction of
EMT. Cell Biol Int. 26:463–476. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Paul I, Bhattacharya S, Chatterjee A and
Ghosh MK: Current understanding on EGFR and Wnt/β-catenin signaling
in glioma and their possible crosstalk. Genes Cancer. 4:427–446.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sandberg CJ, Altschuler G, Jeong J,
Strømme KK, Stangeland B, Murrell W, Grasmo-Wendler UH, Myklebost
O, Helseth E, Vik-Mo EO, et al: Comparison of glioma stem cells to
neural stem cells from the adult human brain identifies
dysregulated Wnt- signaling and a fingerprint associated with
clinical outcome. Exp Cell Res. 319:2230–2243. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Clevers H, Loh KM and Nusse R: Stem cell
signaling. An integral program for tissue renewal and regeneration:
Wnt signaling and stem cell control. Science. 346:12480122014.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gong A and Huang S: FoxM1 and
Wnt/β-catenin signaling in glioma stem cells. Cancer Res.
72:5658–5662. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jin X, Jeon HY, Joo KM, Kim JK, Jin J, Kim
SH, Kang BG, Beck S, Lee SJ, Kim JK, et al: Frizzled 4 regulates
stemness and invasiveness of migrating glioma cells established by
serial intracranial transplantation. Cancer Res. 71:3066–3075.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Brabletz S, Bajdak K, Meidhof S, Burk U,
Niedermann G, Firat E, Wellner U, Dimmler A, Faller G, Schubert J
and Brabletz T: The ZEB1/miR-200 feedback loop controls NOTCH
signalling in cancer cells. EMBO J. 30:770–782. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fan X, Khaki L, Zhu TS, Soules ME, Talsma
CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, et al: NOTCH pathway
blockade depletes CD133-positive glioblastoma cells and inhibits
growth of tumor neurospheres and xenografts. Stem Cells. 28:5–16.
2010.PubMed/NCBI
|
|
59
|
Hu YY, Fu LA, Li SZ, Chen Y, Li JC, Han J,
Liang L, Li L, Ji CC, Zheng MH and Han H: Hif-1α and Hif-2α
differentially regulate NOTCH signaling through competitive
interaction with the intracellular domain of NOTCH receptors in
glioma stem cells. Cancer Lett. 349:67–76. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kristoffersen K, Villingshøj M, Poulsen HS
and Stockhausen MT: Level of NOTCH activation determines the effect
on growth and stem cell-like features in glioblastoma multiforme
neurosphere cultures. Cancer Biol Ther. 14:625–637. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Stockhausen MT, Kristoffersen K and
Poulsen HS: NOTCH signaling and brain tumors. Adv Exp Med Biol.
727:289–304. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Anido J, Sáez-Borderías A, Gonzàlez-Juncà
A, Rodón L, Folch G, Carmona MA, Prieto-Sánchez RM, Barba I,
Martínez-Sáez E, Prudkin L, et al: TGF-β receptor inhibitor target
CD44(high)/Id1(high) glioma-initiating cell population in human
glioblastoma. Cancer Cell. 18:655–668. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kaaijk P, Troost D, Morsink F, Keehnen RM,
Leenstra S, Bosch DA and Pals ST: Expression of CD44 splice
variants in human primary brain tumors. J Neurooncol. 26:185–190.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Merzak A, Koocheckpour S and Pilkington
GJ: CD44 mediates human glioma cell adhesion and invasion in vitro.
Cancer Res. 54:3988–3992. 1994.PubMed/NCBI
|
|
65
|
Xu Y, Stamenkovic I and Yu Q: CD44
attenuates activation of the hippo signaling pathway and is a prime
therapeutic target for glioblastoma. Cancer Res. 70:2455–2464.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wei KC, Huang CY, Chen PY, Feng LY, Wu
TWE, Chen SM, Tsai HC, Lu YJ, Tsang NM, Tseng CK, et al: Evaluation
of the prognostic value of CD44 in glioblastoma multiforme.
Anticancer Res. 30:253–259. 2010.PubMed/NCBI
|
|
67
|
Katsushima K and Kondo Y: Non-coding RNAs
as epigenetic regulator of glioma stem-like cell differentiation.
Front Genet. 5:142014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Montagner S, Dehó L and Monticelli S:
MicroRNAs in hematopoietic development. BMC Immunol. 15:142014.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Piubelli C, Meraviglia V, Pompilio G,
D'Alessandra Y, Colombo GI and Rossini A: MicroRNAs and cardiac
cell fate. Cells. 3:802–823. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Godlewski J, Newton HB, Chiocca EA and
Lawler SE: MicroRNAs and glioblastoma; the stem cell connection.
Cell Death Differ. 17:221–228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Møller HG, Rasmussen AP, Andersen HH,
Johnsen KB, Henriksen M and Duroux M: A systematic review of
microRNA in glioblastoma multiforme: Micro-modulators in the
mesenchymal mode of migration and invasion. Mol Neurobiol.
47:131–144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bullock MD, Sayan AE, Packham GK and
Mirnezami AH: MicroRNAs: Critical regulators of epithelial to
mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in
cancer progression. Biol Cell. 104:3–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Shi Z, Zhang J, Qian X, Han L, Zhang K,
Chen L, Liu J, Ren Y, Yang M, Zhang A, et al: AC1MMYR2, an
inhibitor of dicer-mediated biogenesis of Oncomir miR-21, reverses
epithelial-mesenchymal transition and suppresses tumor growth and
progression. Cancer Res. 73:5519–5531. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhang M, Kleber S, Röhrich M, Timke C, Han
N, Tuettenberg J, Martin-Villalba A, Debus J, Peschke P, Wirkner U,
et al: Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor
LY2109761 enhances radiation response and prolongs survival in
glioblastoma. Cancer Res. 71:7155–7167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Timke C, Zieher H, Roth A, Hauser K,
Lipson KE, Weber KJ, Debus J, Abdollahi A and Huber PE: Combination
of vascular endothelial growth factor receptor/platelet-derived
growth factor receptor inhibition markedly improves radiation tumor
therapy. Clin Cancer Res. 14:2210–2219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhou YC, Liu JY, Li J, Zhang J, Xu YQ,
Zhang HW, Qiu LB, Ding GR, Su XM, Mei-Shi and Guo GZ: Ionizing
radiation promotes migration and invasion of cancer cells through
transforming growth factor-beta-mediated epithelial-mesenchymal
transition. Int J Radiat Oncol Biol Phys. 81:1530–1537. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Theys J, Jutten B, Habets R, Paesmans K,
Groot AJ, Lambin P, Wouters BG, Lammering G and Vooijs M:
E-Cadherin loss associated with EMT promotes radioresistance in
human tumor cells. Radiother Oncol. 99:392–397. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Meng J, Li P, Zhang Q, Yang Z and Fu S: A
radiosensitivity gene signature in predicting glioma prognostic via
EMT pathway. Oncotarget. 5:4683–4693. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Piao Y, Liang J, Holmes L, Henry V, Sulman
E and de Groot JF: Acquired resistance to anti-VEGF therapy in
glioblastoma is associated with a mesenchymal transition. Clin
Cancer Res. 19:4392–4403. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Jain RK: Normalization of tumor
vasculature: An emerging concept in antiangiogenic therapy.
Science. 307:58–62. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Gilbert MR, Dignam JJ, Armstrong TS, Wefel
JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S,
Won M, et al: A randomized trial of bevacizumab for newly diagnosed
glioblastoma. N Engl J Med. 370:699–708. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Chinot OL, Wick W, Mason W, Henriksson R,
Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea
D, et al: Bevacizumab plus radiotherapy-temozolomide for newly
diagnosed glioblastoma. N Engl J Med. 370:709–722. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Behnan J, Isakson P, Joel M, Cilio C,
Langmoen IA, Vik-Mo EO and Badn W: Recruited brain tumor-derived
mesenchymal stem cells contribute to brain tumor progression. Stem
Cells. 32:1110–1123. 2014. View Article : Google Scholar : PubMed/NCBI
|