Introduction
Head and neck squamous cell carcinomas (HNSCCs) are
the sixth most common cause of cancer-associated mortality
worldwide (1). Epidermal growth
factor receptor (EGFR), also known as HER1 or ErbB1, is a member of
the human EGFR tyrosine kinase family, which induces cell growth,
proliferation, survival and dedifferentiation in a variety of
tissues via the EGFR signalling pathway (2). However, aberrant EGFR-signalling occurs
in >90% of HNSCCs during the oncogenic process (3). In addition, overexpression of EGFR
correlates with tumour spread, poor survival and resistance to
radiotherapy and chemotherapy (3,4).
Concurrent chemoradiation (CRT) has been associated with improved
locoregional control and organ preservation, but it causes acute
and chronic toxicity. Molecular target therapies specifically
directed to EGFR may improve the outcomes of patients with HNSCC
while offering lower toxicity than traditional treatments (1). Thus, the development of monoclonal
antibodies or agents that inhibit EGFR or its tyrosine kinase is of
clinical relevance (5). In this
context, cetuximab (Erbitux®, Merck Serono GmbH, Darmstadt,
Germany) and trastuzumab (Herceptin®, Genentech Inc., South San
Francisco, CA, USA), which are monoclonal antibodies against ErbB1
and ErbB2, may be promising agents for the treatment of HNSCC
(6). In addition, gefitinib (Iressa®,
AstraZeneca, London, UK), a monoclonal antibody that inhibits
ErbB1-tyrosine kinase, has demonstrated anti-tumour activity in
small cell lung and HN cancer (2,7). Yoo et
al (8) noticed that the aberrant
expression of E-cadherin and β-catenin in non-small cell lung
cancer harbouring EGFR mutations was associated with poor response
to EGFR-tyrosine kinase inhibitor. Thus, the expression levels of
E-cadherin and β-catenin may affect certain anti-tumour therapies
(9).
Lapatinib, a novel synthetic small molecule
inhibitor of EGF1 and human HER2-tyrosine kinases, is used in the
form of lapatinib ditosylate (Tyverb®, GlaxoSmithKline, Brentford,
UK) as an active drug for breast and other solid tumours (2). In a randomized double-blind phase III
trial with 67 patients, Harrington et al (10) demonstrated that lapatinib combined
with CRT was a well-tolerated and safe therapy in patients with
high risk of recurrence following surgical treatment for stage
III/IV HN cancer. Thus, lapatinib may be used as concomitant and
maintenance therapy during cisplatin-based CRT, since this drug was
able to increase the rate of complete response at 6 months post-CRT
in p16- HNSCC (10).
The metastatic process consists of several steps: i)
The initial step, termed invasion, which requires the epithelial
tumour cells to become motile and degrade the underlying basement
membrane; ii) the second step, known as intravasation, during which
tumour cells invade across the endothelial lamina prior to
penetrating into blood or lymphatic vessels; iii) the third step,
known as systemic transport, during which a small number of tumour
cells appear to be capable of surviving various insults within
circulation; iv) the fourth step, termed extravasation, during
which a number of surviving cells may arrest in the vascular lumen;
and v) the final step, named colonization, which represents the
potential of the surviving tumour cells to proliferate (11).
Epithelial-mesenchymal transition (EMT) is described
as the loss of cell adhesion of non-motile, polarized epithelial
cells, followed by their transformation into a fibroblastoid,
mesenchymal phenotype with a high ability to migrate (12). EMT has been suggested to be crucial
for the development of a metastatic carcinoma cell phenotype with
potential capacity of invasion (12).
In oral SCC, EMT is characterized by the downregulation of
epithelial-specific adhesion proteins such as tight and adherent
junction proteins, including E-cadherin, cytokeratin, claudin and
desmoplakin (13). Furthermore, EMT
induces the expression of mesenchymal proteins such as vimentin,
N-cadherin and fibronectin, and promotes the development of
migratory attributes and alterations in the morphology of the
cells, including cell scattering (13–15).
Matrix metalloproteinases (MMPs) such as MMP-3 and −9 act as EMT
regulators by controlling certain aspects of oncogenesis (16). It has been previously reported that
the selective blockade of MMP-14 appears to abrogate invasion,
tumour growth and angiogenesis in ovarian cancer cells (17). By contrast, Zarrabi et al
(18) have reported that the
inhibition of MMP-14 promotes the migration of cancer cells
(18). The role of c-kit during EMT
remains unclear. Also known as cluster of differentiation (CD)117,
c-kit is a member of the receptor tyrosine kinase family, and acts
as oncogene in several tumours (19).
Tang et al (20) have
previously described an important function for c-kit in the
progression of salivary adenoid cystic cancer by orchestrating EMT.
In addition, the authors observed that the overexpression of c-kit
correlated with poor prognosis in these patients.
Various growth factors are capable of inducing EMT,
including vascular endothelial growth factor, hepatocyte growth
factor, transforming growth factor β 1 (TGFβ1) and EGF (16,21–24).
Epithelial cells may activate a transitory EMT program and its
reverse process, known as mesenchymal-epithelial transition (MET),
in order to continue their differentiation (12). These dynamic EMT/MET events highlight
the remarkable flexibility exhibited by differentiated cells during
morphogenesis and carcinogenesis (11,25).
Brabletz et al (26)observed
an association between nuclear β-catenin and mesenchymal transition
in metastases, as strong expression of nuclear β-catenin was
detected in the central areas of many metastases; however, tumor
cells recapitulated the differentiated epithelial phenotype of the
primary tumor. This indicates an ongoing shift between EMT and MET
during tumor progression.
MET is characterized by a reduction of proliferative
activity in the destroying tumour cells, which express high levels
of nuclear β-catenin (27). Lee et
al (16) reported the findings of
previous studies, which had identified hybrid cells exhibiting
epithelial and mesenchymal phenotype. This metastable phenotype was
characterized by residual expression of cadherin, cytokeratin,
nuclear β-catenin and vimentin, in addition to collective cell
growth.
Previous studies have suggested that the
multifunctional protein β-catenin is one of the most important
factors for reducing cell-cell interactions in malignant epithelial
cells (28,29). However, previous studies have reported
that β-catenin participates in the development of HN cancer
via a nuclear downstream effector of the canonical
wingless-related integration site (WNT)-signalling cascade
(15). Membrane-associated β-catenin
may be released into the cytoplasm by alteration of the degradation
complex, destabilization of cell-cell adhesion and loss of
expression of E-cadherin (12). The
accumulation of β-catenin in the cytoplasm leads to its nuclear
translocation, whereby it acts as cofactor of transcriptional
regulators. In invasive regions, tumour cells undergoing EMT
dissociate into single, disseminating tumour cells, which exhibit a
marked accumulation of nuclear β-catenin. The process of nuclear
accumulation of β-catenin and subsequent EMT is reversed in
metastases, while tumour cells adopt the differentiated epithelial
phenotype (12). This indicates
continuous switching between EMT and MET during the formation of
metastases (26). During the periods
of reduced proliferative activity, high expression levels of
nuclear β-catenin have been observed in dissociating mesenchymal
tumour cells (30). However, the
regulators of the intracellular distribution of β-catenin, which
are responsible for the heterogeneous pattern of expression of
β-catenin observed in tumour cells, remain unknown (31).
Cells undergoing EMT become invasive and develop
resistance to anticancer agents (32). The acquisition of EMT features has
also been associated with chemoresistance following standard
chemotherapy (33). Furthermore, EMT
has been suggested to be associated with drug resistance to
gefitinib and erlotinib (Tarceva®, OSI Pharmaceuticals, Inc.,
Melville, NY, USA) in non-small cell lung cancer (32). However, in pancreatic and ovarian
cancer, stable cell lines resistant to gemcitabine (Gemzar®, Eli
Lilly, Indianapolis, IN, USA) and paclitaxel (Taxol®, Bristol-Myers
Squibb, New York City, NY, USA), which were established by
continuous exposure to these drugs, were able to undergo EMT with
increased expression levels of snail family zinc finger 1 and twist
family bHLH transcription factor 1, which are transcription factors
that regulate the EMT process (34,35).
Therefore, the identification of tumour cell
phenotypes with malignant potential is of clinical interest, since
in order to inhibit the process of EMT or EMT-associated resistance
to anticancer agents, it is necessary to examine the phenotype of
SCC cells. The aim of the present study was to assess in a
quantitative and qualitative manner the expression pattern of
vimentin, β-catenin, E-cadherin, MMP-14 and c-kit in mesenchymal-
and epithelial-associated SCC cells, prior and following exposure
to lapatinib and gefitinib, and to evaluate the invasive metastatic
cell phenotype by detecting potential differences between
mesenchymal- and epithelial- SCC cells.
Materials and methods
Cell lines and culture
The SCC cell lines HNSCC22B (UMSCC22B) and HNSCC11A
(UMSCC11A), which descend from human metastatic HNSCC of the
sinus piriformis (hypopharynx) and supraglottic larynx, were
kindly provided by Dr T. E. Carey (University of Michigan, Ann
Arbor, MI, USA). Cells were cultured at 37°C in humidified
atmosphere with 5% CO2, using 500 ml minimum essential medium
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS; Thermo Fisher
Scientific, Inc.) and antibiotics (5 ml L-glutamine solution, cat
no. 9183.1; Carl Roth GmbH & Co. KG, Karlsruhe, Germany; and
penicillin, streptomycin, fungizone solution, cat no. C42020;
PromoCell GmbH, Heidelberg, Germany). For immunocytochemistry,
1×104 cells/well were seeded in 8-well cell culture slides (BD
Biosciences, Franklin Lakes, NJ, USA). When confluent, cells were
starved using minimum essential medium (MEM) depleted of FBS for 5
h, and then incubated for 5, 24 and 96 h with 0.5 and 2 µg/ml
lapatinib and gefitinib in 0.5% FBS/MEM. The stimulation was
repeated every 24 h. The different drug concentrations and
stimulation times used in the present study were selected upon
performing a cell proliferation assay with alamarBlue® (AbD
Serotec, Oxford, UK). Following incubation with the above drugs,
the cell supernatants were collected together in sterile tubes, and
stored at −20°C until further analysis.
Enzyme-linked immunosorbent assay
(ELISA)
The protein expression levels of β-catenin,
E-cadherin and vimentin were determined by ELISA, using DuoSet® IC
ELISA (R&D Systems, Inc., Minneapolis, MN, USA) and PathScan®
Total Vimentin Sandwich ELISA Kit (cat no. 7789; R&D Systems,
Inc.), respectively. The system utilized anti-human solid-phase
monoclonal antibodies and enzyme-linked monoclonal mouse antibodies
against β-catenin (Human Total β-Catenin DuoSet® IC ELISA, 2 Plate;
cat no. DYC1329; R&D Systems, Inc.) and E-cadherin (Human
E-Cadherin DuoSet® ELISA, 15 Plate; cat no. DY648; R&D Systems,
Inc.) and a mouse monoclonal antibody against vimentin (PathScan®
Total Vimentin Sandwich ELISA Kit; cat no. 7789; Cell Signaling
Technology Inc, Danvers, MA, USA). Following 0, 5, 24 and 96-h
incubation with 2 µg/ml lapatinib and gefitinib, the expression
levels of β-catenin, E-cadherin and vimentin in the supernatants of
the treated and untreated cells were determined by measuring the
optical density at a wavelength of 540 nm in a microplate reader.
Each assay was measured in 100 µl of supernatant, and all analyses
and calibrations were performed in triplicate. The concentration of
β-catenin and E-cadherin, reported in pg/ml, and vimentin, reported
in µg/ml, were defined upon subjecting the HNSCC tumour cell lines
to a quantitative cell proliferation assay with alamarBlue® (AbD
Serotec). The non-stimulated cells were grow to confluence in the
presence of 10% FBS/MEM and then incubated for 5 h with MEM and
phosphate-buffered saline (PBS; Dako, Glostrup, Germany) instead of
drug.
Immunocytochemistry
Immunocytochemical analysis was performed using
anti-human antibodies directed against β-catenin (monoclonal
rabbit; 1:200 dilution; cat no. 32572; Abcam, Cambridge, UK),
E-cadherin (monoclonal mouse; 1:50 dilution; cat no. Ab1416;
Abcam), c-kit (polyclonal rabbit; 1:200 dilution; cat no.
NB100-1766; Novus Biologicals, LLC; Littleton, CO, USA), MMP-14
(polyclonal rabbit; 1:250 dilution; cat no. 73879; Abcam) and
vimentin (monoclonal mouse; 1:50 dilution; cat no. M0725; Dako).
Immunostaining was performed using the streptavidin-biotin complex
method (Amersham, RPN 1051; 1:100 dilution). Prior to being
subjected to immunocytochemistry, SCC cells were cultured in 8-well
chambers overnight, and exposed to different concentrations of
lapatinib (0.5 or 2 mg/ml) for 0, 5, 24 and 96 h while growing to
confluency. Subsequently, the cells underwent fixation with a 2:1
dilution of acetone (cat no. 100014.2511; Merck Serono, GmbH) and
alcohol (denaturedethanol absolute, 99,8%; cat no. K928.4; Carl
Roth GmbH & Co. KG), followed by three washes with PBS (Buffer
kit; Dako) for 5 min each time at room temperature.
Next, the cells were automatically stained with
TechMate™ 500 (Dako), which executed the following steps: i)
Incubation of the cells for 30 min at room temperature with the
corresponding primary antibody solution, using the aforementioned
ratios of antibody:cells; and ii) three washes of the slices with
PBS for 5 min at room temperature. Following incubation with a
mouse antibody against alkaline phosphatase-anti-alkaline
phosphatase (cat no. K5000; Dako), the results of the
immunoreaction were visualized with DAKO ChemMate™ Detection Kit
(Dako) and AxioVision Scan Scope 4.8.3 software (Zeiss AG,
Oberkochen, Germany), according to the manufacturers' protocol. For
this purpose, the cells were incubated in sheep serum (1:10
dilution; cat no. ADI-ALBSH20-S; Linaris Biological Products GmbH,
Dossenheim, Germany) in the presence of the aforementioned
monoclonal antibodies. Next, the cells were incubated with a
specific biotinylated secondary antibody and streptavidin-biotin
horseradish peroxidase complex (GE Healthcare Life Sciences,
Chalfont, UK) for 45 min at room temperature. Aminoethylcarbazol
(cat no. A5754; Sigma-Aldrich, St. Louis, MO, USA) was used as
chromogen in the peroxidase reaction, and the endogenous peroxidase
activity was blocked prior to washing the cells three times with
PBS for 5 min each at room temperature. For the negative controls,
the cells were incubated with all the reagents described above,
except for the primary antibody. The sections underwent
counterstaining by Harris haematoxylin (MSDS code, SSHXHHE; Emergo
Group, The Hague, The Netherlands) for 30 sec, followed by
dehydration in graded ethanol and coverslipping. The protein
expression levels of β-catenin, E-cadherin, vimentin, MMP-14 and
c-kit were determined immunohistochemically using an AxioVision
Scan Scope microscope (Zeiss AG) and AxioVision Scan Scope 4.8.2
software (Zeiss AG). The staining intensity was considered to be
strong if >80% cells were positively stained; moderate, if
50–80% cells were positively stained; and negative, if no
positively stained cells were detected.
Statistical analysis
Statistical analysis was performed in collaboration
with Dr C. Weiss (Institute of Biomathematics, Faculty of Medicine,
Mannheim, Germany). Data are presented as the mean ± standard
deviation. P<0.05 was considered to indicate a statistically
significant difference. Any differences in the protein expression
levels of β-catenin, E-cadherin, vimentin, MMP-14 and c-kit between
the treated and control cultures were analysed using Dunnett's and
t tests for linear models.
Results
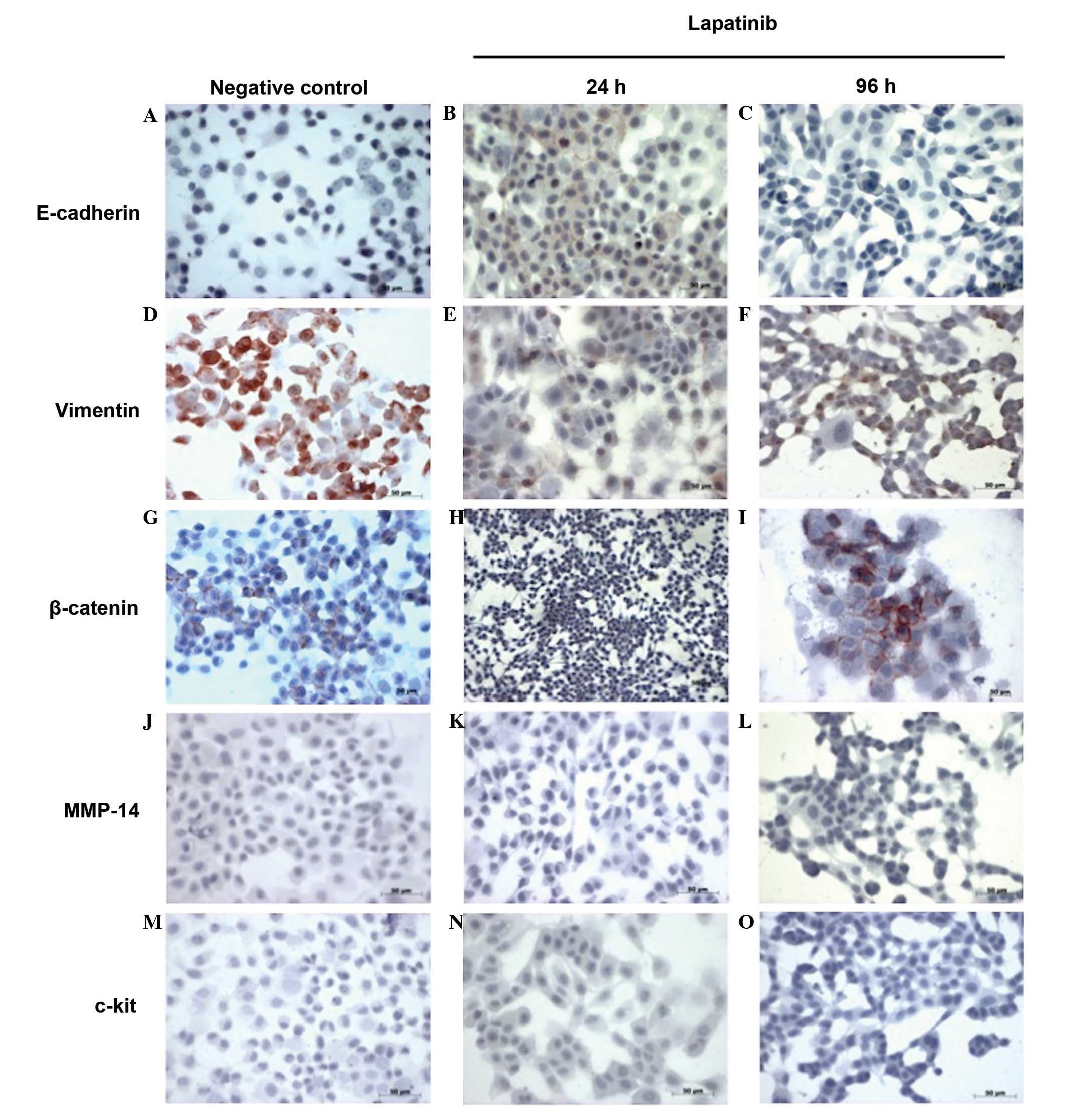
Immunocytochemistry of E-cadherin,
vimentin, β-catenin, MMP-14 and c-kit expression in HNSCC11A and
HNSCC22B cells
Prior to stimulation, the established HNSCC11A and
HNSCC22B cell lines were assessed for morphological alterations and
expression of key EMT markers. The SCC cell line HNSCC22B
(UMSCC22B) exhibited the most pronounced epithelial phenotype
(scattering-, E-cadherin+ and vimentin-) (data not shown).
Contrarily, the HNSCC11A SCC cell line (UMSCC11A) exhibited the
most pronounced mesenchymal phenotype (scattering+, E-cadherin- and
vimentin+), with disaggregated cell growth and spindle shape (data
not shown). Immunocytochemical analysis demonstrated that in the
negative controls of the HNSCC11A cell line, the cells expressed a
moderate level of membrane-associated E-cadherin and a strong
positivity for vimentin. However, no deposition of c-kit and MMP-14
was observed (Fig. 1). In the
lapatinib-treated HNSCC11A cells, an increase in the levels of
membrane-associated E-cadherin and a considerable loss of
deposition of vimentin was observed, particularly at 24 h
post-incubation (Fig. 1). Lapatinib
also induced a reduction in the levels of β-catenin and the
reorganization of membrane-associated β-catenin and its nuclear
deposition in the HNSCC11A cell line, particularly following 24 h
incubation with lapatinib. This implies that the effect of
lapatinib on the expression of β-catenin may be reversible. At 96 h
post-incubation, a stronger expression of β-catenin was observed,
whereas no deposition of MMP-14 and c-kit in the HNSCC11A cells was
detected following treatment with lapatinib (Fig. 1).
By contrast, the non-treated HNSCC22B cells
preferentially exhibited a strong membranous staining for
E-cadherin and β-catenin, but no immunohistochemical positivity for
vimentin and c-kit (Table I).
Following incubation with lapatinib, a slightly reduced deposition
of E-cadherin and a mild increase in the expression levels of
β-catenin were noticed, particularly at 96 h post-incubation, but
no positivity for vimentin and c-kit was observed. In addition, no
increase in immunohistochemical positivity for MMP-14 was detected
at post-incubation with lapatinib compared with the control
(Table I). The differences observed
for control cells at different times may be due to epigenetic
mechanisms that activate plasticity (36), thus affecting cell phenotype (Table I).
 | Table I.Immunostaining results for
E-cadherin, vimentin, β-catenin, MMP-14 and c-kit expression in
HNSCC22B cells. |
Table I.
Immunostaining results for
E-cadherin, vimentin, β-catenin, MMP-14 and c-kit expression in
HNSCC22B cells.
|
| Lapatinib |
|---|
|
|
|
|---|
| Immunostaining | 5 h | 24 h | 96 h |
|---|
| Control group |
|
|
|
|
E-cadherin | ++ | ++ | + |
|
Vimentin | 0 | 0 | 0 |
|
β-catenin | + | +/++ | + |
|
MMP-14 | 0 | 0/+ | 0/+ |
|
c-kit | 0 | 0 | 0 |
| Lapatinib 0.5
µg |
|
|
|
|
E-cadherin | ++ | ++ | +/++ |
|
Vimentin | 0 | 0 | 0 |
|
β-catenin | ++ | ++ | ++ |
|
MMP-14 | 0 | 0/+ | 0 |
|
c-kit | 0 | 0 | 0 |
| Lapatinib 2 µg |
|
|
|
|
E-cadherin | ++ | ++ | ++ |
|
Vimentin | 0 | 0 | 0 |
|
β-catenin | ++ | + | ++ |
|
MMP-14 | 0 | 0 | 0 |
|
c-kit | 0 | 0 | 0 |
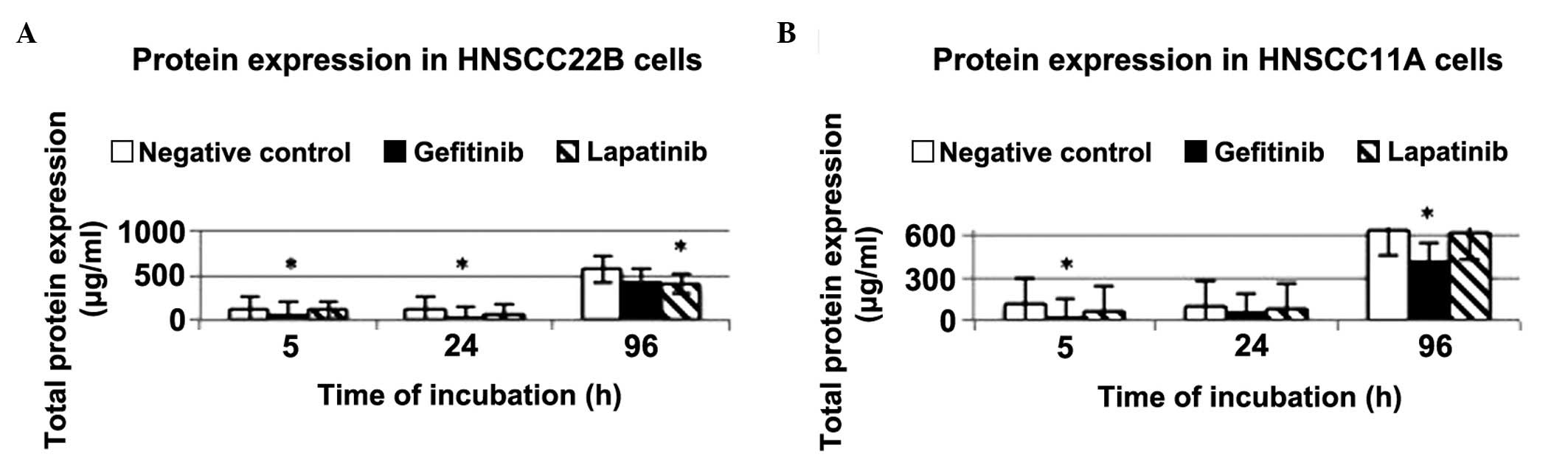
ELISA of total protein expression in
HNSCC11A and HNSCC22B cells
In order to determine the total protein expression
levels in the supernatant of the tumour cell lines HNSCC22B and
HNSCC11A, ELISA analyses were conducted at 5, 24 and 96 h
post-incubation with 2 µg/ml lapatinib and gefitinib. The total
protein content appeared to increase with time in the control and
drug-treated cells, suggesting that none of the drugs tested have a
clear effect on total protein expression levels. By contrast, an
increase in total protein expression levels was detected in
HNSCC22B cells, particularly at 96 h post-stimulation with
lapatinib. In HNSCC11A cells, the total protein expression levels
were markedly downregulated following treatment with 2 µg/ml
gefitinib, particularly at 5 and 96 h post-incubation (total
protein content = 19.38 and 423.06 µg/ml, P=0.0089 and 0.0038,
respectively), compared with the control group (Fig. 2). The concentration of the drug did
not exert a significant influence on the total protein expression
levels (data not shown).
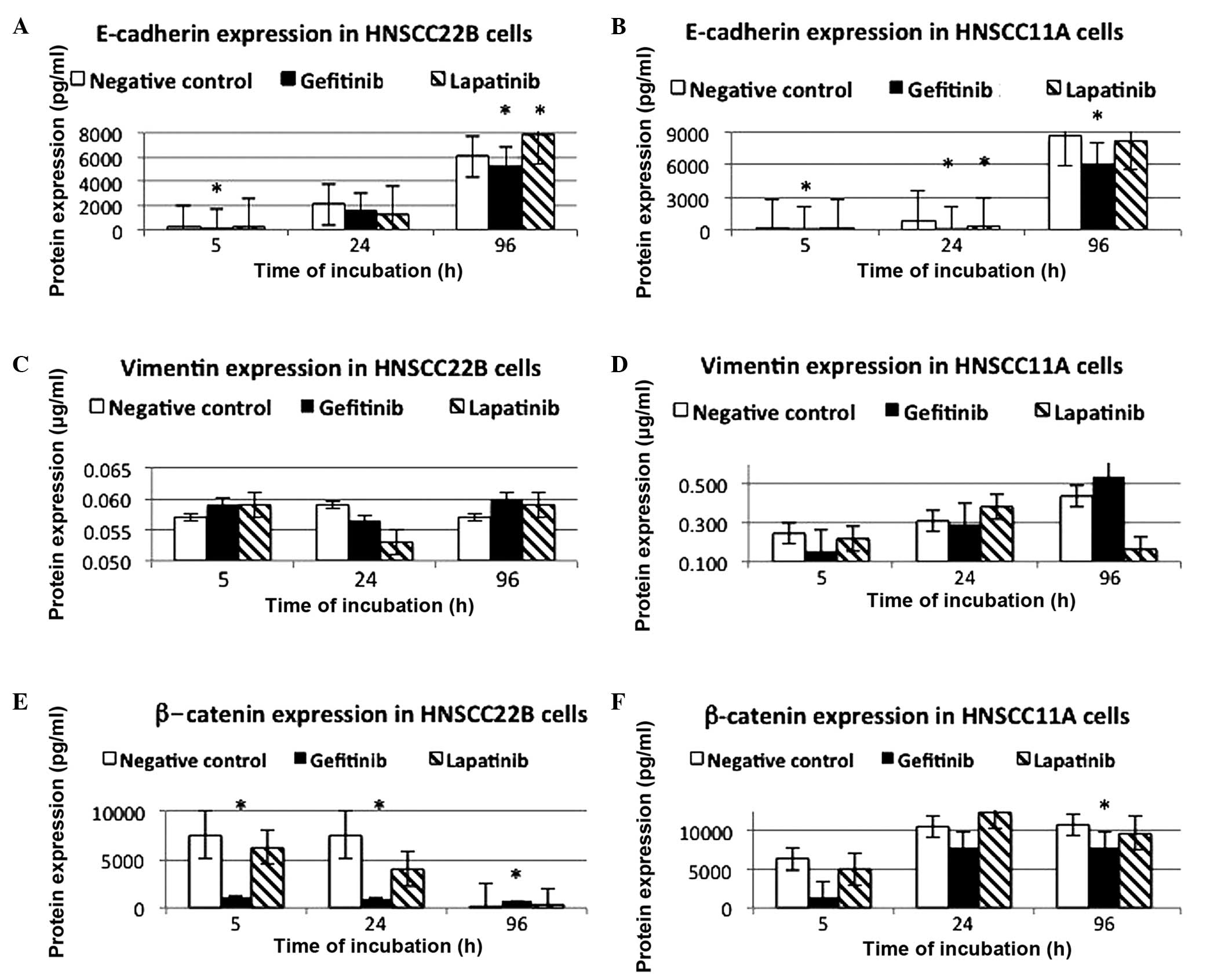
ELISA of E-cadherin, vimentin and
β-catenin expression in HNSCC11A and HNSCC22B cells
In contrast to the HNSCC11A cells in the control
group, high protein expression levels of E-cadherin were exhibited
by the non-stimulated HNSCC22B cells at 5 h incubation (69.919 vs.
227.520 pg/ml, respectively). The increase in the levels of
E-cadherin displayed by these two SCC cell lines upon incubation
with lapatinib was time-dependent. A significant increase in the
expression levels of E-cadherin was detected in HNSCC22B cells
following 96-h treatment with 2 µg/ml lapatinib (7,814.760 pg/ml,
P<0.0001) and 5-h incubation with 2 µg/ml gefitinib (118.700
pg/ml, P<0.0001) (Table II).
Notably, in the SCC HNSCC22B cell line, a marginal increase in the
expression level of E-cadherin was noted following a prolonged
treatment time (96 h) with lapatinib (Fig. 3A and B, Table II). In Table II, the differences observed for
control cells at different times may be due to epigenetic
mechanisms that activate plasticity (36).
| Table II.Expression levels of E-cadherin in
HNSCC11A and HNSCC22B cells following incubation with 2 µg/ml
lapatinib and gefitinib, as determined by enzyme-linked
immunosorbent assay. |
Table II.
Expression levels of E-cadherin in
HNSCC11A and HNSCC22B cells following incubation with 2 µg/ml
lapatinib and gefitinib, as determined by enzyme-linked
immunosorbent assay.
|
| Mean ± SD
expression levels of E-cadherin, pg/ml (P-value)a |
|---|
|
|
|
|---|
| Incubation time
(h) | Control | Lapatinib | Gefitinib |
|---|
| HNSCC11A |
|
|
|
| 5 | 69.919±9.89 | 74.601±4.46
(0.9791) | 105.395±10.98
(0.0197)b |
| 24 | 858.025±184.42 | 324.370±186.53
(<0.0001)b | 96.557±10.89
(<0.0001)b |
| 96 |
8,676.943±367.50 | 8,250.640±1,007.40
(0.9684) | 6,073.640±1,700.32
(0.0355) |
| HNSCC22B |
|
|
|
| 5 | 227.520±19.08 | 220.920±25.09
(0.9594) | 118.700±17.79
(<0.0001)b |
| 24 |
2,109.010±454.91 | 1,233.480±1,041.00
(0.5330) | 1,538.070±1,196.82
(0.8194) |
| 96 |
6,053.370±323.68 | 7,814.760±249.48
(<0.0001)b | 5,260.540±436.67
(0.0111)b |
In contrast to the low protein expression levels of
vimentin in HNSCC22B cells, higher protein expression levels were
observed in control HNSCC11A cells at 5-h incubation (0.247 µg/ml).
The protein expression levels of vimentin in HNSCC22B cells were
reduced following 24-h incubation with lapatinib and gefitinib. The
expression of vimentin was not significantly downregulated in
HNSCC11A cells following 96-h treatment with lapatinib (0.170
µg/ml, P=0.5000), while upon 96-h treatment with 2 µg/ml gefitinib,
the protein expression levels of vimentin in these cells were 0.533
µg/ml (P=0.96). By contrast, increased protein expression levels of
vimentin were observed in HNSCC22B cells following prolonged
treatment with lapatinib and gefitinib, particularly upon 96 and
5-h incubation with these drugs (Fig. 3C
and D).
In summary, low protein expression levels of
vimentin were detected in non-stimulated HNSCC22B cells, and a
time-dependent increase in the protein expression levels of
vimentin was observed in HNSCC11A cells exposed to 2 µg/ml
gefitinib for 5–96 h, whereas the drug treatment at different
concentrations did not exert a significant influence on the protein
expression levels of vimentin (data not shown).
High protein expression levels of β-catenin were
detected in the control HNSCC22B cells upon 5-h incubation
(7,587.00 pg/ml). HNSCC22B cells displayed reduction in the protein
expression levels of β-catenin following incubation with lapatinib
and gefitinib. In HNSCC22B cells, the protein expression levels of
β-catenin were significantly downregulated upon incubation with 2
µg/ml gefitinib for 5 and 24 h (1,051.33 and 960.33 pg/ml, P=0.0021
and 0.0293, respectively). By contrast, HNSCC11A cells displayed a
consistent trend towards an incubation time-dependent increase in
the protein expression levels of β-catenin after 24 h, and then
stabilized in the control and treated cells. Particularly, a
significant impact on the protein expression levels of β-catenin
was observed in HNSCC11A cells following 96-h stimulation with 2
µg/ml gefitinib (P=0.0003). Significant downregulation of the
expression of β-catenin was observed in HNSCC22B cells upon 5, 24
and 96-h incubation with 2 µg/ml gefitinib (1,051.33, 960.33 and
680.53 pg/ml, P=0.0021, 0.0293 and 0.0183, respectively; Fig. 3E and F, Table III). Similarly, a significant
time-dependent reduction in the expression levels of β-catenin was
observed in HNSCC22B cells when exposed to 0.5 µg/ml gefitinib for
5 h (3,496.00 pg/ml; P<0.0400; data not shown). The differences
observed for control cells at different times in Table III may be due to epigenetic
mechanisms that activate plasticity (36).
| Table III.Expression levels of β-catenin in
HNSCC11A and HNSCC22B cells following incubation with 2 µg/ml
lapatinib and gefitinib, as determined by enzyme-linked
immunosorbent assay. |
Table III.
Expression levels of β-catenin in
HNSCC11A and HNSCC22B cells following incubation with 2 µg/ml
lapatinib and gefitinib, as determined by enzyme-linked
immunosorbent assay.
|
| Mean ±SD expression
levels of β-catenin, pg/ml (P-value)a |
|---|
|
|
|
|---|
| Incubation time
(h) | Control | Lapatinib | Gefitinib |
|---|
| HNSCC11A |
|
|
|
| 5 |
6,316.33±2.697.22 | 5,008.33±3,464.97
(0.9288) | 1,262.67±433.66
(0.1231) |
| 24 |
10,557.33±2,958.91 | 12,331.00±1,327.30
(0.8905) | 7,759.67±4,585.48
(0.6567) |
| 96 |
10,774.33±731.52 | 9,692.33±277.49
(0.1350) | 7,774.33±778.50
(0.0003) |
| HNSCC22B |
|
|
|
| 5 |
7,587.00±1,848.14 | 6,308.67±1,830.21
(0.7439) | 1,051.33±141.93
(0.0021)b |
| 24 |
7,546.00±1,653.26 | 4,009.67±3,565.12
(0.3053) | 960.33±140.86
(0.0293)b |
| 96 | 55.07±52.2 | 251.83±86.62
(0.6529) | 680.53±133.54
(0.0183)b |
Discussion
Based on its incidence rate, HNSCC is considered the
sixth most common type of cancer worldwide (37). The consumption of tobacco and alcohol
have been identified as risk factors for HNSCC, and appear to exert
a synergistic effect in the development of the disease (12). In addition, certain types of HNSCC,
particularly oropharyngeal cancer, are caused by infection with
high risk HPV (38). Patients with
HNSCC usually exhibit poor survival rates, due to the high
frequency of local invasion, cervical lymph node dissemination,
distant metastasis and second primary tumours (39). In addition, HNSCC presents a high
propensity to develop local recurrence following treatment
(40). Concomitant CTR treatment has
been previously demonstrated to increase the overall and five-year
survival rate of patients with HNSCC at advanced stages, and offer
better locoregional control rate (29). To further improve the survival rates
for patients with HNSCC, particularly those presenting with
unresectable HNSCC, innovative strategies and targeted therapies
must be explored (41). HNSCC arises
from the accumulation of epigenetic and genetic events at the
cellular level, which result in malignant cells exhibiting
characteristics such as resistance to apoptosis, insensitivity to
anti-growth signals, abnormalities in cancer-associated signalling
pathways, limitless replicative potential, self-sufficiency
regarding growth signals, increased angiogenesis, metastasis and
invasion capacity (40). Although the
tumorigenic pathways and molecular aetiologies of HNSCC have been
studied extensively, a limited number of diagnostic clinical
applications are currently in practice (42).
Thus, to improve the survival rates of patients with
HNSCC, it is important to investigate the phenotypic alterations
that occur in HNSCC cells and enable them to destroy the basement
membrane, invade and metastasize (12). In the past years, there has been an
increased interest in EMT and its role in the progression of HNSCC
(39). EMT is a mechanism by which
solid tumours gain metastatic potential (43). During EMT, cells within the tumour
environment downregulate the expression of adhesion receptors such
as cadherins and integrins, which are involved in cell-cell
attachment. By contrast, the expression of adhesion receptors that
induce cell motility is upregulated during EMT (44). In addition, the expression of
metalloproteases is also upregulated during EMT, which promotes
metastasis (45). Therefore, the
identification of the phenotype responsible for the malignant
potential of SCC tumour cells is of clinical interest (46). Richter et al (47) previously demonstrated that oral SCC
cells with an epithelial phenotype were capable of undergoing a
complete EMT process, including downregulation of E-cadherin,
upregulation of vimentin and scattered cell growth, following
prolonged co-stimulation with EGF and TGFβ1. EMT is a key
developmental program that is often activated during cancer
progression and may promote resistance to anti-tumour therapy.
Thus, the inhibition of EMT appears to be the crucial mechanism to
avoid drug resistance (33). Lee
et al (16) assessed the
morphological alterations displayed by lung and colon cancer cells
resistant to gefitinib chemotherapy, and evaluated their cell
invasion and motility abilities, and the levels of key EMT markers
expressed by these cells. The authors demonstrated that the
transfection of microRNA-147, aimed to induce MET and promote cell
cycle arrest, increased the sensitivity of these tumour cells
towards the EGFR inhibitor gefitinib (48).
β-catenin is involved in WNT-signalling (44). Upon nuclear translocation, β-catenin
may upregulate the transcription genes that promote cytoplasmatic
degradation and cancer progression, thus affecting the progression
of EMT (49). Brabletz et al
(27) detected nuclear accumulation
of β-catenin in dedifferentiated mesenchymal-like tumour cells
during the invasion step of colorectal adenocarcinoma. The
metastatic process of epithelial tumours is characterised by
aberrant expression of E-cadherin and β-catenin, which occurs at
different points in time (46).
Significant downregulation of the expression of
β-catenin was observed in HNSCC22B cells following incubation with
gefitinib for 5 and 24 h. In HNSCC11A cells, nuclear deposition of
β-catenin was detected by histopathological reactivity,
particularly following 96-h incubation with lapatinib. These
results are in agreement with the aforementioned findings by
Brabletz et al (27), who had
described an increase in the levels of nuclear β-catenin in
dedifferentiated mesenchymal-like tumour cells at the invasive
front in colorectal cancer.
The majority of HNSCCs (>90%) display
overexpression of EGFR at the protein level (50), which is associated with local or
regional recurrence and overall reduced survival (51,52).
Lapatinib is a novel synthetic small molecule inhibitor of EGFR and
HER2 tyrosine kinases (53). In the
present study, positivity for vimentin was detected
immunocytochemically in non-stimulated HNSCC11A cells. Following
incubation with lapatinib, reduced deposition of vimentin was
observed in these cells. In addition, increased levels of
E-cadherin were observed in HNSCC11A cells following 24-h
incubation with lapatinib.
Housman et al (45) previously reported several factors
occurring during EMT that participate in the development of drug
resistance. These factors are dependent on the metastatic grade of
the tumour, which is defined as the level of dedifferentiation and
degree of EMT exhibited by the tumour (45). The reverse process of EMT, known as
MET, has also been reported (54).
EMT and MET are involved in organ development, stem cell biology,
wound healing and cancer progression (44). In order to undergo MET, upregulation
of E-cadherin is required, since this protein enables cells to
return to their epithelial phenotype (43).
MET is characterized by the loss of mesenchymal
markers, including vimentin, fibronectin and α smooth muscle
protein, and the enhancement of epithelial markers such as
E-cadherin, occludin and desmoplakin (54). In the present study, a mild
upregulation of the expression levels of E-cadherin, and
downregulation of the expression of vimentin, were observed in
HNSCC11A cells following 96-h incubation with lapatinib. The same
effect on expression of E-cadherin was also observed in the
untreated HNSCC11A cells (Fig. 3D).
These results suggest that the incubation with lapatinib inhibited
a partial EMT and promoted a partial MET in these cells. Previous
studies have indicated that MET may be enhanced by blocking the
activity of certain factors and signalling pathways that activate
EMT (55). Thus, metastatic cells may
revert back via MET to re-acquire epithelial characteristics
similar to exhibited by the cells in the primary tumour (44).
The different effects of gefitinib and lapatinib on
the expression levels of β-catenin and vimentin may be explained by
the different phenotypes of the HNSCC cell lines. The HNSCC22B cell
line exhibited the most pronounced epithelial phenotype
(scattering-, E-cadherin+ and vimentin-). By contrast, the HNSCC11A
cell line exhibited the most pronounced mesenchymal phenotype
(scattering+, E-cadherin-, vimentin+). Only oral SCC cells with an
epithelial phenotype were capable of undergoing a complete EMT.
This may also explain why different tumor cells show different
changes following incubation with gefitinib and lapatinib.
Furthermore, this suggests that inhibiting EMT by gefitinib or
lapatinib cannot be observed in every tumor cell. Therefore, it is
important to characterize the phenotype of the tumor cell line
prior to treating with geftinib or lapatinib.
It has been previously demonstrated that the
activation of EMT promotes local tumour invasion, intravasation and
extravasation of the systemic circulation, while MET is essential
for establishing macrometastases (25). Therefore, targeting EMT and MET may
provide effective therapeutic agents for the treatment of cancer
(25). EMT is associated with reduced
cell proliferation, in contrast to MET, which promotes metastatic
growth. Whether inhibiting EMT is a valid approach to prevent
metastasis remains unknown. Nevertheless, potential therapeutic
interventions aimed to target EMT and MET may be complex, since EMT
occurs at an early stage of metastasis, whereas MET occurs at later
stages (25). In the present study,
aberrant expression levels of β-catenin were detected in HNSCC11A
and HNSCC22B cells. Furthermore, the expression of vimentin was
observed to be upregulated in HNSCC22B cells following incubation
with lapatinib and gefitinib for 96 h.
In order to develop novel anti-HNSCC therapies that
block the progression of EMT, the mechanistic role of EMT markers
associated with cell-cell contact in HNSCC cells should be further
clarified. For this purpose, the cells in the present study were
grown to confluency prior to stimulation. In HNSCC11A,
vimentin-upregulated and small E-cadherin-downregulated SCC cells,
MET was observed, alongside upregulated expression of β-catenin,
particularly following 24-h treatment with lapatinib. Targeting EMT
and MET may provide effective therapeutics for cancer. Thus, future
studies on the molecular interactions associated with the
inhibition of EMT and activation of MET are required, to further
understand the process of drug-induced EMT and resistance of SCC
cells to targeted therapy, and to develop novel anti-tumour
therapies for the treatment of SCC.
Acknowledgements
The authors would like to thank Miss Petra Prohaska
for her excellent technical assistance, and Dr C. Weiss for his
valuable assistance in statistical analysis.
References
|
1
|
Denaro N, Russi EG, Adamo V and Merlano
MC: State-of-the-art and emerging treatment options in the
management of head and neck cancer: News from 2013. Oncology.
86:212–229. 2014.PubMed/NCBI
|
|
2
|
Burris HA III: Dual kinase inhibition in
the treatment of breast cancer: Initial experience with the
EGFR/ErbB-2 inhibitor lapatinib. Oncologist. 9(Suppl 3): 10–15.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kalyankrishna S and Grandis JR: Epidermal
growth factor receptor biology in head and neck cancer. J Clin
Oncol. 24:2666–2672. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jedlinski A, Ansell A, Johansson AC and
Roberg K: EGFR status and EGFR ligand expression influence the
treatment response of head and neck cancer cell lines. J Oral
Pathol Med. 42:26–36. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Califano R, Morgillo F, De Mello RA and
Mountzios G: Role of mesenchymal-epithelial transition
amplification in resistance to anti-epidermal growth factor
receptor agents. Ann Transl Med. 3:812015.PubMed/NCBI
|
|
6
|
Privitera G, Luca T, Musso N, Vancheri C,
Crimi N, Barresi V, Condorelli D and Castorina S: In vitro
antiproliferative effect of trastuzumab (Herceptin®) combined with
cetuximab (Erbitux®) in a model of human non-small cell lung cancer
expressing EGFR and HER2. Clin Exp Med. Feb 26–2015.(Epub ahead of
print). View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Erjala K, Sundvall M, Junttila TT, Zhang
N, Savisalo M, Mali P, Kulmala J, Pulkkinen J, Grenman R and
Elenius K: Signalling via ErbB2 and ErbB3 associates with
resistance and epidermal growth factor receptor (EGFR)
amplification with sensitivity to EGFR inhibitor gefitinib in head
and neck squamous cell carcinoma cells. Clin Cancer Res.
12:4103–4111. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoo SB, Kim YJ, Kim H, Jin Y, Sun PL,
Jheon S, Lee JS and Chung JH: Alteration of the
E-cadherin/β-catenin complex predicts poor response to epidermal
growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI)
treatment. Ann Surg Oncol. 20(Suppl 3): S545–S552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mukherjee S, Mazumdar M, Chakraborty S,
Manna A, Saha S, Khan P, Bhattacharjee P, Guha D, Adhikary A,
Mukhjerjee S and Das T: Curcumin inhibits breast cancer stem cell
migration by amplifying the E-cadherin/β-catenin negative feedback
loop. Stem Cell Res Ther. 5:1162014. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Harrington K, Berrier A, Robinson M, et
al: Randomised Phase II study of oral lapatinib combined with
chemoradiotherapy in patients with advanced squamous cell carcinoma
of the head and neck: Rationale for future randomised trials in
human papilloma virus-negative disease. Eur J Cancer. 49:1609–1618.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Umbreit C, Flanjak J, Weiss C, Erben P,
Aderhold C, Faber A, Stern-Straeter J, Hoermann K and Schultz JD:
Incomplete epithelial-mesenchymal transition in p16-positive
squamous cell carcinoma cells correlates with β-catenin expression.
Anticancer Res. 34:7061–7069. 2014.PubMed/NCBI
|
|
13
|
Krisanaprakornkit S and Iamaroon A:
Epithelial-mesenchymal transition in oral squamous cell carcinoma.
ISRN Oncol. 2012:6814692012.PubMed/NCBI
|
|
14
|
Hay ED: The mesenchymal cell, its role in
the embryo and the remarkable signalling mechanisms that create it.
Dev Dyn. 233:706–720. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumour
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signalling, development and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kaimal R, Aljumaily R, Tressel SL, et al:
Selective blockade of matrix metalloprotease-14 with a monoclonal
antibody abrogates invasion, angiogenesis and tumour growth in
ovarian cancer. Cancer Res. 73:2457–2467. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zarrabi K, Dufour A, Li J, Kuscu C,
Pulkoski-Gross A, Zhi J, Hu Y, Sampson NS, Zucker S and Cao J:
Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer
cell migration. J Biol Chem. 286:33167–33177. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Umbreit C, Aderhold C, Faber A, Sommer JU,
Sauter A, Hofheinz RD, Stern-Sträter J, Hoermann K and Schultz JD:
Unexpected alteration of β-catenin and c-KIT expression by 5-FU and
docetaxel in p16-positive squamous cell carcinoma compared to
HPV-negative HNSCC cells in vitro. Anticancer Res. 33:2457–2465.
2013.PubMed/NCBI
|
|
20
|
Tang YL, Fan YL, Jiang J, Li KD, Zheng M,
Chen W, Ma XR, Geng N, Chen QM, Chen Y and Liang XH: C-kit induces
epithelial-mesenchymal transition and contributes to salivary
adenoid cystic cancer progression. Oncotarget. 5:1491–1501. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Al Moustafa AE, Achkhar A and Yasmeen A:
EGF-receptor signalling and epithelial-mesenchymal transition in
human carcinomas. Front Biosci (Schol Ed). 4:671–684. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang JY, Wright JM and Svoboda KK:
Signall transduction pathways involved in epithelial-mesenchymal
transition in oral cancer compared with other cancers. Cells
Tissues Organs. 185:40–47. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang ZC, Yi MJ, Ran N, Wang C, Fu P, Feng
XY, Xu L and Qu ZH: Transforming growth factor-β1 induces bronchial
epithelial cells to mesenchymal transition by activating the Snail
pathway and promotes airway remodeling in asthma. Mol Med Rep.
8:1663–1668. 2013.PubMed/NCBI
|
|
24
|
Acevedo VD, Gangula RD, Freeman KW, Li R,
Zhang Y, Wang F, Ayala GE, Peterson LE, Ittmann M and Spencer DM:
Inducible FGFR-1 activation leads to irreversible prostate
adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer
Cell. 12:559–571. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guo F, Parker Kerrigan BC, Yang D, Hu L,
Shmulevich I, Sood AK, Xue F and Zhang W: Post-transcriptional
regulatory network of epithelial-to-mesenchymal and
mesenchymal-to-epithelial transitions. J Hematol Oncol. 7:192014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brabletz T, Hlubek F, Spaderna S,
Schmalhofer O, Hiendlmeyer E, Jung A and Kirchner T: Invasion and
metastasis in colorectal cancer: Epithelial-mesenchymal transition,
mesenchymal-epithelial transition, stem cells and beta-catenin.
Cells Tissues Organs. 179:56–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brabletz T, Jung A, Reu S, Porzner M,
Hlubek F, Kunz-Schughart LA, Knuechel R and Kirchner T: Variable
beta-catenin expression in colorectal cancers indicates tumour
progression driven by the tumour environment. Proc Natl Acad Sci
USA. 98:10356–10361. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hajra KM and Fearon ER: Cadherin and
catenin alterations in human cancer. Genes Chromosomes Cancer.
34:255–268. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schultz JD, Sommer JU, Hoedt S, Erben P,
Hofheinz RD, Faber A, Thorn C, Hörmann K and Sauter A:
Chemotherapeutic alteration of β-catenin and c-kit expression by
imatinib in p16-positive squamous cell carcinoma compared to
HPV-negative HNSCC cells in vitro. Oncol Rep. 27:270–280.
2012.PubMed/NCBI
|
|
30
|
Brabletz T: The Rudolf Virchow Prize 2001.
The role of the oncoprotein beta-catenin in the progression of
colorectal cancers. Verh Dtsch Ges Pathol. 85:243–249. 2001.(In
German). PubMed/NCBI
|
|
31
|
Zhang X and Hao J: Development of
anticancer agents targeting the Wnt/β-catenin signaling. Am J
Cancer Res. 5:2344–2360. 2015.(Review). PubMed/NCBI
|
|
32
|
Maseki S, Ijichi K, Tanaka H, Fujii M,
Hasegawa Y, Ogawa T, Murakami S, Kondo E and Nakanishi H:
Acquisition of EMT phenotype in the gefitinib-resistant cells of a
head and neck squamous cell carcinoma cell line through
Akt/GSK-3β/snail signallling pathway. Br J Cancer. 106:1196–1204.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Iwatsuki M, Mimori K, Yokobori T, Ishi H,
Beppu T, Nakamori S, Baba H and Mori M: Epithelial-mesenchymal
transition in cancer development and its clinical significance.
Cancer Sci. 101:293–299. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kajiyama H, Shibata K, Terauchi M,
Yamashita M, Ino K, Nawa A and Kikkawa F: Chemoresistance to
paclitaxel induces epithelial-mesenchymal transition and enhances
metastatic potential for epithelial ovarian carcinoma cells. Int J
Oncol. 31:277–283. 2007.PubMed/NCBI
|
|
35
|
Shah AN, Summy JM, Zhang J, Park SI,
Parikh NU and Gallick GE: Development and characterization of
gemcitabine-resistant pancreatic tumour cells. Ann Surg Oncol.
14:3629–3637. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vig N, Mackenzie IC and Biddle A:
Phenotypic plasticity and epithelial-to-mesenchymal transition in
the behaviour and therapeutic response of oral squamous cell
carcinoma. J Oral Pathol Med. 44:649–655. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Leemans CR, Braakhuis BJ and Brakenhoff
RH: The molecular biology of head and neck cancer. Nat Rev Cancer.
11:9–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Klussmann JP, Preuss SF and Speel EJ:
Human papillomavirus and cancer of the oropharynx. Molecular
interaction and clinical implications. HNO. 57:113–122. 2009.(In
German). View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Scanlon CS, Van Tubergen EA, Inglehart RC
and D'Silva NJ: Biomarkers of epithelial-mesenchymal transition in
squamous cell carcinoma. J Dent Res. 92:114–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Koontongkaew S: The tumour
microenvironment contribution to development, growth, invasion and
metastasis of head and neck squamous cell carcinomas. J Cancer.
4:66–83. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Seiki M: Membrane-type 1 matrix
metalloproteinase: A key enzyme for tumour invasion. Cancer Lett.
194:1–11. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schultz JD, Muhlheim K, Erben P, et al:
Chemotherapeutic alteration of VEGF-/PDGF- and PDGF-Rα/β expression
by imatinib in HPV-transformed squamous cell carcinoma compared to
HPV-negative HNSCC in vitro. Oncology Reports. 26:1099–1109.
2011.PubMed/NCBI
|
|
43
|
Cervantes-Arias A, Pang LY and Argyle DJ:
Epithelial-mesenchymal transition as a fundamental mechanism
underlying the cancer phenotype. Vet Comp Oncol. 11:169–184. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Housman G, Byler S, Heerboth S, Lapinska
K, Longacre M, Snyder N and Sarkar S: Drug resistance in cancer: An
overview. Cancers (Basel). 6:1769–1792. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rodrigues IS, Lavorato-Rocha AM, de Maia
MB, Stiepcich MM, de Carvalho FM, Baiocchi G, Soares FA and Rocha
RM: Epithelial-mesenchymal transition-like events in vulvar cancer
and its relation with HPV. Br J Cancer. 109:184–194. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Richter P, Umbreit C, Franz M and Berndt
A, Grimm S, Uecker A, Böhmer FD, Kosmehl H and Berndt A: EGF/TGFβ1
co-stimulation of oral squamous cell carcinoma cells causes an
epithelial-mesenchymal transition cell phenotype expressing laminin
332. J Oral Pathol Med. 40:46–54. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee CG, McCarthy S, Gruidl M, Timme C and
Yeatman TJ: MicroRNA-147 induces a mesenchymal-to-epithelial
transition (MET) and reverses EGFR inhibitor resistance. PLoS One.
9:e845972014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Valenta T, Hausmann G and Basler K: The
many faces and functions of β-catenin. EMBO J. 31:2714–2736. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sahu N and Grandis JR: New advances in
molecular approaches to head and neck squamous cell carcinoma.
Anticancer Drugs. 22:656–664. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Smilek P, Neuwirthova J, Jarkovsky J,
Dusek L, Rottenberg J, Kostrica R, Srovnal J, Hajduch M, Drabek J
and Klozar J: Epidermal growth factor receptor (EGFR) expression
and mutations in the EGFR signalling pathway in correlation with
anti-EGFR therapy in head and neck squamous cell carcinomas.
Neoplasma. 59:508–515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ciardiello F and Tortora G: Epidermal
growth factor receptor (EGFR) as a target in cancer therapy:
Understanding the role of receptor expression and other molecular
determinants that could influence the response to anti-EGFR drugs.
Eur J Cancer. 39:1348–1354. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fumagalli I, Dugue D, Bibault JE,
Clémenson C, Vozenin MC, Mondini M and Deutsch E: Cytotoxic effect
of lapatinib is restricted to human papillomavirus-positive head
and neck squamous cell carcinoma cell lines. Onco Targets Ther.
8:335–345. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lamouille S, Subramanyam D, Blelloch R and
Derynck R: Regulation of epithelial-mesenchymal and
mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell
Biol. 25:200–207. 2013. View Article : Google Scholar : PubMed/NCBI
|