Introduction
Pulmonary artery sarcoma (PAS) is an extremely rare
and highly malignant tumor that originates in the pulmonary artery
(1). It has an incidence rate of
0.001–0.030% (2). Since 1923, when
the first case of PAS was reported by Mandelstamm (3), ≤300 cases have been reported to date
worldwide (4–7). The etiology of PAS is unknown, and this
disease has a poor prognosis (5).
Early diagnosis followed by radical surgical resection constitutes
the only chance of survival for patients with PAS (5), and patients have an average survival
time of ~1.5 months without surgical treatment (8). However, due to the rare and nonspecific
clinical manifestations and imaging findings, PAS is frequently
misdiagnosed as various pulmonary thromboembolic diseases,
including pulmonary thromboembolism (PTE) or chronic thromboembolic
pulmonary hypertension (CTEPH), and the majority of reported PAS
cases are confirmed by pathological examination subsequent to
surgery or by autopsy (2,9,10). The
present study reports three cases of PAS that were initially
misdiagnosed as PTE or CTEPH, and were later identified as PAS
following surgery. The clinicopathological and immunohistochemical
characteristics of these cases are reported in the present study to
aid the understanding and differential diagnosis of the PAS tumor
in clinical practice.
Case report
Between 2008 and 2012, three adult patients with
PAS, whose diagnoses were confirmed by surgery and pathological
examinations, were managed by the Beijing Anzhen Hospital (Beijing,
China). The characteristics of each patient are summarized in
Table I. The patients, two women and
one man with a mean age of 41.3 years old (range, 36–47 years),
possessed disease histories that ranged between 50 days and 4
years. All the patients experienced dyspnea due to exertion, and
two of them also experienced chest tightness, while one patient
experienced repeated and intermittent syncope. The
electrocardiogram (ECG; ECG-903; Shenzhen Spring Technology
Industry Co., Ltd., Shenzhen, China) revealed that one patient
possessed right ventricular enlargement, in addition to ST segment
and T wave changes (case 1); another patient exhibited SI QIII TIII
pattern (case 2); and the other patient possessed a normal ECG
(case 3). Echocardiography (Sonos 2500 Ultrasound; Hewlett-Packard,
Palo Alto, CA, USA) showed that all the patients presented
pulmonary artery expansion, pulmonary artery hypertension and right
ventricular hypertrophy, and two of the patients (cases 1 and 3)
were suggested to possess PTE. The chest radiographs (MobileDaRt
Evolution; Shimadzu Corporation, Kyoto, Japan) revealed a
thickening of the right or left pulmonary artery in two patients
(cases 2 and 3), and normal findings in one patient (case 1).
Computed tomography pulmonary angiography (CTPA; Aquilion ONE;
Toshiba Medical Systems Corporation, Otawara, Japan) revealed that
two of the patients presented a filling defect in the main, left
and right pulmonary arteries, and were diagnosed as central PTE
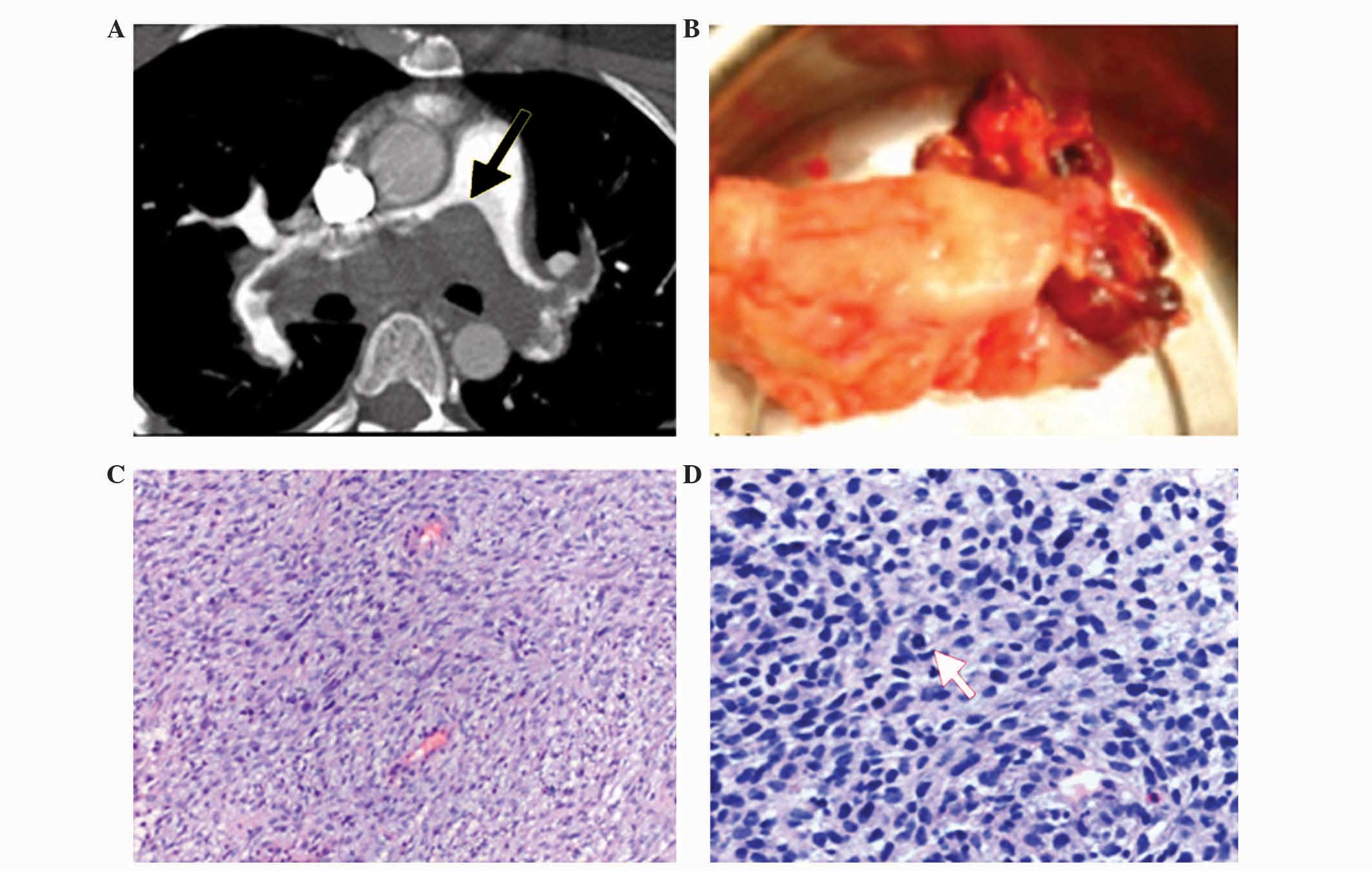
(cases 1 and 3; case 1, Fig. 1A),
while the third patient presented expansion of the main pulmonary
artery and left pulmonary artery lumen, and a filling defect in the
right upper pulmonary artery (case 2). The lung
ventilation/perfusion (V/Q) scan (Technegas Generator; Cyclomedica
Australia, Pty, Ltd., Lucas Heights, NSW, Australia) of one patient
revealed that all the segments of the left lung and the right upper
lung lobe possessed perfusion defects, but the ventilation was
normal (case 1), whereas another patient presented diffuse lung V/Q
mismatch (case 3). The dual-color Doppler ultrasound examination
(iE33 Ultrasound Machine; Philips Medical Systems, Inc., Bothell,
WA, USA) revealed no abnormalities in the deep veins of the legs of
all three patients. Clinically, two of the patients were diagnosed
with CTEPH (cases 1 and 2), while one patient was diagnosed with
PTE (case 3). Pulmonary artery thromboendarterectomies were
performed for the patients diagnosed with CTEPH (cases 1 and 2),
while an embolectomy was performed for the patient diagnosed with
PTE (case 3). During the surgery, tumor tissues were identified in
the main, left and right pulmonary arteries. Lesions were attached
to the vessel wall, grew into the lumen, and were shaped along the
vessel branches in two of the patients (cases 1 and 3), whereas
lesions were located in the right pulmonary artery in one patient
(case 2). The tumor tissues were adhesive to the vessel wall, and
not easily separated from it. Surgery was attempted to maximally
remove the tumor tissue in all the patients.
 | Table I.Patients characteristics. |
Table I.
Patients characteristics.
| Case no./year | Age,
years/gender | Presenting
symptoms | TTE findings | Radiological
findings | Presumptive
diagnosis | Other diagnostic
studies and surgery | Patient outcome |
|---|
| 1/2008 | 36/F | Chest pressure; DOE
for 1 year; syncope, 4 times | PA bifurcation,
abnormal echo image; potential PTE; RA/RV dilatation; tricuspid
regurgitation; SPAP, 69 mmHg | Chest X-ray, normal;
CTPA, MPA/RPA/LPA filling defects | CTEPH | ECG, enlarged RA/RV,
ST segment depression; V/Q, no perfusion in L lung and R upper
lobe; thoracotomy for thromboendarterectomy; neoplasm observed,
size 9.5×4.5 cm; intimal sarcoma | Succumbed to disease
5 months post-surgery |
| 2/2009 | 41/F | DOE; fatigue; leg
edema for 4 years | RA/RV dilatation;
tricuspid regurgitation; SPAP, 90 mmHg | Chest X-ray, enlarged
RPA; R pleural effusion; CTPA, MPA/RPA/LPA dilation; R upper PA
filling defect | CTEPH | ECG, SI QIII TIII
pattern; thoracotomy for thromboendarterectomy; neoplasm observed,
size 5.0×1.2 cm; leiomyosarcoma | Succumbed to disease
1 month post-surgery |
| 3/2012 | 47/M | Chest pressure; DOE;
fatigue for 50 days | MPA, abnormal echo
image; potential PTE; RA/RV dilatation; SPAP, 53 mmHg | Chest X-ray, enlarged
LPA; CTPA, MPA/RPA/LPA filling defects | PTE | ECG, normal; V/Q,
mismatch of V/Q in both lungs; thoracotomy for embolectomy;
neoplasm observed, size 7.0×2.7 cm; leiomyosarcoma | Succumbed to disease
18 months post-surgery |
Macroscopic examination of the tumor specimens
(Leica DM3000; Leica Microsystems, Inc., Buffalo Grove, IL, USA;
camera: AxioCam MRc5, Zeiss AG, Oberkochen, Germany) revealed that
two of the patients presented mucoid or smooth gelatinous tumor
tissue that filled the vascular lumens. Certain regions appeared to
be partially wrapped by a capsule. The tumor surface possessed firm
fibrotic regions with gray or brown cut surface. The sizes of the
tumors were 9.5×4.5 cm (case 1, Fig.
1B) and 7.0×2.7 cm (case 3). The tumor of the patient
corresponding to case 2 was 5.0×1.2 cm in size, and possessed
broken and gray soft tissue with a crisp texture.
The resected tumors were subjected to histological
and immunohistochemical analysis. For that purpose, specimens were
fixed in 4% neutral formalin (Beijing Yili Fine Chemical Co., Ltd.,
Beijing, China), embedded in paraffin (Beijing Yili Fine Chemical
Co., Ltd.), sectioned, and stained with hematoxylin and eosin
(Beijing Yili Fine Chemical Co., Ltd.). Dako EnVision System
(Agilent Technologies, Inc., Santa Clara, CA, USA) (11,12) was
used for immunohistochemical analysis. The following antibodies
were used, which were all mouse monoclonal anti-human, unless
otherwise specified, diluted in phosphate-buffered saline and
purchased from Beijing Zhongshan Golden Bridge Biotechnology Co.,
Ltd. (OriGene Technologies, Inc., Beijing, China): Vimentin (clone,
V9; catalog no., ZM-0260; dilution, 1:150), desmin (clone, D33;
catalog no., ZM-0091; dilution, 1:100), cluster of differentiation
(CD)34 (clone, QBEnd/10; catalog no., ZM-0046; dilution, 1:150),
CD31 (clone, 1A10; catalog no., ZM-0044; dilution, 1:100), α-smooth
muscle actin (α-SMA; clone, HHF35; catalog no., ZM-0001; dilution,
1:150), cytokeratin (clone, AE1/AE3; catalog no., ZM-0069;
dilution, 1:150), epithelial membrane antigen (EMA; clone, GP1.4;
catalog no., ZM-0095; dilution, 1:150), CD68 (clone, 514H12;
catalog no., ZM-0060; dilution, 1:150), CD117 (clone, 2E4; catalog
no., ZM-0437; dilution, 1:100), S-100 (catalog no., ZA-0225;
dilution, 1:150), human melanoma black 45 (clone, HMB45; catalog
no., ZM-0187; dilution, 1:150), myelin basic protein (MBP; clone,
7H11; catalog no., ZM-0488; dilution, 1:200), myogenic
differentiation 1 (MyoD1; rabbit anti-human monoclonal; clone,
Ep212; catalog no., ZA-0585; dilution, 1:150), myosin (clone, MY32;
catalog no., ZM-0196; dilution, 1:150) and myoglobin (clone, MYO18;
catalog no., ZM-0193; dilution, 1:150). The immunohistochemical
assays were performed according to the manufacturers protocol with
an incubation time of 30 min at 25°C. Pathological diagnoses were
established based on the 2004 and 2002 ‘World Health Organization
Classification of Tumors’ (1,13).
The histopathological and immunohistochemical
results revealed that the tumor of case 1 possessed a large number
of spindle cells arranged as bundles and braids or irregularly
(Fig. 1C). Scattered necrosis and
significant atypia of the tumor cells, which were mostly spindle
shaped (like fibroblasts or myofibroblasts), were observed. In
certain regions, the cells were differentiated epithelioid cells,
with large, rod-shaped, round, oval or irregular nuclei. The
chromatin was blocky and coarse. Certain cells contained 1–2
nucleoli. Mitosis was commonly identified in giant tumor cells at
~20/10 high power fields (Fig. 1D).
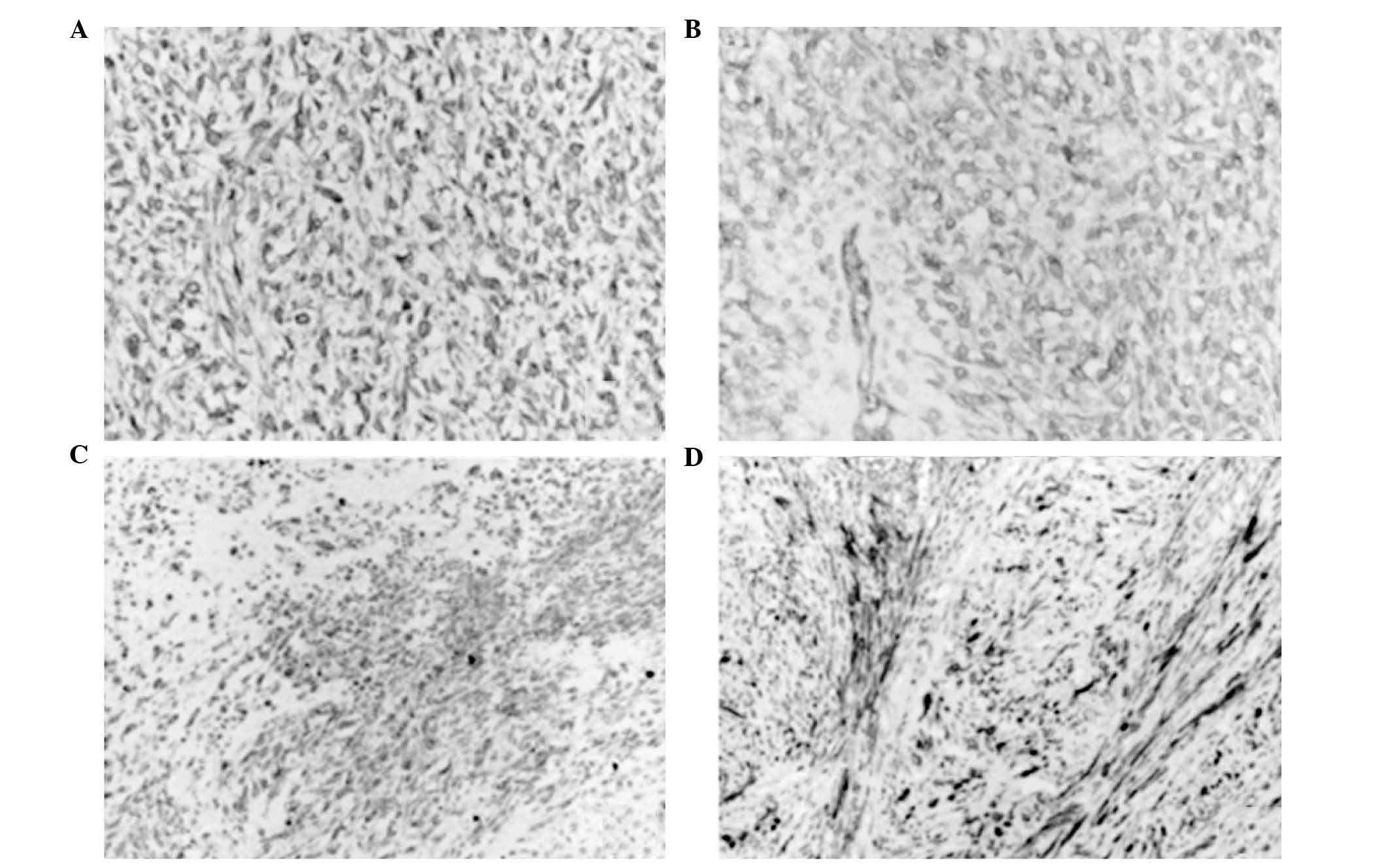
Vimentin (Fig. 2A), CD34 (Fig. 2B), desmin (Fig. 2C) and α-SMA (Fig. 2D) were expressed, while CD31,
cytokeratin AE1/AE3, EMA, CD68, CD117, S-100, HMB-45, MBP, MyoD1,
myosin and myoglobin were not. These findings were in agreement
with the characteristics of intimal sarcoma (13). In two of the patients (cases 2 and 3),
the tumors were composed of spindle cells arranged as interwoven
bundles, and exhibited high density and compactness. Part of the
tumor was mucus. The tumor cells were long and spindle-like, and
contained rich cytoplasm and oblong, blunt-ended nuclei. In poorly
differentiated cells, marked staining revealed that the size
varied, and the nuclei were spindle-shaped and pleomorphic. The
cytoplasm was eosinophilic and displayed no evident pattern.
Vimentin and α-SMA were expressed, while all other marker proteins
were not expressed, with the exception of desmin and CD34 in case
3. The characteristics presented by case 2 and 3 were in agreement
with a diagnosis of leiomyosarcoma (13).
Following surgery, one of the patients (case 2) died
in the hospital a month later, due to septic shock and heart and
lung failure. The other two patients (cases 1 and 3) were
discharged from hospital, and one of them received chemotherapy
(case 3). However, the two patients succumbed to tumor recurrence
and metastasis 5 and 18 months subsequent to surgery,
respectively.
The present study was conducted in accordance with
the declaration of Helsinki, and with the approval of the Ethics
Committee of Capital Medical University (Beijing, China; approval
no., 2008002X). Written informed consent was obtained from all
participants.
Discussion
PAS almost invariably arises from the main pulmonary
arteries or the pulmonary valve (5,14). When
diagnosing PAS, the first step is to rule out the possibility of a
metastatic tumor from other regions of the body (11). PAS is a rare malignant tumor with an
incidence of 0.001–0.030%, and the pathogenesis of the disease
remains unclear (2). PAS has been
previously reported in patients aged 13–86 years, with a mean age
of 52 years old, and a female-to-male ratio of 2:1 (9). The three patients reported in the
present study are consistent with the previously reported age and
gender characteristics of the disease (15). PAS generally occurs insidiously
(15). A tumor that protrudes toward
the pulmonary artery lumen may cause the reduction of pulmonary
blood volume and pulmonary hypertension (16). The most common clinical manifestation
of PAS is dyspnea, which is usually accompanied by chest pain,
cough, hemoptysis, fatigue and weight loss (8). A serious obstruction occurring in the
pulmonary artery may result in decreased cardiac output, leading to
syncope and sudden mortality (9).
During physical examination, the most common signs of PAS are
pulmonary valve noise, a systolic ejection murmur with a loud
pulmonary component of the second heart sound (P2), jugular venous
distension, hepatomegaly and peripheral edema (8).
Imaging examinations are the major tools for the
diagnosis and differential diagnosis of PAS (17). Echocardiography of the large pulmonary
artery and right ventricular outflow tract may reveal projections
with irregular surfaces as spot echoes and hyperechoic regions.
However, distinguishing PAS from pulmonary thromboembolic diseases
such as PTE or CTEPH is challenging (18). Chest X-ray examinations of patients
with PAS are often nonspecific, and may reveal a hilar mass,
protruding pulmonary artery segment, unilateral pulmonary artery,
proximal branch expansion, lung nodules, right ventricular
enlargement and sparse peripheral vasculature (8,19). CTPA
and pulmonary magnetic resonance imaging (MRI) present advantages
for the diagnosis of PAS (7,18,20). CTPA
may clearly reveal the involvement of lesions in the pulmonary
artery, right ventricular outflow tract and pulmonary valve
(17). A lesion with a full, bulged
or lobulated-edged surface may be indicative of PAS (17). An MRI scan may reveal signals from the
abnormal soft tissue in the main pulmonary artery cavity, but these
signals are difficult to distinguish from those caused by pulmonary
thromboembolic diseases (17).
However, the intensity of contrast enhancement following contrast
injection is associated with the degree of tumor differentiation,
which is a good indicator of PAS or other malignant lesions
(18). The three patients with PAS
reported in the present study presented with symptoms and clinical
manifestations similar to those caused by PTE, since the tumors
grew inside the pulmonary artery lumen, leading to pulmonary artery
stenosis or obstruction. Although the clinical and imaging data
were suggestive of PAS, the data were nonspecific. As a result, all
three patients were clinically diagnosed with PTE or CTEPH and
referred for surgery.
According to the 2004 ‘World Health Organization
Classification of Tumours’ (1), PAS
is divided into two types: Intimal and intramural. Intimal sarcoma
presents an intraluminal polypoid growth pattern, and usually
exhibits fibroblastic or myofibroblastic differentiation (21). Intramural sarcoma is considered
distinct from intimal sarcoma, and is classified separately
according to the histological subtypes as in soft tissue sarcoma
(leiomyosarcoma) (4). Since intimal
PAS is more common than intramural PAS, PAS is often referred to as
intimal sarcoma (4). PAS often occurs
in the dorsal region of the main pulmonary artery, mostly with a
polypoid or finger-like form, an extends to the bifurcation of the
main pulmonary artery, and the left and right pulmonary arteries
(22). Certain tumors that involve
the pulmonary valve may also simultaneously involve or spread to
the outflow tract of the right ventricle (3). In total, 90% of PAS cases involve ≥2
regions, and 85% of patients with PAS possess main pulmonary artery
lesions, while 71% possess right pulmonary artery lesions, 65%
possess left pulmonary artery lesions, 32% possess lesions invading
the pulmonary valve and 10% possess lesions invading the outflow
tract of right ventricle (4). An
intimal sarcoma resembles mucoid or gelatinous clots that fill
vascular lumens, and the distal extension of the tumor may possess
firm fibrotic regions, be bony or gritty, and chondromyxoid foci
may be present in mural lesions (23). Hemorrhage and necrosis are common in
high-grade tumors (1,13). The patients in the present study
underwent an embolectomy or thromboendarterectomy due to the
diagnosis as PTE or CTEPH. During the surgery, large emboli were
identified, which closely connected to the base of the pulmonary
valve or the bifurcation of the trunk and branch of the pulmonary
artery. The emboli extended to the pulmonary artery in the same
direction as the pulmonary vascular tree, and accounted for almost
all the lumen. Gross examination revealed that the emboli were gray
and brown or myxoid, but was unable to distinguish the emboli from
thrombi.
Regarding the pathogenesis and origin of PAS, the
majority of studies published thus far hypothesize that an intimal
sarcoma may arise from the pluripotent mesenchymal cells of the
intima artery, that is, the inner membrane of the pulmonary artery
(24). Therefore, PAS may have a
variety of pathological types. Cox et al (4) reported the pathological results of 138
cases of PAS, of which, 43 (~1/3) were undifferentiated sarcoma.
The second highest number of cases of PAS reported by the authors
was leiomyosarcoma, followed by spindle cell sarcoma, malignant
fibrous histiocytoma, fibromyxoid sarcoma, rhabdomyosarcoma,
chondrosarcoma, mesenchymal tumors and osteosarcoma. According to
previous immunohistochemistry and electron microscopy studies, PAS
also presents other pathological types, including angiosarcoma and
epithelioid hemangioendothelioma (23). Therefore, PAS has multiple
histological types and may originate from pluripotent cells. The
typical histological features and immunohistochemical staining for
PAS are important for classifying the different pathological types
(4). Under the microscope, intimal
sarcoma exhibits proliferation of spindle cells in a myxoid
background, alternating with hypocellular collagenized stroma
(25,26). Recanalized thrombi may be intimately
admixed, and considerable nuclear pleomorphisms and varying degrees
of spindle cells are usually present in the tumor tissue (27). The tumor may also be associated with
large regional myxoid tissues and local necrosis, and typical
spindle cells are arranged like a woven mat or striated, as in
leiomyosarcoma. A number of intimal sarcomas and the majority of
intramural sarcomas possess foci of more differentiated sarcomas,
including rhabdomyosarcoma, osteosarcoma or angiosarcoma (27). In numerous intimal sarcomas, the
immunohistochemical and ultrastructural examinations reveal the
existence of fibroblast cells, in which vimentin is diffusely
expressed, and osteopontin and α-SMA may also be expressed
(28). In a tumor that shows evidence
of differentiation from vascular or smooth muscle cells, desmin and
endothelial markers such as CD31, CD34 or factor VIII (FVIII) may
be expressed (13). For the patient
with pulmonary artery intimal sarcoma of the present study (case
1), immunophenotyping revealed that vimentin, desmin and CD34 were
expressed by the tumor cells, while α-SMA was focally expressed.
These results suggest that the patient possessed PAS that exhibited
differentiation into angiosarcoma and leiomyosarcoma. In previous
studies, pulmonary leiomyosarcoma was also categorized as a common
type of PAS, accounting for ~20% of PAS cases (13). The other two patients in the present
study (cases 2 and 3) possessed the leiomyosarcoma phenotype,
expressed vimentin and diffusely expressed α-SMA. In addition, one
of the patients also expressed desmin and CD34. The results of
these two patients suggest a diagnosis of leiomyosarcoma. However,
the diagnosis should not be solely based on phenotype, since
certain markers such as α-SMA and desmin are not smooth
muscle-specific (28). The diagnosis
of leiomyosarcoma may be more accurate if it is based on the
results of two examinations, compared with one examination, and
should be combined with typical morphological characteristics
(23,28).
The ultrastructural features of intimal sarcoma
include actin filaments, the presence of dense bodies in the
cytoplasm surrounded by non-continuous plate bodies; these
structures are similar to those observed in leiomyosarcoma
(29). Regarding the genetic
characteristics of PAS, a previous study using the comparative
genomic hybridization method indicated that 75% (6/8 cases) of
lesions are located in the 12q13-14 region, and a number of genes
were observed to be amplified (1). In
addition, other less common genetic changes occurred in 3p; losses
occurred in 3q, 4q, 9p, 11q, 13q, Xp and Xq; gains occurred in 7p,
17p and 17q; and amplifications occurred in in 4q, 5p, 6p and 11q
(1). Furthermore,
immunohistochemistry identified intimal sarcoma in eight cases that
exhibited focal expression of p53, of which, six cases
overexpressed mouse double minute 2 homolog (Mdm2), which suggests
that the Mdm2 and p53 signaling pathways are also responsible for
the pathogenesis of intimal sarcoma (1). Intimal sarcomas are, in general,
malignant mesenchymal tumors with poorly differentiated fibroblasts
or myofibroblasts (23,30). Intimal sarcoma is not strictly a
histological classification, since the term ‘intimal sarcoma’ is
based on the sites of the lesions and the abnormal multipotency of
the tumors and pleomorphic sarcomas, including leiomyosarcoma,
rhabdomyosarcoma, angiosarcoma, osteosarcoma and malignant fibrous
histiocytoma (23,25,31). In
order to improve the intervention and follow-up examinations of
patients with PAS, determining the specific subtypes of intimal
sarcoma, based on the classification criteria of soft tissue
tumors, clinical manifestations, imaging data, histological
features and immunophenotype, is extremely important (1,2,6,13).
Regarding the differential diagnosis of PAS,
following the exclusion of PAS from metastatic cancer, based on
clinical and pathological examinations, PAS tumors must be
distinguished from common tumors that occur in the heart and large
vessels, such as angiosarcoma, rhabdomyosarcoma and myxoma
(1,13).
Angiosarcomas are mesenchymal malignancies that have
differentiated from or toward vascular endothelial cells (23). Angiosarcomas are composed of spindle
or epithelioid endothelial cells, which form tubular, fissures or
sinus-like structures with red blood cells. The tumor cells are
surrounded by reticular fibers that reveal clear vascular
structures following staining. Immunohistochemically, the tumor
cells express endothelial markers, including FVIII, CD31 and CD34
(23). Although one of the patients
in the present study expressed CD34 (case 2), no histological
differentiation was observed that would enable to confirm the
diagnosis of angiosarcoma.
Rhabdomyosarcoma is rare in the cardiovascular
system, and occurs more often in young people (32). The tumor cells in a rhabdomyosarcoma
possess eosinophilic and granular cytoplasm, and are rich in
longitudinal filaments, but not stripes. The cells are round,
spindle, tadpole-, ribbon- or tennis racquet-shaped, with markedly
stained nuclei. Interstitial mucus is present within the sparsely
packed cells, and the tumor cells express muscle specific actin and
desmin (32). In the present study,
two cases expressed desmin, but the cells were mostly spindle and
epithelioid, not typical of rhabdomyosarcoma.
Myxoma is a primary multipotent tumor that
originates from mesenchymal cells (33). Histologically, fine reticular fibers
and sparsely packed cells are often observed in rich and loose
mucus. The cells have clear borders, and are star- or
bipolar-shaped, but generally do not exhibit the characteristics of
malignant cells, including giant cells or mitotic nuclei. The cells
are positive for Alcian blue and toluidine blue staining, but the
immunohistochemical analysis does not aid the differential
diagnosis of the disease (34,35).
In conclusion, PAS is an extremely rare tumor that
occurs in the cardiovascular system. Due to its nonspecific
clinical manifestations and radiological features, PAS is often
misdiagnosed. Therefore, the diagnosis of PAS is recommended to be
based on the typical morphological features and immunohistochemical
analysis of the tumor tissue.
References
|
1
|
Travis WD, Brambilla E, Müller-Hermelink
HK and Harris CC: World Health Organization Classification of
Tumours. Pathology & Genetics of Tumours of the Lung, Pleura,
Thymus and Heart. IARC Press. (Lyon). 2004.
|
|
2
|
Bhagwat K, Hallam J, Antippa P and
Larobina M: Diagnostic enigma: Primary pulmonary artery sarcoma.
Interact Cardiovasc Thorac Surg. 14:342–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mandelstamm M: Über primäre Neubildungen
des Herzens. Virchows Arch Pathol Anat. 245:43–54. 1923.(In
German). View Article : Google Scholar
|
|
4
|
Cox JE, Chiles C, Aquino SL, Savage P and
Oaks T: Pulmonary artery sarcomas: A review of clinical and
radiologic features. J Comput Assist Tomogr. 21:750–755. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shehatha J, Saxena P, Clarke B, Dunning J
and Konstantinov IE: Surgical management of extensive pulmonary
artery sarcoma. Ann Thorac Surg. 87:1269–1271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huo L, Moran CA, Fuller GN, Gladish G and
Suster S: Pulmonary artery sarcoma: A clinicopathologic and
immunohistochemical study of 12 cases. Am J Clin Pathol.
125:419–424. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gan HL, Zhang JQ, Huang XY and Yu W: The
wall eclipsing sign on pulmonary artery computed tomography
angiography is pathognomonic for pulmonary artery sarcoma. PLoS
One. 8:e832002013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Parish JM, Rosenow EC III, Swensen SJ and
Crotty TB: Pulmonary artery sarcoma. Clinical features. Chest.
110:1480–1488. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dimitrakakis G, Zilidis G, Buchalter M and
Von Oppell U: Pulmonary artery sarcoma - a challenging diagnosis: A
case report. Heart Surg Forum. 9:E897–E899. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cook DJ, Tanser PH, Dobranowski J and
Tuttle RJ: Primary pulmonary artery sarcoma mimicking pulmonary
thromboembolism. Can J Cardiol. 4:393–396. 1988.PubMed/NCBI
|
|
11
|
Etienne-Mastroianni B, Falchero L,
Chalabreysse L, Loire R, Ranchère D, Souquet PJ and Cordier JF:
Primary sarcomas of the lung: A clinicopathologic study of 12
cases. Lung Cancer. 38:283–289. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Régnard JF, Icard P, Guibert L, de
Montpreville VT, Magdeleinat P and Levasseur P: Prognostic factors
and results after surgical treatment of primary sarcomas of the
lung. Ann Thorac Surg. 68:227–231. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fletcher CDM, Unni KK and Mertens F: World
Health Organization Classification of Tumours. Pathology &
Genetics of Tumours of Soft Tissue and Bone. IARC Press. (Lyon).
2002.
|
|
14
|
Bloomberg RD, Butany JW, Cusimano RJ and
Leask RL: Primary cardiac sarcoma involving the pulmonary artery
and valve. Can J Cardiol. 19:843–847. 2003.PubMed/NCBI
|
|
15
|
Mussot S, Ghigna MR, Mercier O, Fabre D,
Fadel E, Le Cesne A, Simonneau G and Dartevelle P: Retrospective
institutional study of 31 patients treated for pulmonary artery
sarcoma. Eur J Cardiothorac Surg. 43:787–793. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Araki Y, Tajima K, Yoshikawa M, Abe T and
Suenaga Y: A case of primary pulmonary intimal sarcoma of the
pulmonary artery. Nihon Kyobu Geka Gakkai Zasshi. 45:1039–1043.
1997.(In Japanese). PubMed/NCBI
|
|
17
|
Wijesuriya S, Chandratreya L and Medford
AR: Chronic pulmonary emboli and radiologic mimics on CT pulmonary
angiography: A diagnostic challenge. Chest. 143:1460–1471. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Attinà D, Niro F, Tchouanté P, Mineo G,
Russo V, Palazzini M, Galiè N, Fanti S, Lovato L and Zompatori M:
Pulmonary artery intimal sarcoma. Problems in the differential
diagnosis. Radiol Med (Torino). 118:1259–1268. 2013. View Article : Google Scholar
|
|
19
|
Hu XP, Xu JP and Liu NN: Primary pulmonary
artery sarcoma: Surgical management and differential diagnosis with
pulmonary embolism and pulmonary valve stenosis. J Card Surg.
24:613–616. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ebner L, Huber A, Ott D and Christe A:
Pulmonary intimal sarcoma: A rare differential diagnosis for
arterial filling defects on a chest CT. Acta Radiol Short Rep.
3:20479816135140522014.PubMed/NCBI
|
|
21
|
Tavora F, Miettinen M, Fanburg-Smith J,
Franks TJ and Burke A: Pulmonary artery sarcoma: A histologic and
follow-up study with emphasis on a subset of low-grade
myofibroblastic sarcomas with a good long-term follow-up. Am J Surg
Pathol. 32:1751–1761. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mayer E, Kriegsmann J, Gaumann A, Kauczor
HU, Dahm M, Hake U, Schmid FX and Oelert H: Surgical treatment of
pulmonary artery sarcoma. J Thorac Cardiovasc Surg. 121:77–82.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Keel SB, Bacha E, Mark EJ, Nielsen GP and
Rosenberg AE: Primary pulmonary sarcoma: a clinicopathologic study
of 26 cases. Mod Pathol. 12:1124–1131. 1999.PubMed/NCBI
|
|
24
|
Mattoo A, Fedullo PF, Kapelanski D and
Ilowite JS: Pulmonary artery sarcoma: A case report of surgical
cure and 5-year follow-up. Chest. 122:745–747. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Burke AP and Virmani R: Sarcomas of the
great vessels. A clinicopathologic study. Cancer. 71:1761–1773.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nonomura A, Kurumaya H, Kono N, Nakanuma
Y, Ohta G, Terahata S, Matsubara F, Matsuda T, Asaka T and Nishino
T: Primary pulmonary artery sarcoma. Report of two autopsy cases
studied by immunohistochemistry and electron microscopy, and review
of 110 cases reported in the literature. Acta Pathol Jpn.
38:883–896. 1988.PubMed/NCBI
|
|
27
|
Yi CA, Lee KS, Choe YH, Han D, Kwon OJ and
Kim S: Computed tomography in pulmonary artery sarcoma:
Distinguishing features from pulmonary embolic disease. J Comput
Assist Tomogr. 28:34–39. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Govender D and Pillay SV: Right pulmonary
artery sarcoma. Pathology. 33:243–245. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Croitoru AG, Klein MJ, Galla JD and Fallon
JT: Primary pulmonary artery leiomyosarcoma. Cardiovasc Pathol.
12:166–169. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Spirtas R, Connelly RR and Tucker MA:
Survival patterns for malignant mesothelioma: The SEER experience.
Int J Cancer. 41:525–530. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Burke AP and Virmani R: Osteosarcomas of
the heart. Am J Surg Pathol. 15:289–295. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bleisch VR and Kraus FT: Polypoid sarcoma
of the pulmonary trunk: Analysis of the literature and report of a
case with leptomeric organelles and ultrastructural features of
rhabdomyosarcoma. Cancer. 46:314–324. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jain D, Maleszewski JJ and Halushka MK:
Benign cardiac tumors and tumorlike conditions. Ann Diagn Pathol.
14:215–230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Burke AP and Virmani R: Cardiac myxoma. A
clinicopathologic study. Am J Clin Pathol. 100:671–680. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hemachandran M, Kakkar N and Khandelwal N:
Giant-cell-rich myxoma of right atrium. An ultrastructural
analysis. Cardiovasc Pathol. 12:287–289. 2003. View Article : Google Scholar : PubMed/NCBI
|