Introduction
Colorectal cancer (CRC) is the third most common
malignancy and third leading causes of cancer-related mortality
(9–10% incidence and mortality rates) in the United States. The
risk of developing invasive CRC within a lifetime is estimated to
be ~5% (1). Due to increased early
detection and precancerous lesion removal using screening methods
in recent years, the rate of CRC-related mortality is being reduced
in United States, however, this rate is increasing in developing
countries (2). For example, previous
studies have shown that the incidence of CRC is increasing in Iran
(3–5).
Furthermore, due to the high proportion of young people in the
Iranian population, the incidence rate of different types of
cancer, such as CRC, is expected to increase in the coming years as
the population grows older.
It has been suggested that three major pathways are
involved in colorectal tumorigenesis, including chromosomal
instability, microsatellite instability and the CpG island
methylator phenotype (CIPM) (6).
Previous studies have demonstrated that epigenetic modifications,
such as aberrant DNA methylation, have important roles during CRC
development. CIPM-positive CRC cases, which account for ~15% of
sporadic cases, have hypermethylation in the promoter regions of
certain tumor suppressor genes (7,8). CpG
islands are genomic regions with a high percentage of CpG
dinucleotides. DNA methylation occurs at CpG dinucleotides, and has
been demonstrated to suppress the expression of nearby genes
(9). There are ~25,500 CpG islands in
the human genome, half of which are associated with constitutively
expressed genes; a number of these constitutively expressed genes
are methylated in CRC (10). In a
study by Kim et al, the previously reported methylation
markers has been reviewed in CRC (11). Epigenetic modifications, such as
promoter hypermethylation, commonly precede disease pathology,
making them valuable diagnostic biomarkers for the early detection
of diseases or for determining clinical response to the
therapeutics (12,13). Furthermore, DNA methylation markers
are more stable compared with RNA and protein markers, thus, making
them more suitable biomarkers for detection in different biological
substances, such as blood and stool samples (14).
Apoptosis or programmed cell death is an important
homeostatic process; its failure is considered to contribute to
cancer development and resistance to cancer therapies (15). Apoptosis occurs through two
interconnected pathways; the extrinsic and the intrinsic pathways.
The extrinsic pathway initiates by the binding of death ligands to
their respective cell surface death receptors: Tumor necrosis
factor receptor 1, Fas cell surface death receptor (Fas), and tumor
necrosis factor receptor superfamily members 10a and 10b. Ligand
binding leads to caspase activation and subsequent apoptotic
signaling cascades (16). By
contrast, the intrinsic or mitochondrial pathway is triggered by
oxidative stress and DNA damage, and progresses through proteolytic
caspase cascades. The key elements of apoptosis include
pro-apoptotic and anti-apoptotic proteins that are involved in the
progression and regulation of apoptosis (17,18).
Molecular alterations, such as genetic mutations or epigenetic
silencing of apoptotic genes, frequently occur in tumor progression
and drug resistance (19). As an
epigenetic mechanism of gene inactivation, aberrant DNA
methylation, in addition to genetic mutations, may act as a second
hit to turn off important tumor suppressor genes, and initiate or
progress cancer development (20).
Proapoptotic genes that have important roles in
apoptosis initiation and progression may contain CpG islands in
their promoter sequences; hence, these genes are prone to be
silenced by DNA hypermethylation. In our previous study, two
proapoptotic genes, FAS and BCL2-associated X protein (BAX), were
demonstrated to be downregulated in adjacent normal colorectal
tissue compared with CRC tissue samples (21). As a cell surface death receptor, Fas
protein leads to the initiation of programmed cell death following
binding to its ligand. By contrast, Bax is considered to have a
significant effect on mitochondrial outer membrane
permeabilization; this permeabilization allows cytochrome c
to be directed from the mitochondrial intermembrane space into the
cytoplasm, and eventually leads to activation of caspases during
apoptosis initiation and progression (22). Both the FAS and BAX genes have CpG
islands in their upstream sequences according to the MethPrimer
criteria (23). The cell death
receptor FAS is involved in initiating the extrinsic pathway of
apoptosis, while the BAX protein has important roles in the
intrinsic and extrinsic apoptotic pathways (24). Therefore, the aim of the present study
was to investigate the methylation status of CpG islands in two
essential downregulated proapoptotic genes, FAS and BAX, in CRC
samples.
Materials and methods
Patient samples
Fresh-frozen patient samples obtained from hospitals
were provided by the Iran National Tumor Bank (Tehran, Iran), which
is funded by the Cancer Institute of Tehran University (Tehran,
Iran; Table I). A total of 30
colorectal tumor samples and their adjacent normal tissues were
collected from Baqiyatallah Hospital (Tehran, Iran) and Imam
Khomeini Hospital Complex (Tehran, Iran). Samples were collected
during surgical resection between March 2011 and September 2012.
The criteria for inclusion of patient samples was the sporadic
colon cancer, and rectum mucinous and non-mucinous adenocarcinoma.
Tumors were classified based on the pathological diagnostic
criteria of the WHO classification (25). No patients underwent chemotherapy
prior to surgery and had no other forms of cancer. The adjacent
normal tissues were obtained from at least 6 cm away from the tumor
sites. The samples were snap-frozen and stored in liquid nitrogen
until extraction. The study was approved by Shahid Beheshti
University of Medical Sciences ethical committee (Tehran, Iran) and
written informed consent was obtained from all patients or close
relatives for sampling.
 | Table I.Clinical data of 25 colorectal cancer
samples. |
Table I.
Clinical data of 25 colorectal cancer
samples.
| Characteristic | Cases, n (%) |
|---|
| Gender |
|
| Male | 14 (56) |
|
Female | 11 (44) |
| Age, years |
|
|
<60 | 15 (60) |
| ≥60 | 10 (40) |
| Site of primary
tumor |
|
|
Colon | 13 (52) |
|
Rectum | 12 (48) |
| Histological
type |
|
|
Non-mucinous
adenocarcinoma | 20 (80) |
| Mucinous
adenocarcinoma | 5 (5) |
| Tumor grade |
|
| I | 15 (60) |
| II | 5
(20) |
| III | 4
(16) |
| IV | 1 (4) |
| T classification |
|
| T2 | 4
(16) |
| T3 | 21 (84) |
Cell culture and treatment
The HT-29 colorectal adenocarcinoma cell line (ATCC
HTB-38; American Type Culture Collection, Manassas, VA, USA) was
cultured in RPMI-1640 medium (Biosera, Sussex, UK) supplemented
with 10% heat-inactivated fetal bovine serum and 1%
penicillin-streptomycin (Life Technologies; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 37°C in a humidified
atmosphere with 5% CO2. The cells were cultured in
6-well plate at equal concentrations of ~1×104 cells per
well. Subsequently, the cells were exposed media containing 10 µM
5-aza-2′-deoxycytidine (Sigma-Aldrich Chemie GmbH, Hamburg,
Germany) for 48 h to induce DNA demethylation, while control cells
were left untreated. After 24 h, the media was changed and replaced
with fresh media containing 10 µM 5-aza-2′-deoxycytidine.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
To minimize gene expression variations, lysis buffer
was poured directly on the cells in the wells. Total RNA from the
5-aza-2′-deoxycytidine-treated and untreated control cells was
extracted after 48 h exposure using an AllPrep DNA/RNA Mini kit
(Qiagen, Valencia, CA, USA). cDNA was synthesized using a
PrimeScript RT Reagent kit (Takara Bio, Inc.), and the expression
of the FAS and BAX genes were measured in the
5-aza-2′-deoxycytidine-treated and untreated HT-29 cells. qPCR was
performed under the following conditions: 30 sec initial
denaturation step at 95°C followed by 40 amplification cycles at
94°C for 5 sec and 60°C for 30 sec. Melting curve analysis was then
performed. Relative quantification of the FAS and BAX genes was
performed by RT-qPCR in a Rotor-Gene 6000 cycler (Corbett Life
Science, Sydney, Australia) using SYBR Premix Ex Taq II (Takara
Bio, Inc.), and the GAPDH gene was used as a positive internal
control, whilst no template control NTC reactions served as
negative controls for nucleic acid contamination and primer dimer
formation. The primer sequences for RT-qPCR were extracted from
PrimerBank (http://pga.mgh.harvard.edu/primerbank/) (Table II). RT-qPCR was repeated twice for
relative quantification analysis, which was performed using the
∆∆Cq method (26).
| Table II.Reverse transcription-quantitative
polymerase chain reaction primer sets, extracted from the
PrimerBank database. |
Table II.
Reverse transcription-quantitative
polymerase chain reaction primer sets, extracted from the
PrimerBank database.
| Gene name | Primer sequence,
5′→3′ |
|---|
| GAPDH | F:
AAGGTGAAGGTCGGAGTCAAC |
|
| R:
GGGGTCATTGATGGCAACAATA |
| FAS | F:
CACCCGGACCCAGAATACC |
|
| R:
TGTTGCTGGTGAGTGTGCATT |
| BAX | F:
CCCGAGAGGTCTTTTTCCGAG |
|
| R:
CCAGCCCATGATGGTTCTGAT |
Methylation-sensitive restriction
enzyme PCR
DNA and RNA were extracted simultaneously from
tumoral and normal adjacent tissues using an AllPrep DNA/RNA Mini
kit. The extracted DNA samples were digested using
methylation-sensitive HpaII and methylation-insensitive
MspI restriction enzymes (Fermentas EpiJET DNA Methylation
Analysis kit; Thermo Fisher Scientific, Inc.). The primers for the
PCR reactions were designed using Primer3 software v0.4.0
(http://frodo.wi.mit.edu/primer3),
wherein the primers flank the HpaII/MspI enzyme
cutting site (5′-CCGG-3′) in the sequences. The primers used for
amplification were as follows: Forward, 5′-ACTTCCTGCCTCTGGCACT-3′
and reverse, 5′-AGGCTGGGCCTGTATCCTAC-3′ for BAX gene (GenBank:
AF339054.1); forward, 5′-ACGAACCCTGACTCCTTCCT-3′ and reverse,
5′-TCAGAGACGAGCTCACGAAA-3′ for FAS gene (GenBank: X82279.1). For
PCR amplification, a 25-µl reaction volume, including 12.5 µl Taq
DNA Polymerase Master Mix Red from Ampliqon (Odense M, Denmark), 2
µl digested DNA, 8.5 µl ddH2O and 10 pmol of each
primer, was added to 0.2-ml Eppendorf microtubes. The reactions
were performed for 30 cycles on a Mastercycler® Nexus
(Eppendorf, Hamburg, Germany); 95°C predenaturation for 4 min
followed by denaturation at 94°C for 35 sec, annealing at 64°C for
30 sec, extension at 72°C for 30 sec and final extension at 72°C
for 5 min. The amplified products were visualized on 1.5% agarose
gel electrophoresis and gel band pixel quantification was performed
using ImageJ software v1.47p (National Institutes of Health,
Bethesda, MD, USA).
Bisulfite conversion and
methylation-specific PCR (MSP)
Sodium bisulfite conversion of genomic DNA was
performed on 1 µg DNA using an EpiTect Bisulfite kit, according to
the manufacturer's protocol (Qiagen, Hilden, Germany). Human
control DNA, containing bisulfite-converted methylated and
unmethylated DNA (EpiTect PCR Control DNA Set; Qiagen), was used as
the control for MSP reactions. The specific primer sets for PCR
were designed by MethylPrimer software v1.0 (Thermo Fisher
Scientific, Inc.) and/or extracted from studies previously
published in the literature (25,27). The
primers were as follows: Forward, 5′-AGTGATATATAGGTGTTTAAAGACGT-3′
and reverse, 5′-AACTAACCTCAAAATATATTCCGTA-3′ for methylated
sequences; forward, 5′-AGTGATATATAGGTGTTTAAAGATGT-3′ and reverse,
5′-AACTAACCTCAAAATATATTCCATA-3′ for unmethylated sequences. The PCR
protocol was the same as performed for methylation-sensitive
restriction enzyme PCR.
Statistical analysis
GraphPad Prism 5 software (GraphPad Software, Inc.,
San Diego, CA, USA) was used to perform statistical analyses.
Student's t-test was applied to compare mean gene expression value
between tumor samples and adjacent normal samples. The experiments
were repeated twice or in cases of discrepancy they were repeated
three times, and all data are expressed as mean ± standard
deviation. P<0.05 was considered to indicate a statistically
significant difference.
Results
RT-qPCR
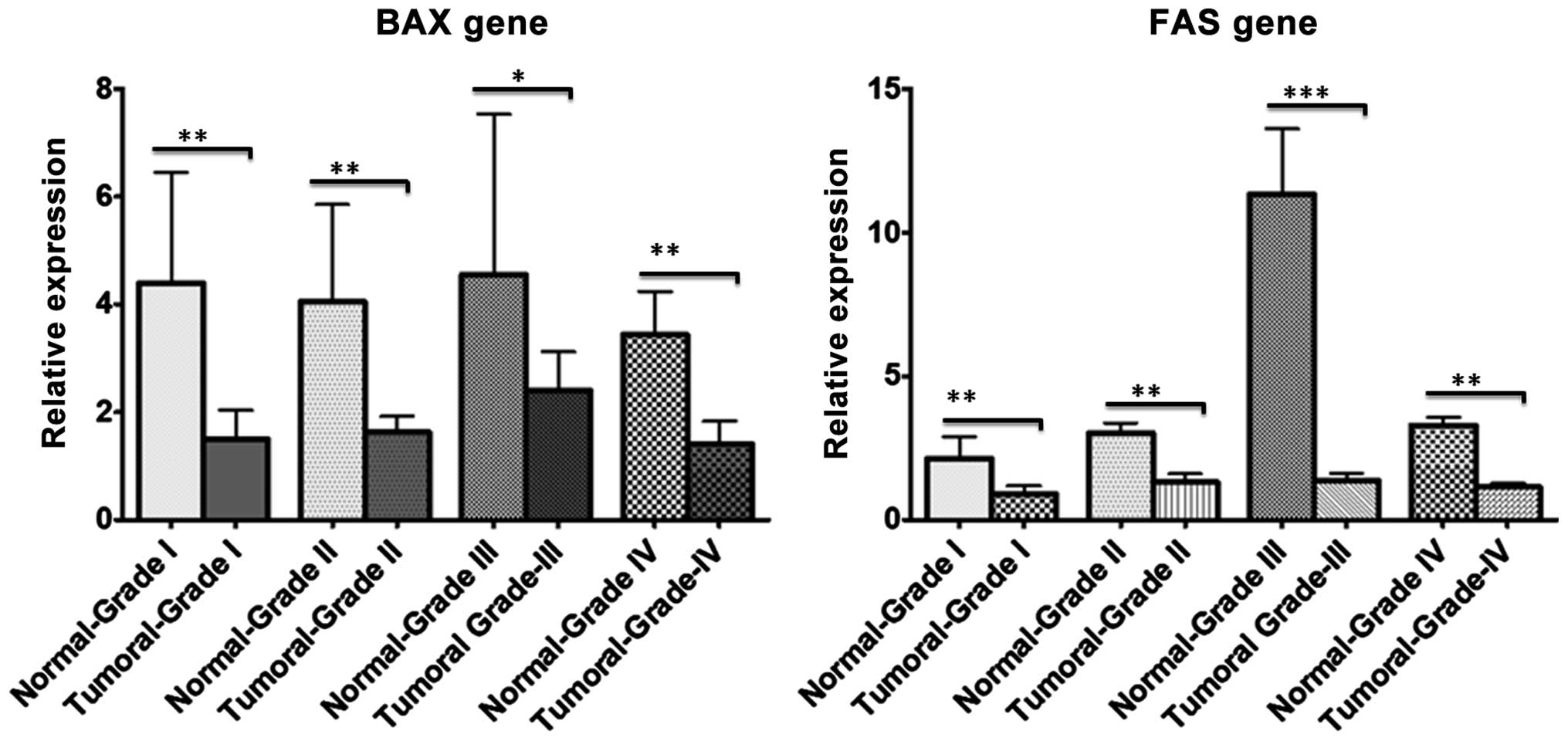
Using RT-qPCR, BAX and FAS gene expression were
analyzed in CRC and normal samples according to their pathological
grade (I–IV). RT-qPCR revealed downregulation of the two genes (19
out of 30 FAS samples and 18 out of 30 BAX samples) in CRC tissues
compared with adjacent normal samples in almost all pathological
grades; however, this decrease in expression was more significant
in tumor grades I and III (Fig. 1).
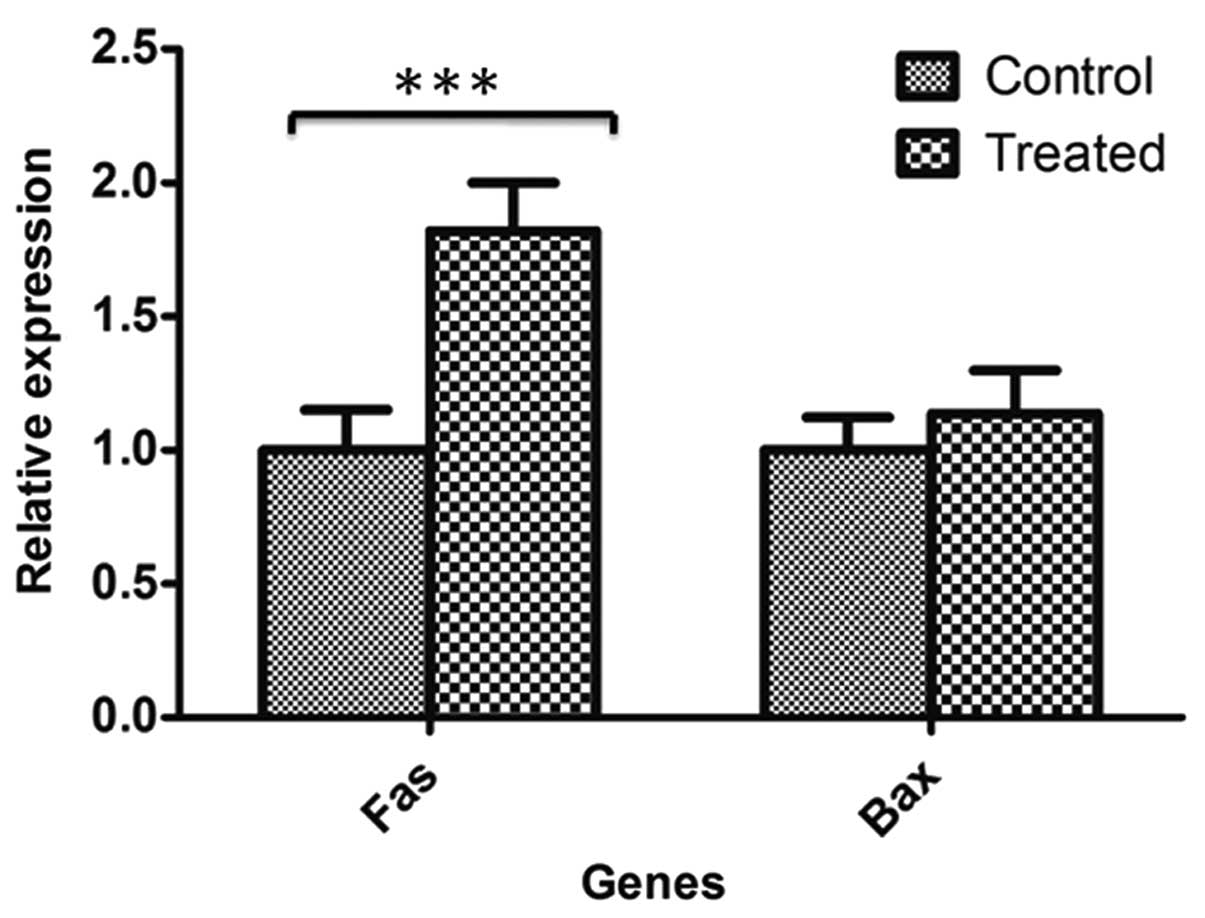
Furthermore, RT-qPCR demonstrated that FAS gene expression was
significantly upregulated in 5-aza-2′-deoxycytidine-treated HT-29
cells compared with untreated HT-29 control cells (P=0.00518);
however, the difference in BAX gene expression was not significant
between treated and untreated control cells (Fig. 2).
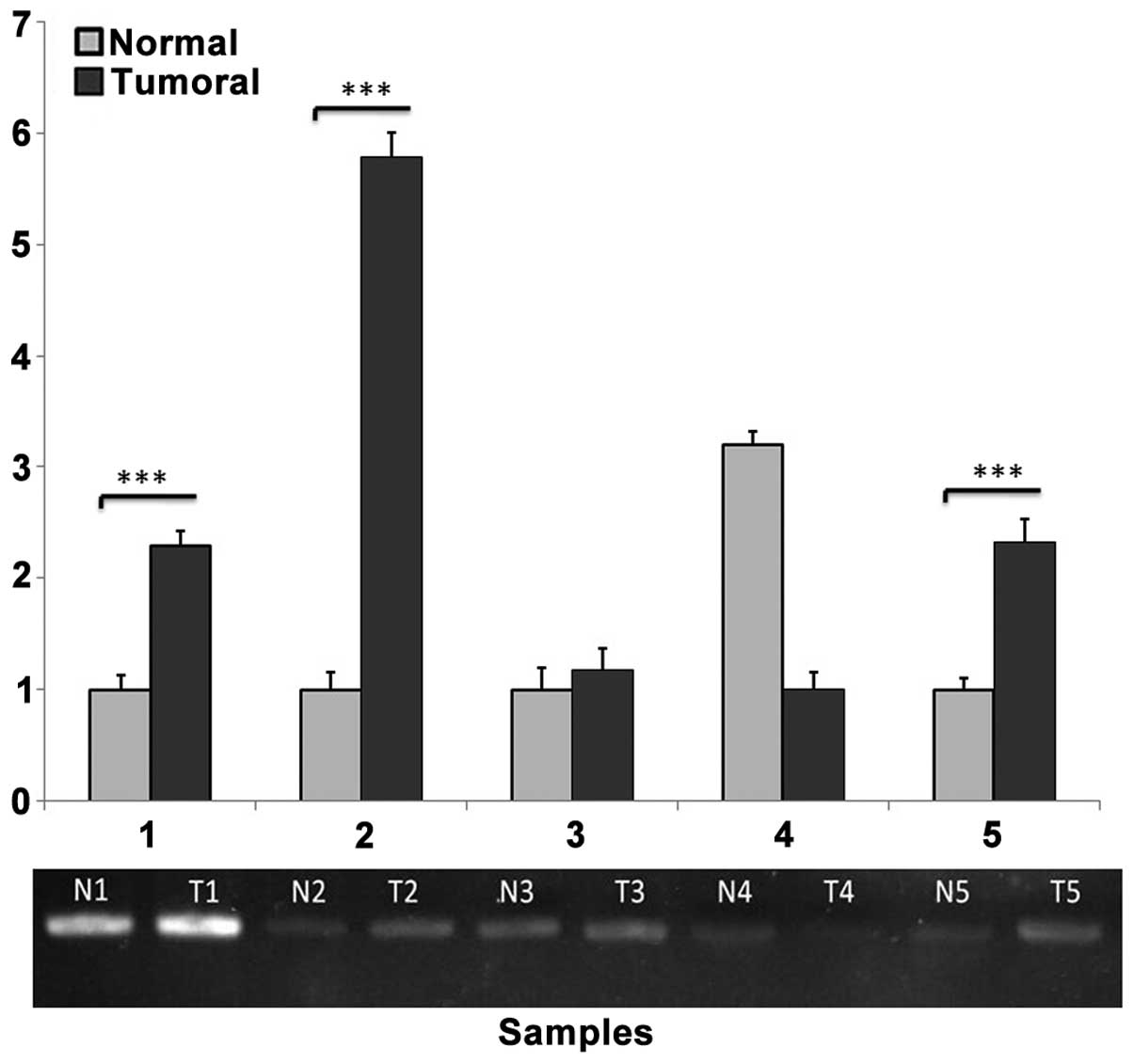
Methylation-sensitive restriction
enzyme PCR
PCR amplification of the selected region (−166 to −6)
was performed in the upstream region of the FAS gene to yield a
160-bp product containing two HpaII/MspI cutting
sites. Semi-quantification of agarose gel bands using ImageJ
software revealed significant methylation of the desired region of
the FAS gene in 19 out of 30 tumor samples compared with adjacent
normal samples (Fig. 3).
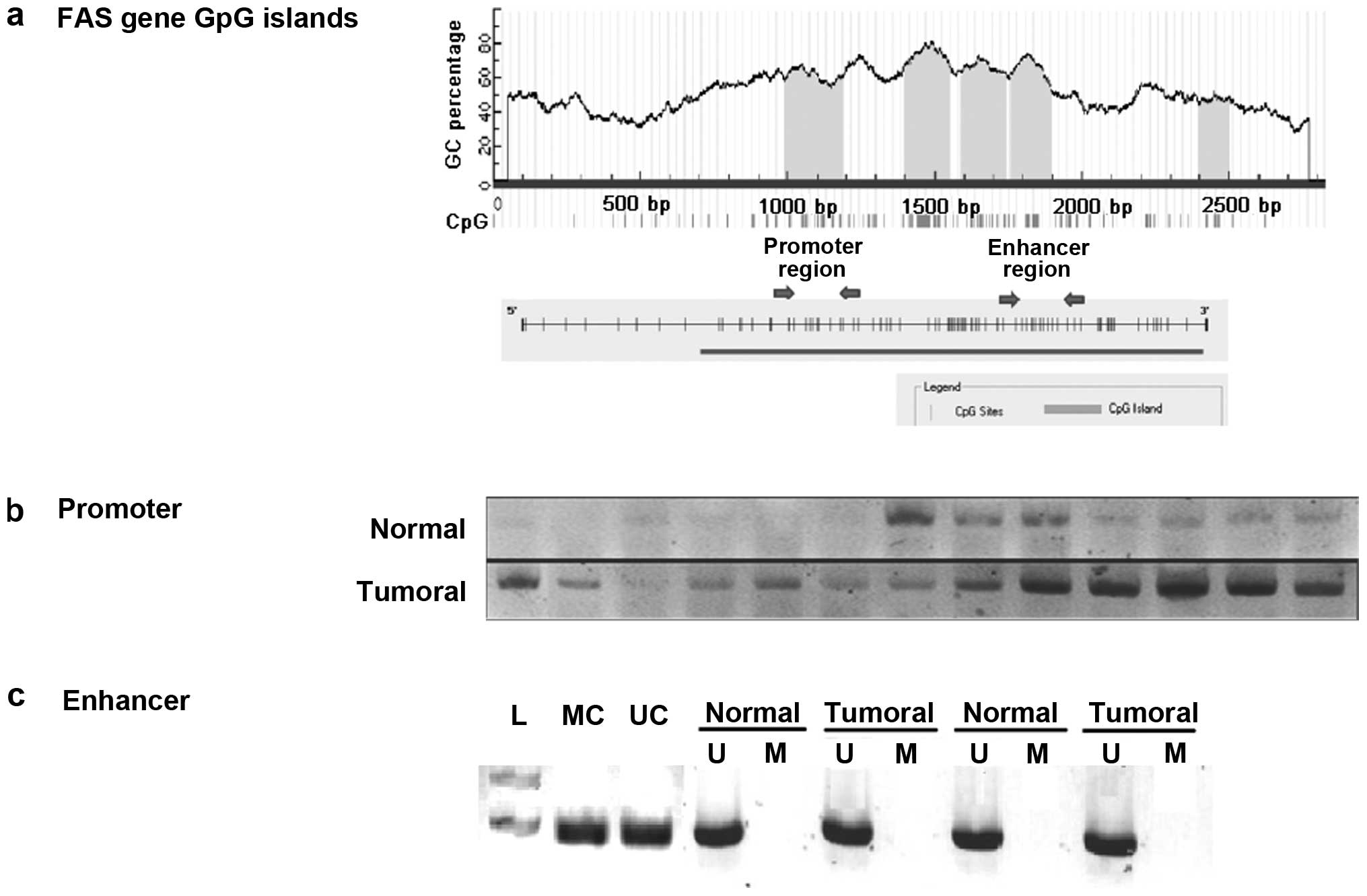
MSP
Two different CpG rich sections in the regulatory
regions of the FAS gene were analyzed by MSP (Fig. 4A). MSP analysis of the promoter region
revealed marked methylation in the tumor samples compared with the
normal adjacent samples (Fig. 4B).
The results identified that 16/30 (53.3%) tumoral samples were
markedly methylated in the FAS promoter region compared with the
normal samples. To determine if the hypermethylation in the FAS
promoter is correlated with its downregulation, the expression of
FAS in these 16 hypermethylated samples were compared and 12/16
samples showed significantly lower expression in tumor samples than
in normal adjacent samples. The first intron of the FAS gene has a
putative enhancer containing a P53 response element. To determine
the methylation in the enhancer region of the FAS gene in CRC
tissue samples, the MSP technique was performed with sodium
bisulfate-treated DNA from tumor and normal adjacent tissues.
Methylation status analysis of the FAS enhancer region using MSP
indicated no methylation in this region in CRC and normal samples
(Fig. 4C).
Discussion
Numerous efforts have been made to identify
epigenetic biomarkers that can be used for CRC early detection,
progression, tendency to metastasis, prognosis and response to
chemotherapeutics (28–30). DNA methylation has been investigated
for application in cancer screening of high-risk populations due to
its stability compared with protein and RNA biomarkers, and its
easy detection using PCR-based approaches. Methylation marks that
are methylated more frequently in tumoral samples compared with
normal samples may have potential as methylation biomarkers
(31). A previous study revealed that
the number of neoplasms containing methylated genes is not
significantly different between advanced colorectal adenoma and
colorectal adenocarcinoma; however, these methylated genes are
highly different between early colorectal adenoma and colorectal
adenocarcinoma (32). Therefore, the
methylation of specific gene promoters appears to occur during the
early stages of CRC tumorigenesis, and these DNA marks may be
classified as useful non-invasive early detection biomarkers if
they can be easily detected in available body specimens, such as
blood and stool.
In the present study, we hypothesized that DNA
methylation of proapoptotic genes that are considered to be tumor
suppressor genes may lead to resistance to apoptosis and the
development of CRC. Thus, two proapoptotic genes with CpG islands
in their promoter regions and flanking their transcription
initiation sites were selected for analysis in the present study.
Gene expression evaluation of tumoral and normal adjacent samples
demonstrated that the two genes, FAS and BAX, exhibited
significantly lower expression in tumoral compared with normal
samples in the majority of specimens (Fig. 1). These genes contain CpG islands
flanking the initiation site; hence, methylation and resultant
transcriptional silencing may occur in carcinogenesis. Furthermore,
methylation inhibition of the HT-29 CRC cell line using
5-aza-2′-deoxycytidine resulted in overexpression of the FAS gene
due to DNA demethylation (Fig.
2).
To analyze the CpG island methylation status, the
present study first performed methylation-sensitive restriction
enzyme PCR using HpaII and MspI enzymes. In the
HpaII assays, no significant difference in methylation
status was detected for the BAX gene (data not shown); however, a
significant difference in the methylation status of the FAS gene
promoter was identified between tumoral and normal specimens in the
majority of current samples. To confirm the methylation of CpG
island regions in the FAS gene, the MSP technique was also
performed. Two hotspot CG-rich regions in intron 1 and the promoter
of the FAS gene were examined for methylation marks. As well as CpG
islands, intron 1 of the FAS gene contains a P53-response element,
and is, therefore, considered to be an enhancer region (27).
The MSP results indicated no methylation in the
tumoral or normal samples in the enhancer region of the FAS gene.
Therefore, methylation of the enhancer region in intron 1 of the
FAS gene does not appear to be responsible for FAS downregulation
and, thus, based on the current study, is not a causative risk
factor for colorectal carcinogenesis (Fig. 4C). This result is in contradiction
with a previous study, which identified partial methylation in 4/10
colon carcinoma tumor samples (27).
However, the FAS gene has a CpG island around its 5′-flanking
region; therefore, DNA methylation in this region may be
responsible for the decreased expression of the FAS gene observed
in the present tumoral samples. In the present study, MSP was
performed using a methylation-specific primer set to confirm the
methylation status of the CpG dinucleotides in the promoter region;
the results revealed that the majority of tumoral samples (53.3%)
contained a markedly higher proportion of methylated FAS compared
with the normal samples (Fig. 4B). To
determine if the hypermethylation in the FAS promoter is correlated
with its downregulation, the expression of FAS in these 16
hypermethylated samples were compared and 12/16 samples showed
significantly lower expression in tumor samples than in normal
samples (P=0.0219). Therefore, we propose that methylation in the
promoter region may be responsible for downregulating FAS gene
expression during CRC carcinogenesis. The current findings are in
accordance with a previous study, which demonstrated that FAS
promoter methylation is associated with the levels of FAS
expression in different CRC cell lines; increased methylation of
CpG dinucleotides was associated with lower FAS gene expression
(27). This GC-rich promoter region
contains binding sites for specificity protein-1, activator protein
(AP)-1, AP-2, GAGA factor, nuclear factor of activated T-cells and
nuclear factor-κB transcription factors; and methylation of CpG
dinucleotides in these binding sites may affect transcription of
the FAS gene (33). By contrast, in
another previous study, southern blotting was performed using
methylation-sensitive restriction enzymes for FAS promoter
methylation evaluation, and revealed no correlation between
promoter hypermethylation and loss of FAS expression during
colorectal carcinogenesis (34).
In conclusion, the present results demonstrated that
FAS promoter hypermethylation is associated with FAS downregulation
in CRC but not normal adjacent samples in the investigated
population. Furthermore, the FAS promoter exhibited higher levels
of methylation in tumoral tissues compared with the normal adjacent
tissues. Therefore, FAS promoter methylation levels may be used as
an early detection biomarker for patients with CRC; however,
confirmatory studies using larger sample sizes non-invasive body
specimens, such as blood and/or stool, are required.
Acknowledgements
We thank Iran National Tumor Bank personnel for
their cooperation in providing biological materials. The present
study was supported by a grant from Shahid Beheshti University of
Medical Sciences (grant no. 9729; Tehran, Iran) and Baqyiatallah
University of Medical Sciences (grant no. 1390.22; Tehran,
Iran).
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mousavi SM, Gouya MM, Ramazani R, Davanlou
M, Hajsadeghi N and Seddighi Z: Cancer incidence and mortality in
Iran. Ann Oncol. 20:556–563. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Safaee A, Fatemi SR, Ashtari S, Vahedi M,
Moghimi-Dehkordi B and Zali MR: Four years incidence rate of
colorectal cancer in Iran: a survey of national cancer registry
data - implications for screening. Asian Pac J Cancer Prev.
13:2695–2698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dolatkhah R, Somi MH, Kermani IA,
Ghojazadeh M, Jafarabadi MA, Farassati F and Dastgiri S: Increased
colorectal cancer incidence in Iran: A systematic review and
meta-analysis. BMC Public Health. 15:9972015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Worthley DL and Leggett BA: Colorectal
cancer: Molecular features and clinical opportunities. Clin Biochem
Rev. 31:31–38. 2010.PubMed/NCBI
|
|
7
|
Takayama T, Miyanishi K, Hayashi T, Sato Y
and Niitsu Y: Colorectal cancer: Genetics of development and
metastasis. J Gastroenterol. 41:185–192. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Issa JP: Colon cancer: It's CIN or CIMP.
Clin Cancer Res. 14:5939–5940. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deaton AM and Bird A: CpG islands and the
regulation of transcription. Genes Dev. 25:1010–1022. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Illingworth RS, Gruenewald-Schneider U,
Webb S, Kerr AR, James KD, Turner DJ, Smith C, Harrison DJ, Andrews
R and Bird AP: Orphan CpG islands identify numerous conserved
promoters in the mammalian genome. PLoS Genet. 6:e10011342010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim MS, Lee J and Sidransky D: DNA
methylation markers in colorectal cancer. Cancer Metastasis Rev.
29:181–206. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kelly TK, De Carvalho DD and Jones PA:
Epigenetic modifications as therapeutic targets. Nat Biotechnol.
28:1069–1078. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Portela A and Esteller M: Epigenetic
modifications and human disease. Nat Biotechnol. 28:1057–1068.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fleischhacker M and Schmidt B: Circulating
nucleic acids (CNAs) and cancer-a survey. Biochim Biophys Acta.
1775:181–232. 2007.PubMed/NCBI
|
|
15
|
Iannolo G, Conticello C, Memeo L and De
Maria R: Apoptosis in normal and cancer stem cells. Crit Rev Oncol
Hematol. 66:42–51. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oliver L and Vallette FM: The role of
caspases in cell death and differentiation. Drug Resist Updat.
8:163–170. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qiao L and Wong BC: Targeting apoptosis as
an approach for gastrointestinal cancer therapy. Drug Resist Updat.
12:55–64. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hector S and Prehn JH: Apoptosis signaling
proteins as prognostic biomarkers in colorectal cancer: A review.
Biochim Biophys Acta. 1795:117–129. 2009.PubMed/NCBI
|
|
20
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
21
|
Manoochehri M, Karbasi A, Bandehpour M and
Kazemi B: Down-regulation of BAX gene during carcinogenesis and
acquisition of resistance to 5-FU in colorectal cancer. Pathol
Oncol Res. 20:301–307. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rupnarain C, Dlamini Z, Naicker S and
Bhoola K: Colon cancer: genomics and apoptotic events. Biol Chem.
385:449–464. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li LC and Dahiya R: MethPrimer: Designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol:. 35:495–516. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aaltonen LA and Hamilton SR: World Health
Organization classification of tumours: Pathology and genetics of
tumours of the digestive system. IARC Press. Lyon: 2000.
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Petak I, Danam RP, Tillman DM, Vernes R,
Howell SR, Berczi L, Kopper L, Brent TP and Houghton JA:
Hypermethylation of the gene promoter and enhancer region can
regulate Fas expression and sensitivity in colon carcinoma. Cell
Death Differ. 10:211–217. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Coppedè F: Epigenetic biomarkers of
colorectal cancer: Focus on DNA methylation. Cancer Lett.
342:238–247. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Choong MK and Tsafnat G: Genetic and
epigenetic biomarkers of colorectal cancer. Clin Gastroenterol
Hepatol. 10:9–15. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Binefa G, Rodríguez-Moranta F, Teule A and
Medina-Hayas M: Colorectal cancer: From prevention to personalized
medicine. World J Gastroenterol. 20:6786–6808. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shivapurkar N and Gazdar AF: DNA
methylation based biomarkers in non-invasive cancer screening. Curr
Mol Med. 10:123–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lao VV and Grady WM: Epigenetics and
colorectal cancer. Nat Rev Gastroenterol Hepatol. 8:686–700. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Singh R, Pradhan V, Patwardhan M and Ghosh
K: APO-1/Fas gene: Structural and functional characteristics in
systemic lupus erythematosus and other autoimmune diseases. Indian
J Hum Genet. 15:98–102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Butler LM, Dobrovic A, Bianco T and Cowled
PA: Promoter region methylation does not account for the frequent
loss of expression of the Fas gene in colorectal carcinoma. Br J
Cancer. 82:131–135. 2000.PubMed/NCBI
|