Introduction
It is widely appreciated that cancer cells in a
growing tumor are heterogeneous (1,2). Only a
subset of these cells possesses invasive qualities that drive their
detachment from the primary tumor and movement through the
surrounding microenvironment. Among these invasive cells, an even
smaller subset has the capacity to fully metastasize (3,4). However,
it remains to be elucidated how phenotypic differences between
individual cells or cell populations translate to variations in
metastatic ability.
Among the myriad cellular functions that contribute
to cell phenotype is autophagy. Autophagy, here connoting
macroautophagy, is a catabolic process during which a cell encloses
cytoplasmic components within double-membrane vesicles
(autophagosomes) that subsequently fuse with the lysosomal
compartment, where the autophagosomal cargo is degraded
enzymatically (5). Under normal
conditions, a low level of basal autophagy serves to eliminate
invading microorganisms and unnecessary, old or damaged proteins
and organelles (6,7). During times of stress and starvation,
autophagy is upregulated and the degraded autophagic cargo is
recycled, generating substrates that enable cells to preserve
intra- and extracellular homeostasis. The function of autophagy as
a stress response makes it particularly notable in the framework of
metastasis, an inherently stressful process that requires enhanced
survival capabilities in the very select few cells that complete it
(3,8).
A necessary characteristic of invasive cancer cells that
metastasize successfully is an exceptional ability to weather
stress; throughout the multi-stage metastatic cascade, these cells
must survive a wide range of factors, including nutrient
deprivation, hypoxia, acidosis, extracellular matrix (ECM)
detachment and shear force in the vasculature (4).
The present study investigated heterogeneity among
breast cancer cell populations in terms of their autophagic
capacities, proposing that perhaps the cancer cells with the most
robust abilities to respond to autophagic induction are those that
have the ability to withstand the pressures associated with the
metastatic process. Thus, the present study hypothesized that
distinct populations of breast cancer cells have increased
autophagic potential that, in turn, leads to increased metastatic
potential. The focus of the present study was the ability of breast
cancer cells to endure nutrient deprivation, which is one of the
pressures encountered throughout the metastatic process. The
present study revealed that the metastatic breast cancer cell line
that was sensitive to autophagic induction was also the cell line
that maintained its ability to proliferate following nutrient
deprivation. Furthermore, a subpopulation of this cell line
comprised of cells that survived multiple exposures to nutrient
deprivation was more responsive to autophagic induction compared
with its parent population. The results of the present study
suggest that, within a growing tumor, the autophagic response may
contribute to the capacity of breast cancer cell subpopulations to
endure starvation stress and better weather the metastatic
process.
Materials and methods
Cells and cell culture
The human breast metastatic carcinoma cells lines,
MDA-MB-231 (231) and MDA-MB-435 (435) (9); non-metastatic carcinoma cell line,
MDA-MB-436 (436) (9); and epithelial
cell line, MCF10A (10A) (10) were
cultured as previously described. All cell lines were maintained at
37°C with 5% CO2 in a humidified atmosphere, and were
tested regularly and confirmed as negative for Mycoplasma spp.
contamination (PlasmoTest kit; InvivoGen, San Diego, CA).
Cell subpopulation selection
To select a distinct subpopulation of 231 cells that
exhibited the capacity to survive nutrient deprivation, parental
231 cells were cultured to 80–90% confluency in complete culture
medium consisting of a 1:1 (v/v) mixture of Dulbecco's modified
Eagle's medium and Ham's F12 nutrient mixture (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 5% fetal
bovine serum (Thermo Fisher Scientific, Inc.), 2 mM L-glutamine
(Thermo Fisher Scientific, Inc.), and 0.02 mM non-essential amino
acids (Thermo Fisher Scientific, Inc.). Cells were subsequently
washed twice with phosphate-buffered saline (PBS; Thermo Fisher
Scientific, Inc.), incubated in Earle's balanced salt solution
(EBSS; Thermo Fisher Scientific, Inc.) for 24 h, rewashed in PBS
and cultured again to 80–90% confluency in complete culture medium.
This procedure was repeated 4 additional times. The final
subpopulation (231.EB5) was comprised of adherent cells that had
survived all 5 starvation rounds.
Proliferation assay
Proliferation assays were performed as described
previously (11). Cells were seeded
(5×102 cells/well) in complete culture medium in a
96-well tissue culture plate. To assess innate proliferation
capacity, fluorescence was measured after 1, 2, 3, 4 and 6 days
with the addition of AlamarBlue reagent (Thermo Fisher Scientific,
Inc.; DAL 1025). To assess proliferation capacity upon nutrient
depletion, the complete medium in each well was replaced with EBSS
24 h after seeding and cells were incubated for 24 h, after which
complete medium was returned to the wells. Proliferation was
measured 1, 2, 3, 4 and 6 days following EBSS treatment.
Fluorescence intensity at 570/580 nm excitation/emission was
determined using a Hitachi F-7000 fluorescence
spectrophotometer.
Immunoblot analysis
Cells were seeded in complete medium and treated for
0.5, 3, 6 and/or 12 h with 100 nM rapamycin (Sigma-Aldrich, St.
Louis, MO, USA) and 50 µm chloroquine (Sigma-Aldrich), individually
or in combination. Whole-cell lysates were collected with 1X
radioimmunoprecipitation assay lysis buffer (EMD Millipore,
Billerica, MA, USA) containing 1X Halt™ protease and phosphatase
single-use inhibitor cocktail (Thermo Fisher Scientific, Inc.).
Proteins (30 µg) were separated by SDS-PAGE (4–12% criterion XT
precast gel; Bio-Rad Laboratories, Inc., Hercules, CA, USA) and
transferred to nitrocellulose membranes. Membranes were blocked for
1 h at room temperature with 5% non-fat milk in Tris-buffered
saline supplemented with 0.05% Tween 20 (TBST; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and incubated overnight at
4°C with primary antibody. Membranes were washed in TBST, and
incubated with the corresponding monoclonal donkey anti-rabbit (cat
no. NA934V) or sheep anti-mouse (cat no. NA931V) IgG secondary
antibody (GE Healthcare Life Sciences, Chalfont, UK; dilution,
1:10,000) for 1 h at room temperature, and rewashed before blots
were developed with ECL Western Blotting Substrate (ThermoFisher
Scientific, Inc.) and Supersignal West Dura Extended Duration
Substrate (Thermo Fisher Scientific, Inc.). Results were quantified
with Image Studio version 4.0 software (LI-COR Biosciences,
Lincoln, NE, USA). The following primary antibodies were used:
Anti-LC3B (Abcam, Cambridge, MA, USA; dilution, 1:3,000; cat no.
ab51520), anti-β-actin (Abcam; dilution, 1:10,000), and anti-GAPDH
(Cell Signaling Technology, Inc., Danvers, MA, USA; dilution,
1:2,000; cat no. 2118S).
Immunofluorescence studies
Cells were seeded and grown to 90% confluence on
glass coverslips that were pretreated with 0.01% poly-L-lysine and
placed in 6-well culture plates. Cells were incubated for 3 and 12
h in complete culture medium containing 100 nM rapamycin
individually or in combination with 50 µM chloroquine. Each
condition was performed in triplicate. Cells were subsequently
washed 4 times for 3 min each in PBS, fixed in 3% formaldehyde in
PBS for 45 min at room temperature, permeabilized with 0.5% Triton
X-100 (Sigma-Aldrich) for 3 min at room temperature, blocked with
1% bovine serum albumin in PBS for 1 h and incubated with rabbit
polyclonal anti-human LC3B antibody (Abcam; dilution, 1:2,000; cat
no. ab51520) in 1% BSA in PBS at 4°C overnight. Secondary
incubation was performed for 1 h at room temperature using a goat
anti-rabbit immunoglobulin G fluorescein isothiocyanate-conjugated
secondary antibody (Abcam; dilution, 1:2,000; cat no. ab6717).
Vectashield with DAPI H-1200 (Vector Laboratories, Inc.,
Burlingame, CA, USA) was used to mount the coverslips on glass
slides. Immunofluorescent images were captured with a Nikon Eclipse
TE2000-U fluorescent microscope and archived using QCapture Pro
software, version 5.1.1.14 (QImaging, Surrey, BC, Canada).
Three-dimensional (3D) cell morphology
studies
For 3D cell culture assays, 400 µl/well of reduced
growth factor Matrigel (BD Biosciences, Franklin Lakes, NJ, USA)
was placed in 24-well plates and allowed to solidify at 37°C for 40
min. Cells were plated (2×103 cells/well) in complete
culture medium supplemented with 2% Matrigel and incubated at 37°C.
After 4 d, the media was replaced with a fresh layer of
Matrigel-supplemented medium only, or medium containing 100 nM
rapamycin or 50 µM chloroquine. Media was replaced every 3 d. Each
condition was performed in triplicate. Images were captured at 10 d
with a Nikon Eclipse TE2000-U fluorescent microscope and archived
using TSView software (Tucsen photonics Co., Ltd., Fuzhou,
China).
Results
MDA-MB-231 cells are sensitive to
autophagic induction
The presence of LC3B in autophagosomes and the
conversion of LC3B-I to its lipidated form, LC3B-II, are used
routinely as indicators of autophagic induction (12). The present study evaluated the levels
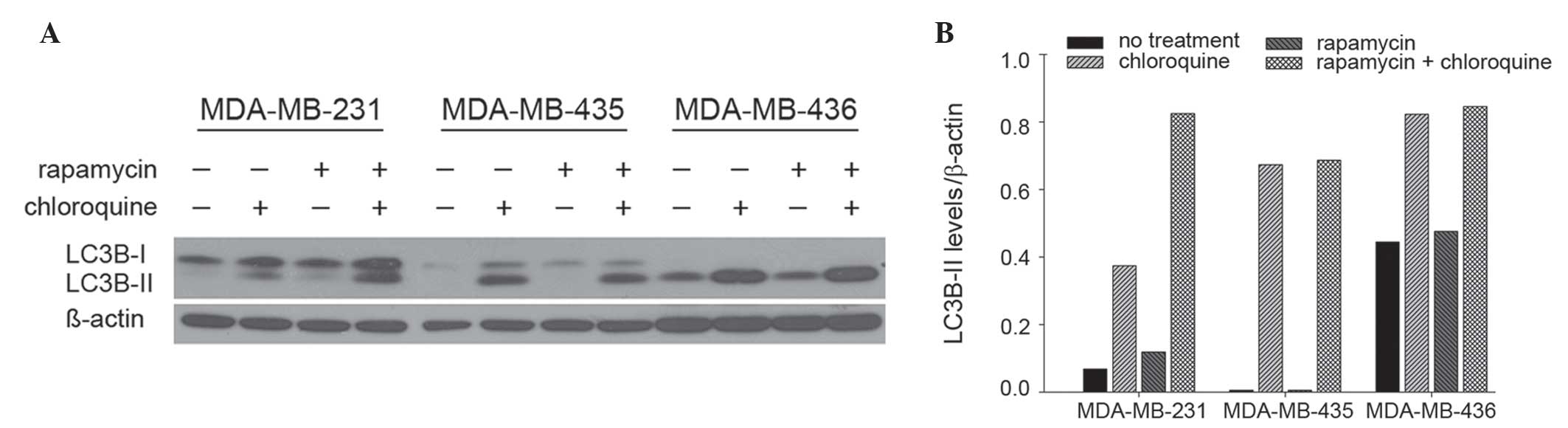
of LC3B-II in 231, 435 and 436 cells by immunoblot (Fig. 1A). Treatment with rapamycin, a
well-accepted autophagy inducer via inhibition of mammalian target
of rapamycin (mTOR), altered LC3B-II levels in 231 cells, but not
435 and 436 cells. Specifically, in 231 cells, LC3B-II levels were
74% higher following rapamycin treatment compared with no
treatment, and 120% higher following co-treatment with rapamycin
and chloroquine, an autophagy-blocking agent (lysosomal inhibitor),
compared with chloroquine treatment alone (Fig. 1B). By contrast, rapamycin did not
appreciably modulate LC3B-II levels in either 435 or 436 cells.
Notably, 231 cells exhibited the lowest levels of basal
autophagosome formation, indicated by LC3B-II levels apparent only
upon the addition of chloroquine. By contrast, basal autophagosome
levels were relatively high in 435 and 436 cells. Overall, these
results indicate that 231 cells are most sensitive to autophagic
induction compared with 435 or 436 cells, which suggests that 435
and 436 cells have intrinsically high levels of autophagy that an
additional stimulus may not robustly alter.
MDA-MB-231 cells proliferate following
nutrient depletion
For cells to metastasize successfully, the capacity
to survive in suboptimal environments is essential. To begin
investigating the association between variations in autophagic
capacity and abilities to withstand challenging conditions, the
present study used the same cell lines to test how nutrient
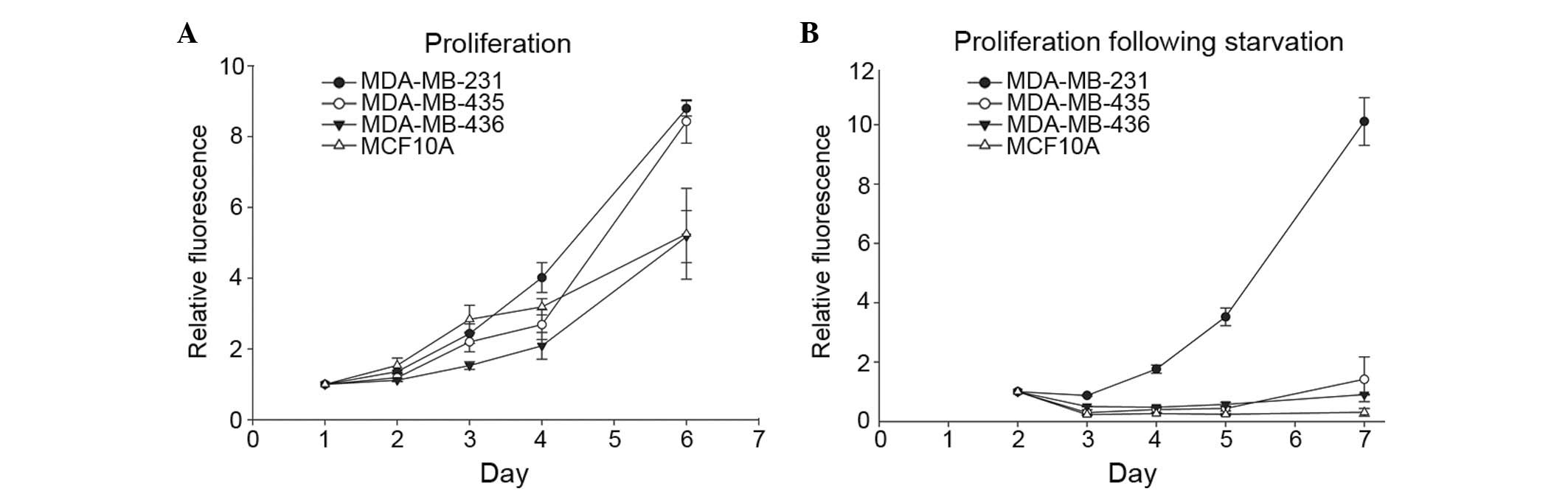
deprivation altered cell proliferation. Cells were grown in
complete medium to determine their innate proliferation rate. As
expected, the metastatic cell lines, 231 and 435, exhibited a
distinctly increased proliferation rate compared with the
non-metastatic 436 breast cancer cell line and the immortalized,
but otherwise normal, MCF10A breast epithelial cell line (Fig. 2A). Proliferation rates following
nutrient deprivation were subsequently assessed by culturing cells
in EBSS for 24 h and then returning them to complete medium. The
231 cells maintained their proliferative ability, whereas 435, 436
and MCF10A cells did not recover from starvation conditions
(Fig. 2B). Together with the results
presented in Fig. 1, these results
suggest that the cells that are most responsive to autophagic
induction are also able to best withstand the stress of nutrient
deprivation.
Autophagic induction is more efficient
in a select MDA-MB-231 subpopulation
Noting that 231 cells were unique in both their
sensitivity to autophagic induction and their sustained
proliferation following stress, the present study assessed whether
a subpopulation of these cells possessed a capacity for autophagic
induction distinct from parental cells. It was reasoned that it may
be this type of subset of invasive cells that has the ability to
fully metastasize. Parental 231 cells were subjected to 5 rounds of
culture in EBSS for 24 h followed by 24 h of culture in complete
medium. The surviving cells were selected (231.EB5). The five
rounds of selection were performed to ensure the generation of a
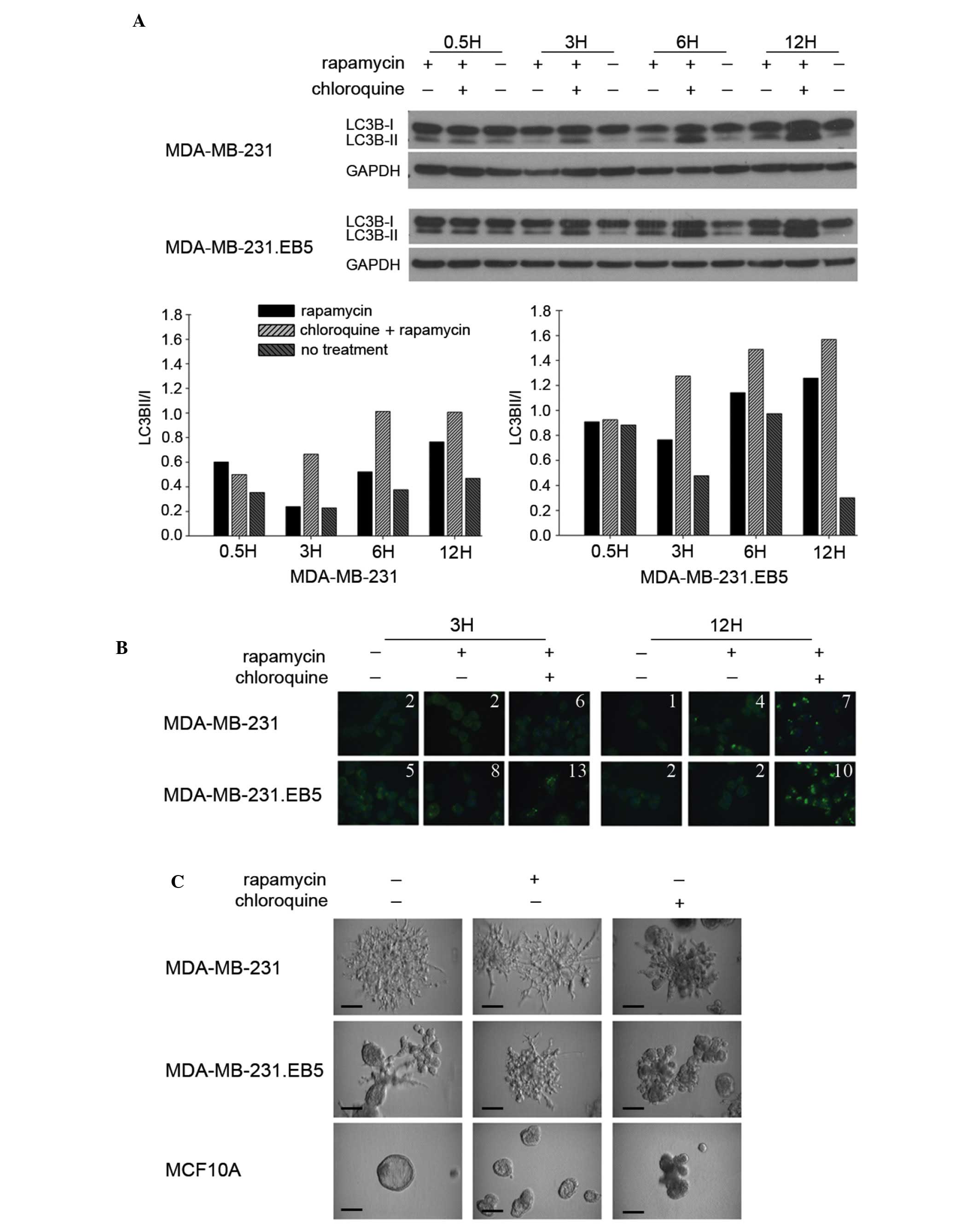
distinct cell subpopulation. LC3B-II levels by immunoblot (Fig. 3A) and LC3B puncta formation by
immunofluorescence staining (Fig. 3B)
were subsequently compared between the parental and selected cell
lines. Notably, 231.EB5 cells displayed the most marked changes in
LC3B-II levels in response to rapamycin treatment compared with
untreated conditions, particularly at 3 and 12 h (Fig. 3A). Supporting the immunoblot results,
the number of LC3B puncta was increased with no treatment and in
response to rapamycin in 231.EB5 cells compared with parent 231
cells (Fig. 3B).
Parent 231 cells grown in 3D Matrigel with complete
medium exhibit spindled branches characteristic of highly invasive
cells (Fig. 3C). Notably, 231.EB5
cells were rounded with few branched spikes, which is typical of
less invasive cells. However, in response to rapamycin treatment,
231.EB5 cells became substantially more invasive, developing a
phenotype reminiscent of the parent population. Minimal changes
were observed in parent 231 cells and MCF10A control cells
following rapamycin treatment compared with no treatment.
Chloroquine reduced the invasive nature of both 231 and 231.EB5
cells, eliciting smaller and more rounded colonies. Overall, the
results of the present study demonstrate distinct phenotypic
differences between a select population of 231 cells compared with
the parent population following a stimulus of autophagic induction,
and reiterate prior observations that responsiveness to autophagic
induction may be correlated with the ability to endure nutrient
deprivation.
Discussion
It is well-acknowledged that cancer cells within a
tumor vary widely in their functions and attributes, yet how traits
of individual cells or cell populations drive tumor progression,
including invasion and metastasis, remains to be fully elucidated
(1). In the present study, the
association between the response to autophagic induction and the
capacity to endure a type of stress encountered during the
metastatic process was investigated in various breast cancer cell
populations.
The role of autophagy is particularly multifarious
in the framework of cancer, in which the matter of whether
autophagy is anti- or protumorigenic is associated with that of
whether it is cytoprotective or cytotoxic (13). It is increasingly appreciated that
autophagic functioning is dependent on numerous factors, notably
cancer stage (14). While it has been
demonstrated that autophagy may act to suppress tumor growth during
tumorigenesis (6,15,16),
autophagy is able to promote tumor growth in transformed cells,
which may exploit autophagy as a protective mechanism against
stresses in the tumor microenvironment, including glucose
deprivation and hypoxia, as well as against therapeutic insult
(5,6,17–20). Activation of autophagy additionally
has been observed in response to stresses characteristic of the
metastatic cascade (21). For
instance, autophagy is upregulated following loss of ECM contact
(22–24), which typically induces programmed cell
death (anoikis) in normal cells, yet is an essential event for
metastasizing tumor cells (3).
The present study observed that 231 cells were
sensitive to autophagic induction (that is, increases in markers of
autophagosome formation in response to an agent, rapamycin, known
to induce autophagy were observed), and that autophagic induction
was associated with increased metastatic potential as assessed by
the ability to weather nutrient deprivation stress. Rapamycin
treatment increased LC3B-II levels in 231 cells, but LC3B-II levels
did not change appreciably in 435 or 436 cells. Furthermore, 231
cells, but not 435 or 436 cells, maintained their innate ability to
grow following a period of nutrient deprivation. The use of
nutrient deprivation to investigate whether cells with sensitivity
to autophagic induction are able to overcome difficult conditions
is consistent with a previous study by Wojtkowiak et al
(19), who demonstrated that an
acidic environment triggered chronic upregulation of autophagy as a
survival mechanism. While direct links cannot be made between the
autophagic response to rapamycin and the response to nutrient
deprivation, there is logic in juxtaposing rapamycin treatment with
starvation. These conditions may promote autophagic induction via
similar signaling pathways: Rapamycin by inhibiting mTORC1
activity, and starvation by upregulating 5′ AMP-activated protein
kinase (AMPK), which in turn may inhibit mTORC1 activity
downstream. Inhibition of mTORC1 and upregulation of AMPK may
promote unc-51 like autophagy activating kinase 1 complexing and
thus autophagy initiation (25–28).
As it cannot be concluded that the presently
investigated cell lines had an identical response to autophagic
induction by nutrient deprivation as they did to that by rapamycin,
there exists an alternative possibility to consider. The 436 cells,
followed by 435 cells, had the highest levels of basal
autophagosome formation compared with the lowest observed in 231
cells; 436 and 435 cells additionally had the lowest numbers of
cells that proliferated following starvation. Autophagy has been
linked not only to cell survival, but also to cell death (13,29,30). Thus,
it cannot be ruled out that the additional stimulation of nutrient
deprivation in the cells with high basal levels of autophagosome
formation resulted in excessive levels that provoked a cytotoxic
response. The importance of considering basal autophagy was
underscored in an investigation by Maycotte et al (31), although their findings associated
autophagy with cell survival, not death. These investigators
reported that breast cell lines differed in their dependency on
autophagy for survival in complete medium under conditions with no
added stress; upon autophagy inhibition with chloroquine and small
hairpin RNA knockdown of autophagy protein 5 (ATG5), ATG7 and
Beclin 1, triple negative breast cancer cell lines (including 231)
displayed the greatest decreases in proliferation and cell
viability. Whether basal dependency on autophagy under normal
conditions is associated with dependency on autophagy for survival
under stress was not assessed.
The results of the present study in 231.EB5 cells, a
subpopulation of 231 cells that survived multiple rounds of
starvation, provide additional evidence in support of the
association between sensitivity to autophagic induction and
metastatic potential. While previous studies have reported the link
between autophagy and nutrient deprivation (32–34), to
the best of our knowledge, the present study is the first to
generate a subpopulation through nutrient deprivation and to
investigate differences in autophagic capacity based on this
factor. These 231.EB5 cells with apparently superior aptitude for
survival exhibited the highest overall ability to respond to
rapamycin, as seen by increases in LC3B-II conversion at 3 h and
particularly at 12 h. The fact that sensitivity to rapamycin varied
by time point may be associated with the observation that autophagy
is regulated through a negative feedback loop based on the
sensitivity of mTORC1 to nutrient levels (7). Yu et al (35) reported that the intracellular
nutrients produced during the autophagic process may reactivate
mTORC1 signaling and inhibit autophagy over time even in the event
of ongoing starvation, a condition that initially inhibits mTORC1
activity and upregulates autophagy. Similar to starvation,
rapamycin treatment upregulates autophagy through mTORC1
inhibition. Therefore, perhaps the fluctuations in LC3B-II
conversion that were observed following prolonged rapamycin
treatment represent cycles of rapamycin-induced autophagic
upregulation counteracted by the subsequent generation of
nutrients. This requires additional investigation, particularly
considering the fact that consistent changes in LC3B-II conversion
in response to rapamycin were noted in parental 231 cells. However,
this observation in itself is in line with the fact that 231.EB5
cells were most sensitive to autophagic induction, which would in
turn incite more robust negative feedback and variation by time
point.
The fact that 231.EB5 cells exhibited a less
invasive phenotype in 3D when cultured in complete medium with no
treatment was unexpected, as it was predicted that these cells
would have heightened invasive qualities based on their high
survival capacity. It was notable that these cells displayed a
markedly more invasive phenotype following autophagic induction. At
this point, the factors responsible for these observations remain
to be elucidated. One key avenue to be investigated is that cancer
stem-like cells (CSCs) may have been enriched during cell
subpopulation selection. That greater changes were observed in
LC3B-II and increased numbers of LC3B puncta were observed in
231.EB5 cells is in line with an observation reported by Gong et
al (36), who identified
significantly increased autophagic flux in breast CSC-enriched
MCF-7 breast cancer cells compared with non-enriched adherent cells
in basal and starvation (EBSS culture) conditions. If the present
study did enrich CSCs in the cell subpopulation, it is possible
that this is the reason for the higher baseline levels of LC3B-II
and that the increased invasive 3D morphology that was observed
upon autophagic induction is associated with generation of
migrating CSCs (37). Additional
potential explanations include stimulation of quiescence and
triggering of an epithelial-to-mesenchymal switch (5). All of these have been associated with
autophagy but none have been completely characterized. Additional
experiments will be required to fully elucidate the reasons for the
difference in phenotype between 231.EB5 and parent 231 cells.
In conclusion, the present study demonstrated that
the populations of metastatic cells that displayed the most robust
reactions to autophagic induction additionally best withstood
starvation stress. It must be acknowledged that nutrient
deprivation and autophagy are not the only factors governing the
progression of a growing tumor. It must also be acknowledged that
autophagic induction does not denote completion of the entire
autophagic process, and thus that future studies should establish
the contribution of autophagic flux. However, the present study is
significant, noting that autophagy inducers and inhibitors are
among the developing therapeutic approaches in breast cancer
(38,39). The results of the present study
suggest that these agents may cause differential responses
depending on the properties of the unique cancer cell populations
and their surrounding environments.
Acknowledgements
Funding for the present study was provided in part
by the American Cancer Society (grant no., RSG-11-259-01-CSM) and
METAvivor Research and Support, Inc., as well as the University of
Alabama at Birmingham Cancer Prevention and Control Training
Program (grant no., R25 CA047888).
Glossary
Abbreviations
Abbreviations:
|
231
|
MDA-MB-231
|
|
231.EB5
|
MDA-MB-231.EB5
|
|
435
|
MDA-MB-435
|
|
436
|
MDA-MB-436
|
|
3D
|
three-dimensional
|
References
|
1
|
Almendro V, Marusyk A and Polyak K:
Cellular heterogeneity and molecular evolution in cancer. Annu Rev
Pathol. 8:277–302. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heppner GH, Loveless SE, Miller FR,
Mahoney KH and Fulton AM: Mammary tumor heterogeneity. Symp Fundam
Cancer Res. 36:209–221. 1983.PubMed/NCBI
|
|
3
|
Talmadge JE and Fidler IJ: AACR centennial
series: The biology of cancer metastasis: Historical perspective.
Cancer Res. 70:5649–5669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Welch DR, Steeg PS and Rinker-Schaeffer
CW: Molecular biology of breast cancer metastasis. Genetic
regulation of human breast carcinoma metastasis. Breast Cancer Res.
2:408–416. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: Therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weiss L: Metastatic inefficiency:
Intravascular and intraperitoneal implantation of cancer cells.
Cancer Treat Res. 82:1–11. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hurst DR, Edmonds MD, Scott GK, Benz CC,
Vaidya KS and Welch DR: Breast cancer metastasis suppressor 1
up-regulates miR-146, which suppresses breast cancer metastasis.
Cancer Res. 69:1279–1283. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hurst DR, Xie Y, Edmonds MD and Welch DR:
Multiple forms of BRMS1 are differentially expressed in the MCF10
isogenic breast cancer progression model. Clin Exp Metastasis.
26:89–96. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cody JJ, Markert JM and Hurst DR: Histone
deacetylase inhibitors improve the replication of oncolytic herpes
simplex virus in breast cancer cells. PLoS One. 9:e929192014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:1845–1846. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Roy S and Debnath J: Autophagy and
tumorigenesis. Semin Immunopathol. 32:383–396. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh
H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al:
Promotion of tumorigenesis by heterozygous disruption of the beclin
1 autophagy gene. J Clin Invest. 112:1809–1820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yue Z, Jin S, Yang C, Levine AJ and Heintz
N: Beclin 1, an autophagy gene essential for early embryonic
development, is a haploinsufficient tumor suppressor. Proc Natl
Acad Sci USA. 100:15077–15082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu YL, DeLay M, Jahangiri A, Molinaro AM,
Rose SD, Carbonell WS and Aghi MK: Hypoxia-induced autophagy
promotes tumor cell survival and adaptation to antiangiogenic
treatment in glioblastoma. Cancer Res. 72:1773–1783. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wojtkowiak JW, Rothberg JM, Kumar V,
Schramm KJ, Haller E, Proemsey JB, Lloyd MC, Sloane BF and Gillies
RJ: Chronic autophagy is a cellular adaptation to tumor acidic pH
microenvironments. Cancer Res. 72:3938–3947. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nedjic J, Aichinger M, Emmerich J,
Mizushima N and Klein L: Autophagy in thymic epithelium shapes the
T-cell repertoire and is essential for tolerance. Nature.
455:396–400. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kenific CM, Thorburn A and Debnath J:
Autophagy and metastasis: Another double-edged sword. Curr Opin
Cell Biol. 22:241–245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bertrand K: Survival of exfoliated
epithelial cells: A delicate balance between anoikis and apoptosis.
J Biomed Biotechnol. 2011:5341392011.PubMed/NCBI
|
|
23
|
Lock R and Debnath J: Extracellular matrix
regulation of autophagy. Curr Opin Cell Biol. 20:583–588. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Debnath J: Detachment-induced autophagy
during anoikis and lumen formation in epithelial acini. Autophagy.
4:351–353. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nazio F, Strappazzon F, Antonioli M,
Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J,
Piacentini M, Fimia GM and Cecconi F: mTOR inhibits autophagy by
controlling ULK1 ubiquitylation, self-association and function
through AMBRA1 and TRAF6. Nat Cell Biol. 15:406–416. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Alers S, Löffler AS, Wesselborg S and
Stork B: Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy:
Cross talk, shortcuts, and feedbacks. Mol Cell Biol. 32:2–11. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee EJ and Tournier C: The requirement of
uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of
autophagy. Autophagy. 7:689–695. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kohli L, Kaza N, Carroll SL and Roth KA:
Protector turns predator: Autophagic death via selective
degradation of KRAS. Autophagy. 9:1438–1439. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maycotte P, Gearheart CM, Barnard R, Aryal
S, Mulcahy Levy JM, Fosmire SP, Hansen RJ, Morgan MJ, Porter CC,
Gustafson DL and Thorburn A: STAT3-mediated autophagy dependence
identifies subtypes of breast cancer where autophagy inhibition can
be efficacious. Cancer Res. 74:2579–2590. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wong PM, Puente C, Ganley IG and Jiang X:
The ULK1 complex: Sensing nutrient signals for autophagy
activation. Autophagy. 9:124–137. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li L, Chen Y and Gibson SB:
Starvation-induced autophagy is regulated by mitochondrial reactive
oxygen species leading to AMPK activation. Cell Signal. 25:50–65.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Y, Zhang Q, Tian R, Wang Q, Zhao JJ,
Iglehart JD, Wang ZC and Richardson AL: Lysosomal transmembrane
protein LAPTM4B promotes autophagy and tolerance to metabolic
stress in cancer cells. Cancer Res. 71:7481–7489. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu L, McPhee CK, Zheng L, Mardones GA,
Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al: Termination of
autophagy and reformation of lysosomes regulated by mTOR. Nature.
465:942–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gong C, Bauvy C, Tonelli G, Yue W,
Deloménie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V,
Tharinger H, et al: Beclin 1 and autophagy are required for the
tumorigenicity of breast cancer stem-like/progenitor cells.
Oncogene. 32:2261–2272. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brabletz T, Jung A, Spaderna S, Hlubek F
and Kirchner T: Opinion: Migrating cancer stem cells - an
integrated concept of malignant tumour progression. Nat Rev Cancer.
5:744–749. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gewirtz DA: The four faces of autophagy:
Implications for cancer therapy. Cancer Res. 74:647–651. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vinayak S and Carlson RW: mTOR inhibitors
in the treatment of breast cancer. Oncology (Williston Park).
27:38–44. 2013.PubMed/NCBI
|