Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
sixth most common type of cancer, and is associated with a low
5-year survival rate, despite recent improvements in the
understanding of its molecular basis (1). It is known that HNSCC development and
progression depends on several aberrations in signaling molecular
pathways, including epidermal growth factor receptor (EGFR), Ras,
nuclear factor-κB, signal transducer and activator of transcription
(STAT), Wnt/β-catenin, transforming growth factor (TGF)-β and
phosphoinositide 3-kinase-AKT-mammalian target of rapamycin (mTOR)
(2). Understanding the role of these
pathways will provide valuable information on the mechanisms
controlling oral cancer, thus providing clinically useful
therapeutic targets.
STAT signaling has been involved in oncogenesis
(3,4).
A number of previous findings strongly suggest that persistent
activation of STAT3 in HNSCC, accompanied by increases in STAT3
tyrosine phosphorylation, is linked to cell proliferation,
differentiation and apoptosis (5,6). Aberrant
activation of upstream signaling pathways, particularly TGF-α/EGFR,
has been associated with constitutive activation of STAT3 molecules
in HNSCC (3,7). STAT3 is also activated through other
pathways such as the alpha-7-nicotinic receptor, interleukin
(IL)-6, IL-10 and IL-22 receptor, and erythropoietin receptor
pathways in several types of cells, including HNSCC (8,10). In
general, ligand binding leads to the phosphorylation of tyrosine
residues within the receptor, resulting in STAT3 tyrosine
phosphorylation, dimer formation, nuclear translocation, DNA
binding and transcriptional activity (3,11–13).
STATs could also become phosphorylated at the Ser727
residue in response to growth factor and cytokine stimulation
(14). However, the role of STAT3
serine phosphorylation remains controversial (14). Previous studies have proposed a
negative impact of Ser727 phosporylation on STAT3 activity
(13,15–17), while
others have indicated that STAT3 serine phosphorylation by several
kinases may upregulate the transcriptional activity and nuclear
translocation of STAT3 in cancer (18,19).
Mitogen-activated protein kinases (MAPKs) are a
family of protein kinases that link extracellular stimuli with
intracellular responses through phosphorylation of specific
downstream target molecules in serine and threonine residues
(20). MAPKs have been implicated in
various cellular processes, including growth, proliferation,
differentiation, inflammation and apoptosis (21,22). In
HNSCC, MAPKs constitute downstream targets of several molecules,
including EGFR, Raf kinase and reactive oxygen species, whose
aberrant regulation has been studied in this type of cancer
(21,23,24).
c-Jun N-terminal kinases (JNKs) are members of the
MAPK family, and become activated by a stressful cellular
environment, including ultraviolet irradiation, oxidative stress,
changes in osmolarity or metabolism, DNA damage, heat shock and
inflammatory cytokine signaling (25). JNK signaling has been studied in HNSCC
with controversial findings; thus, while certain studies support a
pro-oncogenic function of JNK, others provide evidence that JNKs
act as tumor suppressors in HNSCC (26–28).
Previous studies have supported an association
between the activation of specific members of the MAPK family and
negative regulation of STAT3 signaling (29–34). For
example, inhibition of STAT3 activities through extracellular
signal-regulated kinase (ERK) and p38-dependent pathways in human
lung adenocarcinoma has been observed (35). Lim and Cao (15) have reported that activation of JNK by
various stresses or by its upstream kinase MAPK kinase 7 (MKK7) may
be linked to STAT3 inhibition, but the knowledge about the
association between STAT3 and JNK remains insufficient and requires
further investigation. Overall, there is evidence to suggest that
MAPKs may modulate the activation status of STAT3 in various cell
types, including cancer cells (15,
29–34). The role of MAPKs in cancer and STAT3
regulation may vary according to the type and status of the studied
cells, indicating the requirement to investigate this association
in a cell-specific manner.
The aim of the present study is to evaluate the
activation status of JNK and STAT3 in HNSCC cell lines and to
assess the effects of JNK modulation on STAT3 expression, as well
as on cell proliferation and viability. Understanding the role of
these molecules and their potential crosstalk in oral cancer cells
may pave the way for the development of alternative treatment
strategies for patients with HNSCC.
Materials and methods
Cell lines and cell culture
Experiments were performed using established cell
lines of human OSCC (SCC9 and SCC25), which were obtained from the
American Type Culture Collection (Manassas, VA, USA). Cells were
cultured in a 1:1 mixture of Ham's F-12 and Dulbecco's modified
Eagle's medium containing 10% fetal bovine serum, 100 U penicillin
and 400 ng/ml hydrocortisone (Sigma-Aldrich, St. Louis, MO, USA) at
37°C in a 5% CO2 air atmosphere. Cells were subcultured
by disaggregation with trypsin (0.1%) and
ethylenediaminetetraacetic acid (0.01%) in phosphate-buffered
saline (PBS) at pH 7.5.
Selective inhibition of JNK1/2
Cells were plated in 6-well plates at a density of
5×104 cells/well and were allowed to grow to 80%
confluency. Then, cells were either treated with vehicle alone
[dimethyl sulfoxide (DMSO) at a maximum concentration of 0.1%] or
with the selective JNK1/2 inhibitor SP600125 (Calbiochem; EMD
Millipore, Billerica, MA, USA) at concentrations of 20–40 µM for 24
h.
Selective induction of JNK1/2
MAPK
Cells were plated in 6-well plates at a density of
2×105 cells/well and were allowed to grow to 80%
confluency. Cells were then either treated with vehicle alone (DMSO
at a maximum concentration of 0.1%) or with the selective JNK1/2
inducer (active MKK7; Sigma-Aldrich) at concentrations of 5 and 10
µM for 48 h.
Small interfering RNA (siRNA)
transfection
JNK1 and JNK2 siRNA and scrambled control siRNA were
purchased from Qiagen, Inc. (Valencia, CA, USA). All siRNA
transfections were performed using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's protocol, with a final siRNA concentration of 2 and
7 µM. OSCC cells were grown to mid-logarithmic phase and
transiently transfected (2×106 cells) with 50 µg siRNA
control or siRNA against JNK1/2 using Nucleofector™ reagent (Amaxa
Biosystems; Lonza Group AG, Basel, Switzerland). Cells were
collected at 48 h and whole lysates were analyzed by western
blotting.
Western blot analysis
Cells were washed twice with ice-cold PBS, followed
by lysis with radioimmunoprecipation assay buffer (50 mM Tris pH
7.4, 150 mM NaCl, 1% Triton X-100, 1% deoxycholic acid sodium salt,
0.1% sodium dodecyl sulfate, 100 mg/ml phenylmethylsulfonyl
fluoride, 1 mg/ml aprotinin, 1 mM dichlorodiphenyltrichloroethane
and 1 mM sodium orthovanadate) for 10 min at 4°C. The wells were
scraped, and the recovered cell products were centrifuged at 40,000
× g for 15 min at 4°C. The concentration of the recovered proteins
was measured and equalized using the Bio-Rad Protein Assay (Bio-Rad
Laboratories, Inc., Hercules, CA, USA), according to the
manufacturer's protocol.
Proteins in the total cell lysate were separated by
SDS-PAGE (10% separation gel and 5% spacer gel) and
electrotransferred to polyvinylidene difluoride films (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Blotted films were placed
in blocking solution for 1 h at room temperature. Subsequently,
they were probed with indicated primary antibodies overnight at 4°C
(mouse monoclonal STAT3, diluted 1:250, cat no. 9139; rabbit
polyclonal anti-phospho-STAT3 Tyr705, diluted 1:250, cat no. 9131;
rabbit polyclonal phospho-STAT3 Ser727, diluted 1:200, cat no.
9134; rabbit polyclonal anti-JNK1/2, diluted 1:250, cat no. 9252;
rabbit polyclonal anti-p-c-jun ser63, diluted 1:200, cat no. 9261.
and rabbit polyclonal cyclin-D1, diluted 1:250, cat no. 2922. All
antibodies were purchased from Cell Signalling, Beverly, MA, USA.
The film was washed thoroughly, incubated with goat polyclonal
anti-rabbit IgG horse radish peroxidase secondary antibody
(1:3.000; Santa Cruz Biotechnology, Santa Cruz, CA, USA, cat no.
sc-2301) or anti-mouse IgG antibody (dilution, 1:3.000; Santa Cruz
Biotechnology, cat no. sc-2031) with shaking at room temperature
for 1 h at 25°C; β-actin was used as control (Santa Cruz
Biotechnology, cat no. sc-47778). Proteins were visualized using an
enhanced chemiluminescence
Cell proliferation and viability
Cells were counted with a Neubauer hemocytometer
under an inverted microscope. Cell viability upon treatment was
determined by the Trypan blue dye exclusion test. All assays were
performed in quadruplicate and the results are reported as the mean
± standard deviation.
Statistical Analysis
Results of protein expression levels, cell viability
and cell number of treated cells were compared with the results of
untreated (control) cells respectively. Statistical analysis was
performed using statistical Packages for the Social Sciences (SPSS)
version 20. Paired groups were compared with the Student's t test
and P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of SP600125 inhibitor on
JNK1/2 and STAT3 protein expression and activation and cyclin D1
levels
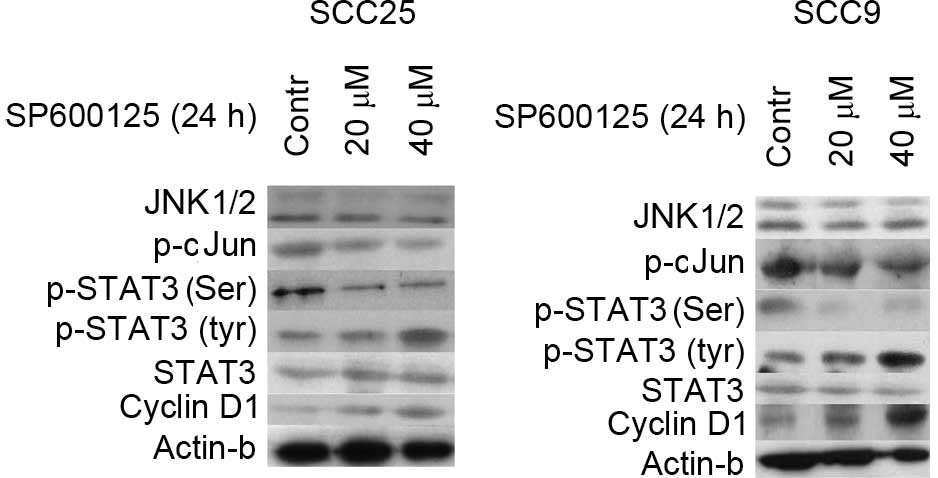
The expression and activation status of JNK1/2 was
examined in OSCC cells. According to the western blotting results,
total protein levels of JNK1/2 were detected in the two OSCC cell
lines tested (SCC9 and SCC25). Protein levels of p-c-Jun (immediate
downstream of JNK), total STAT3, tyrosine phosphorylated (p-Tyr)
STAT3, serine phosphorylated (p-Ser) STAT3 and cyclin D1 were also
observed in both cell lines (Fig.
1).
The effectiveness of JNK1/2 inhibition was next
assessed. Treatment of SCC25 cells with the JNK1/2 inhibitor
SP600125 for 24 h resulted in a dose-dependent inhibition of c-Jun
phosphorylation. Total JNK1/2 protein expression levels remained
steady despite SP600125 treatment (Fig.
1).
Next, the effectiveness of JNK1/2 inhibition on
STAT3 protein expression and activation was examined. In SCC25
cells, a significant reduction (P=0.03) of p-Ser STAT3 could be
detected following 24 h of SP600125 treatment in a dose-dependent
manner. By contrast, an increase in the levels of p-Tyr STAT3 was
observed, which was more pronounced at the highest concentration
used (40 µM). Treatment of SCC9 cells with SP600125 caused a
decrease in p-Ser STAT3 only when a concentration of 40 µM was
used, along with a significant increase (P>0.05)in p-Tyr STAT3
levels. In both cell lines, total STAT3 levels were not affected by
SP600125 treatment (Fig. 1).
Western blotting demonstrated that inhibition of
JNK1/2 was associated with increased expression levels of cyclin D1
in a dose-dependent manner. By contrast, the protein levels of
actin remained stable throughout the treatment, indicating that the
observed effects on the aforementioned proteins were not caused by
a nonspecific reduction of protein expression (Fig. 1).
In summary, inhibition of JNK1/2 activity by
SP600125 treatment was detected in both cell lines and was
associated with increased levels of p-Tyr STAT3 and cyclin D1, and
decreased levels of p-Ser STAT3, particularly at the highest
concentration used.
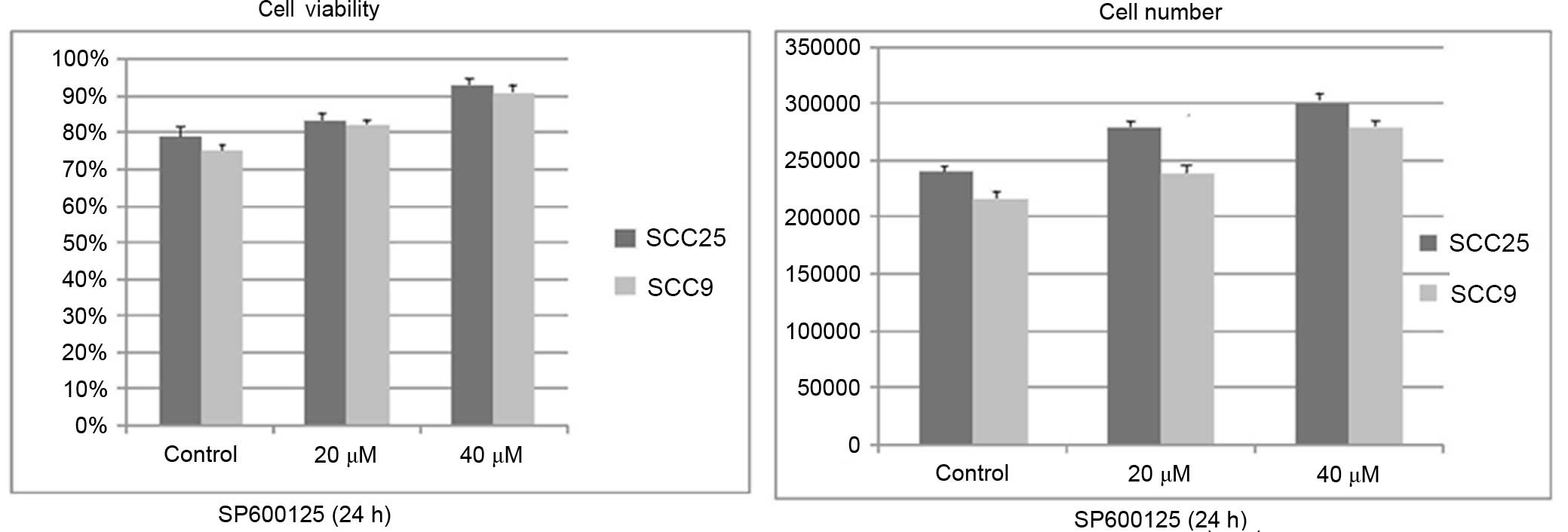
Effects of SP600125 inhibitor on cell
growth and viability
Treatment with SP600125 for 24 h resulted in a
dose-dependent increase in cell growth (total number of cells) and
cell viability (number of viable cells) in the two cell lines
tested. The increase appeared to be more prominent in the SCC25
cell line than in the SCC9 cell lines. However, the observed
changes were not significant (Fig.
2).
Effects of JNK1/2 siRNA silencing on
JNK and STAT3 protein expression and activation and cyclin D1
levels
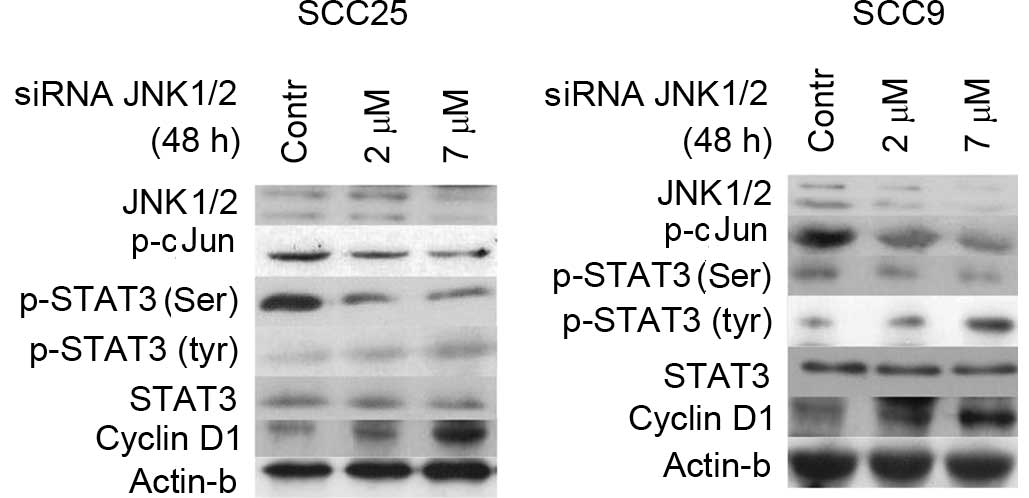
In order to corroborate the results from the
pharmacological inhibition of JNK1/2, specific inhibition was
performed by siRNA-targeting of JNK1/2 in SCC25 and SCC9 cell
lines. Following 48 h of transfection with a specific siRNA against
JNK1/2, western blot analysis revealed that JNK1/2 was efficiently
silenced, which was accompanied by a dose-dependent decrease in the
protein levels of p-c-Jun compared with the control in both cell
lines (Fig. 3).
Decreases in JNK1/2 protein expression and c-Jun
phosphorylation following 48 h of treatment with 2 and 7 µΜ
specific siRNA against JNK1/2 correlated with a decrease in the
levels of p-Ser STAT3 in both cell lines. More prominent was this
decrease at higher concentration (7 µM). Regarding STAT3 tyrosine
phosphorylation, an upregulation in the levels of p-Tyr STAT3 was
detected, particularly at the highest concentration tested. Total
STAT3 protein levels remained steady in both cell lines (Fig. 3).
Furthermore, western blot analysis demonstrated that
silencing of JNK1/2 was associated with markedly increased levels
of cyclin D1 protein expression in a dose-dependent manner in both
cell lines, while the protein levels of β-actin remained stable
(Fig. 3).
In summary, specific silencing of JNK1/2 resulted in
decreases in p-Ser STAT3 and cyclin D1 levels and increases in
p-Tyr-STAT3 levels in both cell lines.
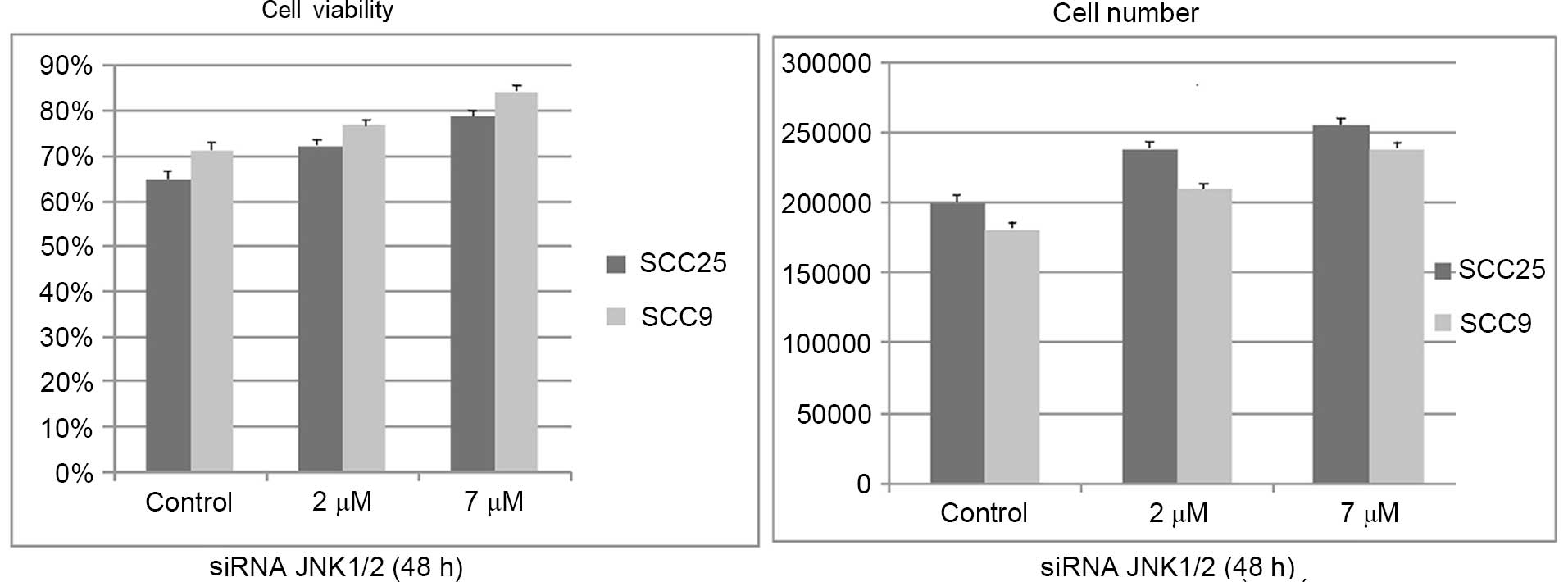
Effects of silencing JNK1/2 on cell
growth and viability
Similar to the effects of the chemical inhibitor
SP600125, siRNA treatment against JNK1/2 for 48 h resulted in a
dose-dependent increase in cell growth and viability in both cell
lines, although it was not significant (Fig. 4).
Effects of JNK1/2 induction on JNK1/2
and STAT3 protein expression and activation and cyclin D1
levels
To further investigate the significance of JNK1/2 in
the modulation of STAT3, pharmacological induction of JNK/2 was
performed using active MKK7 to further induce JNK1/2 expression.
Treatment of cells with selective MKK7 inducer efficiently
upregulated the levels of p-c-Jun in a dose-dependent manner in
both cell lines without affecting the total JNK1/2 levels (Fig. 5).
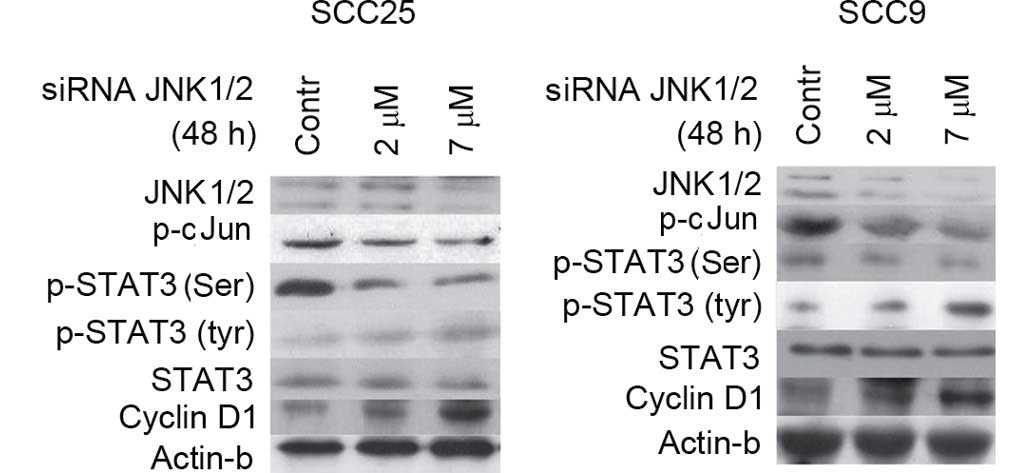 | Figure 5.Effect of JNK1/2 induction on STAT3
phosphorylation and cellular activity. Induction of JNK1/2 with
active MKK7 for 48 h enhanced the levels of p-c-Jun in a
dose-dependent manner in the two cell lines tested without
affecting the levels of total JNK1/2. Furthermore, induction of
JNK1/2 resulted in the upregulation of p-STAT3 (Ser727) at both
concentrations in SCC25 cells, and particularly at the highest
concentration assayed in SCC9 cells. By contrast, the levels of
p-Tyr STAT3 appeared to decrease upon treatment with active MKK7,
particularly at the highest concentration used. Active MKK7
treatment of both cell lines for 48 h caused a downregulation in
cyclin D1 expression levels in a dose-dependent manner. Contr,
control; STAT, signal transducer and activator of transcription;
JNK, c-Jun N-terminal kinase; p-, phosphorylated; siRNA, small
interfering RNA; MKK7, mitogen-activated protein kinase kinase
7. |
Treatment of both cell lines with JNK1/2 inducer for
48 h resulted in induced phosphorylation of STAT3 at Ser727 at the
two concentrations tested in SCC25 cells, and particularly at the
highest concentration assayed in SCC9 cells. By contrast, p-Tyr
STAT3 levels appeared to decrease following treatment with active
MKK7, particularly at the highest concentration tested. Total STAT3
levels were not affected by JNK1/2 induction in either cell line
(Fig. 5).
With regards to cyclin D1, active MKK7 treatment of
both cell lines for 48 h caused a downregulation in cyclin D1
expression levels in a dose-dependent manner. The levels of β-actin
protein remained stable despite the treatment (Fig. 5).
In summary, JNK/2 induction caused an upregulation
of p-Ser STAT3 levels in the two cell lines analyzed, as well as
decreases in the levels of p-Tyr-STAT3 and cyclin D1.
Effects of JNK1/2 induction on cell
growth and viability
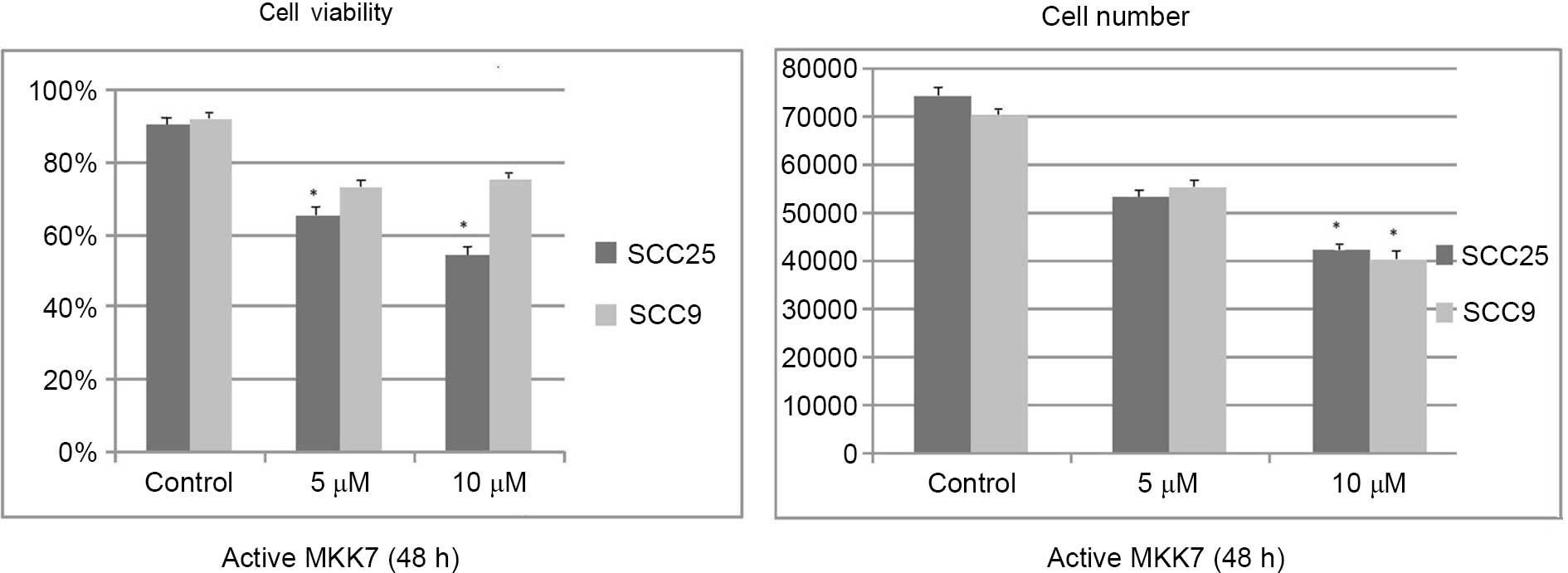
Active MKK7 treatment at the highest concentration
tested for 48 h resulted in a significant dose-dependent decrease
in cell growth and viability, which were more prominent in the
SCC25 cell line than in the SCC9 cell line. Cell growth changes
were significant at the highest concentration assayed in both cell
lines (SCC25 P=0.039; SCC9 P=0.044), compared with the control,
while cell viability changes were significant at the two
concentrations tested in the SCC25 cell line, compared with the
control (5 µM P=0.041; 10 µM P=0.03 (Fig.
6).
Discussion
The role of JNKs in cancer development is still
under investigation. Activation of the JNK signaling pathway in
response to several extracellular stimuli has been viewed as a
critical event that leads to apoptotic or non-apoptotic cell death
in several cells, including bronchial epithelial, human hepatoma,
hepatocellular carcinoma and osteosarcoma cells (36–39). By
contrast, JNK has also been reported to promote cell transformation
and proliferation in cancer stem cells (40). Recent data suggest that the role of
JNK1 and JNK2 in cancer is diverse, resulting in either promotion
or suppression of tumor formation or size in different types of
cancer, which emphasizes the significance of understanding the dual
role of JNKs and the molecular background of these distinct
functions in different tumors (41).
For example, Seki et al (42)
highlighted the relevant role of JNK signaling in the initiation
and progression of liver cancer, and Leventaki et al
(43) reported that JNK activation
induces tumor cell proliferation in classical Hodgkin lymphoma. Jia
et al (44) reported that the
activation of JNK contributes to dihydroartemisinin-induced
autophagy in pancreatic cancer cells, and Shi et al
(45) suggested that JNK enhances the
tumor suppressive role of p53 and promotes apoptosis in several
cell cancer lines, including colon, breast carcinoma and
osteosarcoma.
The functional role of JNK signaling has also been
studied in HNSCC, with conflicting findings. Gross et al
(27) observed that inhibition of
c-Jun with SP600125 suppresses the growth of HNSCC cells. In
addition, Yunoki et al (28)
proposed that combined silencing of B-cell lymphoma 2-associated
athanogene 3 (a co-chaperone of heat shock protein 70) and
inhibition of the JNK signaling pathway may be an option in
hyperthermia therapy in OSCC. However, several studies highlighted
the role of activated JNK in apoptosis and tumor suppression in
HNSCC. For example, Boivin et al (26) demonstrated that JNK mediates
radiotherapy-induced apoptosis in human HNSCC cell lines, and Chen
et al (46) demonstrated that
the apoptotic effect of cisplatin and cordycepin act
synergistically through the activation of the JNK/caspase-7/poly
(ADP-ribose) polymerase signaling pathway in the human oral cancer
cell line OC3. Furthermore, Noutomi et al (47) reported that JNK activation is involved
in the molecular mechanism of tumor necrosis factor-related
apoptosis-inducing ligand-induced cell death in HNSCC.
The present results appear to support a rather
onco-suppressive role of JNK in oral cancer, since JNK1/2
inhibition was associated with a noticeable (although not
significant) increase in the number of living OSCC cells in a
dose-dependent manner, which was accompanied by a corresponding
upregulation in the levels of cyclin D1. Previous studies examined
the role of JNK in mediating the apoptotic effect of several
pharmacological agents or anticancer drugs in HNSCC cell lines,
including N-(4-hydroxyphenyl) retinamide (4HPR), pepsin-digested
bovine lactoferrin, 5-aminolevulinic acid, MLN4924, 6-(N, N
-dimethylamino)-2-(naphthalene-1-yl) −4-quinazolinone,
fomitoside-K, mevastatin and AZD8055 (an mTOR inhibitor), and their
biological mechanisms of apoptosis (48–55).
Consistent with the present results, an antitumor role of JNK has
been proposed upon treatment of HNSCC cells with the JNK inhibitor
SP600125, which diminished the aforementioned pharmacological
agents-induced apoptosis (47–54).
Similarly, Li and Johnson (56)
demonstrated that pharmacological inhibition of JNK with SP600125
markedly inhibited bortezomib-induction of autophagy regulatory
proteins and autophagosome formation in HNSCC.
In the present study, selective siRNA-mediated JNK
inhibition induced similar results to those of JNK pharmacological
inhibition in a dose-dependent manner in the two cell lines
evaluated, followed by a non-significant increase in the total
number of cells and cell viability levels. Using similar siRNA
techniques, Kim et al (49)
demonstrated that suppression of JNK1 and JNK2 decreased, whereas
overexpression of wild-type JNK1 enhanced, 4HPR-induced apoptosis.
In addition, Li et al (48)
described that JNK inhibition by RNA interference alleviated
AZD8055-induced cell death in HNSCC.
By contrast, the present study has demonstrated that
pharmacological induction of JNK appears to have the opposite
effects to those aforementioned in the pharmacological and
selective siRNA-mediated JNK inhibition. Notably, the decrease in
the total cell number follows the relative decrease in cyclin D1
levels. In agreement with these results, Schramek et al
(57) indicated that MKK7 functions
as a major tumor suppressor in lung and mammary cancer in mice,
while Tang et al (37)
reported that alpinetin decreases proliferation of human hepatoma
cells through the activation of MKK7, and Dai et al
(58) reported MKK7/JNK1-dependent
apoptosis in human acute myeloid leukemia cells.
Regarding a potential crosstalk between STAT3 and
JNK, constitutive activation of STAT3, accompanied by increases in
STAT3 tyrosine phosphorylation, is known to regulate cell
proliferation, differentiation and apoptosis in various types of
cancer (5,6). However, the role of serine
phosphorylation is less well understood. Although there is
significant evidence in favor of the important role of STAT3 in
head and neck cancer, the mechanism of STAT3 hyperactivation in
this type of cancer remains to be completely understood (59). STAT proteins are important downstream
targets of MAPKs, which are involved in the regulation of STAT
proteins through crosstalk signaling (12).
The role of specific MAPKs, including JNK, in STAT3
regulation is not completely understood. In a previous study
(60), the present authors
investigated the role of ERK1/2, and demonstrated that ERK induced
the upregulation of STAT3 Ser727 phosphorylation, while
phosphorylation of Tyr705 did not appear to experience major
changes.
The present study focused on the role of JNK in the
regulation of STAT3 signaling, and investigated whether changes in
the expression and activation status of JNK1/2 affect STAT3
tyrosine and/or serine phosphorylation and the total protein
expression levels of STAT3 in HNSCC cell lines. Chemical inhibition
(via SP600125) or selective targeting (via siRNA) of the MAPK
JNK1/2 downregulated STAT3 serine phosphorylation, which was
accompanied by a moderate increase in p-Tyr STAT3 levels. Induction
of JNK had the opposite effects, resulting in upregulation of STAT3
serine phosphorylation and downregulation of STAT3 tyrosine
phosphorylation.
Crosstalk between JNK and STATs has been described
in several cases. For example, Kim et al (61) indicated that the development of
doxorubicin resistance in cancer cell lines is correlated with the
activation of STAT3 through the JNK signaling pathway. In addition,
Guo et al (62) observed that
JNK inhibition leads to reduced STAT3 Ser727 phosphorylation in
breast cancer cells, and noticed that the antitumor activity of
oncrasin-72 was followed by JNK activation and inhibition of Janus
kinase 2/STAT3 phosphorylation. Similar results were obtained in
previous studies in human bronchial epithelial cells, where JNK
inhibition through SP600125 or JNK silencing reduced Ser727
phosphorylation of STAT3 (39,63).
Additionally, Lim and Cao (15)
noticed that STAT3 is negatively regulated through JNK-induced
serine phosphorylation by JNK in monkey kidney cells. Shirakawa
et al (64) studied STAT6,
another member of the STAT family, and suggested that JNK
phosphorylates Ser707 of STAT6 and leads to deactivation and
inhibition of the transcription of STAT6-responsive genes in human
HeLa and HEK293 cells.
In summary, the present findings support a potential
onco-suppressive role of JNK1/2 in OSCC, as the activation of JNK
appears to downregulate cell proliferation and viability, and
decreases cyclin D1 expression levels. It appears that oncogenic
STAT3 constitutive signaling in OSCC cells is negatively regulated
by JNK. The JNK-STAT3 crosstalk is mediated mostly through
JNK-induced upregulation of STAT3 phosphorylation at Ser727, and
downregulation of STAT3 phosphorylation at Tyr705, which
collectively may cause the inhibition of STAT3 activity.
It is possible that the role of JNK in STAT3
modulation varies according to the type and status of the studied
cells, indicating the requirement for identifying the role of MAPK
activation in relation to STAT3 signaling in specific cell types.
Understanding the complexity of the MAPK signaling pathway and the
crosstalk between other major molecules such as STAT3 will be
helpful to design pharmacological therapies for cancer
prevention.
In conclusion, the present results indicate that JNK
may be a potential significant molecular target of novel anticancer
therapies for patients with OSCC.
Acknowledgements
The present study was co-financed by the European
Union [Brussels, Belgium; European Social Fund (ESF), and Greek
national funds through the Operational Program ‘Education and
Lifelong Learning’ of the National Strategic Reference
Framework-Research Funding Program ‘Heracleitus II. Investing in
knowledge society through the ESF’ (Athens, Greece) grant no.
388/1/359.
References
|
1
|
Chin D, Boyle GM, Porceddu S, Theile DR,
Parsons PG and Coman WB: Head and neck cancer: Past, present and
future. Expert Rev Anticancer Ther. 6:1111–1118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Molinolo AA, Amornphimoltham P, Squarize
CH, Castilho RM, Patel V and Gutkind JS: Dysregulated molecular
networks in head and neck carcinogenesis. Oral Oncol. 45:324–334.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rane SG and Reddy EP: Janus kinases:
Components of multiple signaling pathways. Oncogene. 19:5662–5679.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Song JI and Grandis JR: STAT signaling in
head and neck cancer. Oncogene. 19:2489–2495. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kijima T, Niwa H, Steinman RA, Drenning
SD, Gooding WE, Wentzel AL, Xi S and Grandis JR: STAT3 activation
abrogates growth factor dependence and contributes to head and neck
squamous cell carcinoma tumor growth in vivo. Cell Growth Differ.
13:355–362. 2002.PubMed/NCBI
|
|
6
|
Leeman RJ, Lui VW and Grandis JR: STAT3 as
a therapeutic target in head and neck cancer. Expert Opin Biol
Ther. 6:231–241. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bromberg J: Stat proteins and oncogenesis.
J Clin Invest. 109:1139–1142. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jewett A, Head C and Cacalano NA: Emerging
mechanisms of immunosuppression in oral cancers. J Dent Res.
85:1061–1073. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lai SY and Johnson FM: Defining the role
of the JAK-STAT pathway in head and neck and thoracic malignancies:
Implications for future therapeutic approaches. Drug Resist Updat.
13:67–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Naher L, Kiyoshima T, Kobayashi I, Wada H,
Nagata K, Fujiwara H, Ookuma YF, Ozeki S, Nakamura S and Sakai H:
STAT3 signal transduction through interleukin-22 in oral squamous
cell carcinoma. Int J Oncol. 41:1577–1586. 2012.PubMed/NCBI
|
|
11
|
Bromberg J and Darnell JE Jr: The role of
STATs in transcriptional control and their impact on cellular
function. Oncogene. 19:2468–2473. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Decker T and Kovarik P: Serine
phosphorylation of STATs. Oncogene. 19:2628–2637. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chung J, Uchida E, Grammer TC and Blenis
J: STAT3 serine phosphorylation by ERK-dependent and -independent
pathways negatively modulates its tyrosine phosphorylation. Mol
Cell Biol. 17:6508–6516. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reich NC and Liu L: Tracking STAT nuclear
traffic. Nat Rev Immunol. 6:602–612. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lim CP and Cao X: Serine phosphorylation
and negative regulation of Stat3 by JNK. J Biol Chem.
274:31055–31061. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Venkatasubbarao K, Choudary A and Freeman
JW: Farnesyl transferase inhibitor (R115777)-induced inhibition of
STAT3 (Tyr705) phosphorylation in human pancreatic cancer cell
lines require extracellular signal-regulated kinases. Cancer Res.
65:2861–2871. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wakahara R, Kunimoto H, Tanino K, Kojima
H, Inoue A, Shintaku H and Nakajima K: Phospho-Ser727 of STAT3
regulates STAT3 activity by enhancing dephosphorylation of
phospho-Tyr705 largely through TC45. Genes Cells. 17:132–145. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sakaguchi M, Oka M, Iwasaki T, Fukami Y
and Nishigori C: Role and regulation of STAT3 phosphorylation at
Ser727 in melanocytes and melanoma cells. J Invest Dermatol.
132:1877–1885. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aggarwal BB, Kunnumakkara AB, Harikumar
KB, Gupta SR, Tharakan ST, Koca C, Dey S and Sung B: Signal
transducer and activator of transcription-3, inflammation, and
cancer: How intimate is the relationship? Ann N Y Acad Sci.
1171:59–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maggioni D, Gaini R, Nicolini G, Tredici G
and Garavello W: MAPKs activation in head and neck squamous cell
carcinomas. Oncol Rev. 5:223–231. 2011. View Article : Google Scholar
|
|
22
|
Kim TW, Michniewicz M, Bergmann DC and
Wang ZY: Brassinosteroid regulates stomatal development by
GSK3-mediated inhibition of a MAPK pathway. Nature. 482:419–422.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim JY, An JM, Chung WY, Park KK, Hwang
JK, Kim DS, Seo SR and Seo JT: Xanthorrhizol induces apoptosis
through ROS-mediated MAPK activation in human oral squamous cell
carcinoma cells and inhibits DMBA-induced oral carcinogenesis in
hamsters. Phytother Res. 27:493–498. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li B, Lu L, Zhong M, Tan XX, Liu CY, Guo Y
and Yi X: Terbinafine inhibits KSR1 and suppresses Raf-MEK-ERK
signaling in oral squamous cell carcinoma cells. Neoplasma.
60:406–412. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boivin A, Hanot M, Malesys C, Maalouf M,
Rousson R, Rodriguez-Lafrasse C and Ardail D: Transient alteration
of cellular redox buffering before irradiation triggers apoptosis
in head and neck carcinoma stem and non-stem cells. PLoS One.
6:e145582011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gross ND, Boyle JO, Du B, Kekatpure VD,
Lantowski A, Thaler HT, Weksler BB, Subbaramaiah K and Dannenberg
AJ: Inhibition of Jun NH2-terminal kinases suppresses the growth of
experimental head and neck squamous cell carcinoma. Clin Cancer
Res. 13:5910–5917. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yunoki T, Kariya A, Kondo T, Hayashi A and
Tabuchi Y: The combination of silencing BAG3 and inhibition of the
JNK pathway enhances hyperthermia sensitivity in human oral
squamous cell carcinoma cells. Cancer Lett. 335:52–57. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ahmed ST, Mayer A, Ji JD and Ivashkiv LB:
Inhibition of IL-6 signaling by a p38-dependent pathway occurs in
the absence of new protein synthesis. J Leukoc Biol. 72:154–162.
2002.PubMed/NCBI
|
|
30
|
Jain N, Zhang T, Fong SL, Lim CP and Cao
X: Repression of Stat3 activity by activation of mitogen-activated
protein kinase (MAPK). Oncogene. 17:3157–3167. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Quadros MR, Peruzzi F, Kari C and Rodeck
U: Complex regulation of signal transducers and activators of
transcription 3 activation in normal and malignant keratinocytes.
Cancer Res. 64:3934–3939. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tkach M, Rosemblit C, Rivas MA, Proietti
CJ, Díaz Flaqué MC, Mercogliano MF, Beguelin W, Maronna E, Guzmán
P, Gercovich FG, et al: p42/p44 MAPK-mediated Stat3Ser727
phosphorylation is required for progestin-induced full activation
of Stat3 and breast cancer growth. Endocr Relat Cancer. 20:197–212.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sengupta TK, Talbot ES, Scherle PA and
Ivashkiv LB: Rapid inhibition of interleukin-6 signaling and Stat3
activation mediated by mitogen-activated protein kinases. Proc Natl
Acad Sci USA. 95:11107–11112. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kovarik P, Stoiber D, Eyers PA, Menghini
R, Neininger A, Gaestel M, Cohen P and Decker T: Stress-induced
phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated
protein kinase whereas IFN-gamma uses a different signaling
pathway. Proc Natl Acad Sci USA. 96:13956–13961. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xue P, Zhao Y, Liu Y, Yuan Q, Xiong C and
Ruan J: A novel compound RY10-4 induces apoptosis and inhibits
invasion via inhibiting STAT3 through ERK-, p38-dependent pathways
in human lung adenocarcinoma A549 cells. Chem Biol Interact.
209:25–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Song IS, Jun SY, Na HJ, Kim HT, Jung SY,
Ha GH, Park YH, Long LZ, Yu DY, Kim JM, et al: Inhibition of
MKK7-JNK by the TOR signaling pathway regulator-like protein
contributes to resistance of HCC cells to TRAIL-induced apoptosis.
Gastroenterology. 143:1341–1351. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tang RX, Kong FY, Fan BF, Liu XM, You HJ,
Zhang P and Zheng KY: HBx activates FasL and mediates HepG2 cell
apoptosis through MLK3-MKK7-JNKs signal module. World J
Gastroenterol. 18:1485–1495. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sau A, Filomeni G, Pezzola S, D'Aguanno S,
Tregno FP, Urbani A, Serra M, Pasello M, Picci P, Federici G and
Caccuri AM: Targeting GSTP1-1 induces JNK activation and leads to
apoptosis in cisplatin-sensitive and -resistant human osteosarcoma
cell lines. Mol Biosyst. 8:994–1006. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen B, Liu J, Chang Q, Beezhold K, Lu Y
and Chen F: JNK and STAT3 signaling pathways converge on
Akt-mediated phosphorylation of EZH2 in bronchial epithelial cells
induced by arsenic. Cell Cycle. 12:112–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen F: JNK-induced apoptosis,
compensatory growth, and cancer stem cells. Cancer Res. 72:379–386.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bubici C and Papa S: JNK signalling in
cancer: In need of new, smarter therapeutic targets. Br J
Pharmacol. 171:24–37. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Seki E, Brenner DA and Karin M: A liver
full of JNK: Signaling in regulation of cell function and disease
pathogenesis, and clinical approaches. Gastroenterology.
143:307–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Leventaki V, Drakos E, Medeiros LJ, Lim
MS, Elenitoba-Johnson KS, Claret FX and Rassidakis GZ: NPM-ALK
oncogenic kinase promotes cell-cycle progression through activation
of JNK/cJun signaling in anaplastic large-cell lymphoma. Blood.
110:1621–1630. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jia G, Kong R, Ma ZB, Han B, Wang YW, Pan
SH, Li YH and Sun B: The activation of c-Jun
NH2-terminal kinase is required for
dihydroartemisinin-induced autophagy in pancreatic cancer cells. J
Exp Clin Cancer Res. 33:82014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shi Y, Nikulenkov F, Zawacka-Pankau J, Li
H, Gabdoulline R, Xu J, et al: ROS-dependent activation of JNK
converts p53 into an efficient inhibitor of oncogenes leading to
robust apoptosis. Cell Death Differ. 21:612–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen YH, Hao LJ, Hung CP, Chen JW, Leu SF
and Huang BM: Apoptotic effect of cisplatin and cordycepin on OC3
human oral cancer cells. Chin J Integr Med. 20:624–632. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Noutomi T, Itoh M, Toyota H, Takada E and
Mizuguchi J: Tumor necrosis factor-related apoptosis-inducing
ligand induces apoptotic cell death through c-Jun NH2-terminal
kinase activation in squamous cell carcinoma cells. Oncol Rep.
22:1169–1172. 2009.PubMed/NCBI
|
|
48
|
Li Q, Song XM, Ji YY, Jiang H and Xu LG:
The dual mTORC1 and mTORC2 inhibitor AZD8055 inhibits head and neck
squamous cell carcinoma cell growth in vivo and in vitro. Biochem
Biophys Res Commun. 440:701–706. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim HJ, Chakravarti N, Oridate N, Choe C,
Claret FX and Lotan R: N-(4-hydroxyphenyl)retinamide-induced
apoptosis triggered by reactive oxygen species is mediated by
activation of MAPKs in head and neck squamous carcinoma cells.
Oncogene. 25:2785–2794. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sakai T, Banno Y, Kato Y, Nozawa Y and
Kawaguchi M: Pepsin-digested bovine lactoferrin induces apoptotic
cell death with JNK/SAPK activation in oral cancer cells. J
Pharmacol Sci. 98:41–48. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhao L, Yue P, Lonial S, Khuri FR and Sun
SY: The NEDD8-activating enzyme inhibitor, MLN4924, cooperates with
TRAIL to augment apoptosis through facilitating c-FLIP degradation
in head and neck cancer cells. Mol Cancer Ther. 10:2415–2425. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang S, Wang XL, Gan YH and Li SL:
Activation of c-Jun N-terminal kinase is required for
mevastatin-induced apoptosis of salivary adenoid cystic carcinoma
cells. Anticancer Drugs. 21:678–686. 2010.PubMed/NCBI
|
|
53
|
Hour MJ, Lee KH, Chen TL, Lee KT, Zhao Y
and Lee HZ: Molecular modelling, synthesis, cytotoxicity and
anti-tumour mechanisms of 2-aryl-6-substituted quinazolinones as
dual-targeted anti-cancer agents. Br J Pharmacol. 169:1574–1586.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen HM, Liu CM, Yang H, Chou HY, Chiang
CP and Kuo MY: 5-aminolevulinic acid induce apoptosis via NF-κB/JNK
pathway in human oral cancer Ca9-22 cells. J Oral Pathol Med.
40:483–489. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bhattarai G, Lee YH, Lee NH, Lee IK, Yun
BS, Hwang PH and Yi HK: Fomitoside-K from Fomitopsis nigra
induces apoptosis of human oral squamous cell carcinomas (YD-10B)
via mitochondrial signaling pathway. Biol Pharm Bull. 35:1711–1719.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li C and Johnson DE: Bortezomib induces
autophagy in head and neck squamous cell carcinoma cells via JNK
activation. Cancer Lett. 314:102–107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Schramek D, Kotsinas A, Meixner A, Wada T,
Elling U, Pospisilik JA, Neely GG, Zwick RH, Sigl V, Forni G, et
al: The stress kinase MKK7 couples oncogenic stress to p53
stability and tumor suppression. Nat Genet. 43:212–219. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dai Y, Guzman ML, Chen S, Wang L, Yeung
SK, Pei XY, Dent P, Jordan CT and Grant S: The NF (nuclear
factor)-κB inhibitor parthenolide interacts with histone
deacetylase inhibitors to induce MKK7/JNK1-dependent apoptosis in
human acute myeloid leukaemia cells. Br J Haematol. 151:70–83.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lui VW, Peyser ND, Ng PK, Hritz J, Zeng Y,
Lu Y, Li H, Wang L, Gilbert BR, General IJ, et al: Frequent
mutation of receptor protein tyrosine phosphatases provides a
mechanism for STAT3 hyperactivation in head and neck cancer. Proc
Natl Acad Sci USA. 111:1114–1119. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gkouveris I, Nikitakis N, Karanikou M,
Rassidakis G and Sklavounou A: Erk1/2 activation and modulation of
STAT3 signaling in oral cancer. Oncol Rep. 32:2175–2182.
2014.PubMed/NCBI
|
|
61
|
Kim JH, Lee SC, Ro J, Kang HS, Kim HS and
Yoon S: Jnk signaling pathway-mediated regulation of Stat3
activation is linked to the development of doxorubicin resistance
in cancer cell lines. Biochem Pharmacol. 79:373–380. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Guo W, Wu S, Wang L, Wei X, Liu X, Wang J,
Lu Z, Hollingshead M and Fang B: Antitumor activity of a novel
oncrasin analogue is mediated by JNK activation and STAT3
inhibition. PLoS One. 6:e284872011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu J, Chen B, Lu Y, Guan Y and Chen F:
JNK-dependent Stat3 phosphorylation contributes to Akt activation
in response to arsenic exposure. Toxicol Sci. 129:363–371. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Shirakawa T, Kawazoe Y, Tsujikawa T, Jung
D, Sato S and Uesugi M: Deactivation of STAT6 through serine 707
phosphorylation by JNK. J Biol Chem. 286:4003–4010. 2011.
View Article : Google Scholar : PubMed/NCBI
|