Introduction
Parathyroid hormone is the principal physiologic
regulator of calcium homeostasis. Hyperparathyroidism is a result
of increased activity of the parathyroid glands, either from an
intrinsic change altering excretion of parathyroid hormone (primary
hyperparathyroidism, pHPT) or from an extrinsic change affecting
calcium homeostasis stimulating production of parathyroid hormone
(secondary hyperparathyroidism, sHPT) (1). Clinically, hyperparathyroidism leads to
skeletal and renal complications in addition to an impairment in
quality of life (2–4). Prolonged oversecretion of parathyroid
hormone is accompanied by histologically abnormal parathyroid
glands that are typically enlarged and hypercellular with decreased
stromal fat (5,6).
Parathyroid glands are derived from the third and
fourth pharyngeal pouches and are endodermal in origin (7). Generally, pHPT is caused by a single
adenoma (80–85%) or four-gland hyperplasia (10–15%). Parathyroid
carcinomas are rare and account for <1% of pHPT cases. By
contrast, four-gland hyperplasia is the rule in sHPT, ranging from
diffuse chief cell hyperplasia to nodular formations. Significantly
increased proliferation and apoptosis were both observed in pHPT
and sHPT in comparison with normal parathyroids (8). Nonetheless, different genetic
alterations are implicated in the development of different types of
hyperparathyroidism (9,10).
Although pHPT mainly occurs as a sporadic disease,
it may be part of a hereditary syndrome (e.g., multiple endocrine
neoplasia types 1 and 2A). On the other hand, chronic renal failure
is the primary cause of sHPT. The phenomenon that different
pathophysiology and genetic alterations of hyperparathyroidism lead
to partially overlapping histological phenotype is notable.
However, to the best of our knowledge, there have been no reports
comparing transcriptional alterations in different types of
hyperparathyroidism. In the present study, the gene expression
differences between pHPT and sHPT were analyzed and molecular
pathways that are dysregulated in different contexts of
hyperparathyroidism were identified.
Materials and methods
Patients and tissue samples
The present study was approved by the Institutional
Review Board of MacKay Memorial Hospital (Taipei, Taiwan; approval
no. 11MMHIS194), and all patients gave written informed consent.
Parathyroid samples were obtained from patients undergoing surgical
treatment of hyperparathyroidism at MacKay Memorial Hospital,
Taipei, Taiwan (11). All samples
were snap frozen in liquid nitrogen within 10 min of resection and
stored at −80°C. Diagnosis was histologically confirmed by a senior
endocrine pathologist using hematoxylin-eosin staining.
RNA extraction
Total RNA was extracted from homogenized frozen
tissue samples using TRIzol reagent (Life Technologies,
ThermoFisher Scientific, Inc., Waltham, MA, USA) and purified using
the RNeasy Plus Mini Kit (Qiagen, Valencia, CA, USA) according to
the manufacturer's recommendations (12). Sample purity was confirmed by
measuring ratios of sample absorbance at 260 and 280 nm (ranging
from 1.8 to 2.2). The quality of RNA was determined before labeling
using the 2,100 Bioanalyzer (Agilent Technologies, Santa Clara, CA,
USA).
Microarray hybridizations
A total of 200 µg of total RNA was amplified by a
Low Input Quick Amp Labeling Kit (Agilent Technologies) and labeled
with Cy3 during the in vitro transcription process.
Cy3-labled cRNA (600 µg) was fragmented to an average size of
~50–100 nucleotides by incubation with fragmentation buffer at 60°C
for 30 min. Fragmented labeled cRNA was then pooled and hybridized
to Agilent SurePrint G3 Human Gene Expression v2 8×60K Microarray
at 65°C for 17 h. After washing and drying, microarrays were
scanned with an Agilent microarray scanner at 535 nm for Cy3.
Scanned images were analyzed by Feature Extraction software version
10.5.1.1 (Agilent Technologies) to quantify signal and background
intensity.
Data analysis and comparison with
public microarray data
The microarray data were subjected to linear
normalization to allow comparison between arrays. Hierarchical
cluster analysis was performed with Cluster 3.0 (bonsai.hgc.jp/~mdehoon/software/cluster/software.htm),
and heat maps were constructed with Java Treeview software
(www.princeton.edu/~abarysh/treeview/). A public
microarray dataset (GSE10317) was retrieved from the National
Center for Biotechnology Information Gene Expression Omnibus
(www.ncbi.nlm.nih.gov/geo/). GSE10317
comprises gene expression data of a case of pHPT (13). Gene expression levels of the
parathyroid tumor and normal parathyroid tissue were analyzed using
Affymetrix Human Genome U133 Plus 2.0 Arrays. (Affymetrix, Inc.,
Santa Clara, CA, USA)
t statistics were used to estimate the
significance of expression difference between pHPT and sHPT. R
software version 3.0.2 (www.r-project.org) was used for Bayes-regularized
t tests. Associated P-values were adjusted for multiple
testing by controlling for a false discovery rate <5% using the
Benjamini-Hochberg procedure (14),
and adjusted P<0.05 was considered to indicate a statistically
significant difference. For the GSE10317 data, probes with a
differential expression of at least 2-fold were considered to be
significant. A meta-signature that characterized the intersection
of differentially expressed genes from both datasets were
constructed. Genes that demonstrated significantly altered
expression changes in the same direction for both dataset were
considered to be pHPT-associated. The intersection of
differentially expressed genes of the two dataset in the opposite
direction was considered to be sHPT-associated.
Gene Ontology (GO) terms and Kyoto Encyclopedia of
Genes and Genomes (KEGG; www.genome.jp/kegg/) pathway analyses were performed
to annotate the biological functions and pathways in which the
aberrantly expressed genes of pHPT and sHPT were involved.
Results
Microarray gene expression analyses were performed
in parathyroid tissues from 2 pHPT and 3 sHPT patients. The 2 pHPT
patients were female and had single parathyroid adenoma. All sHPT
patients including 2 women and 1 man had four-gland nodular
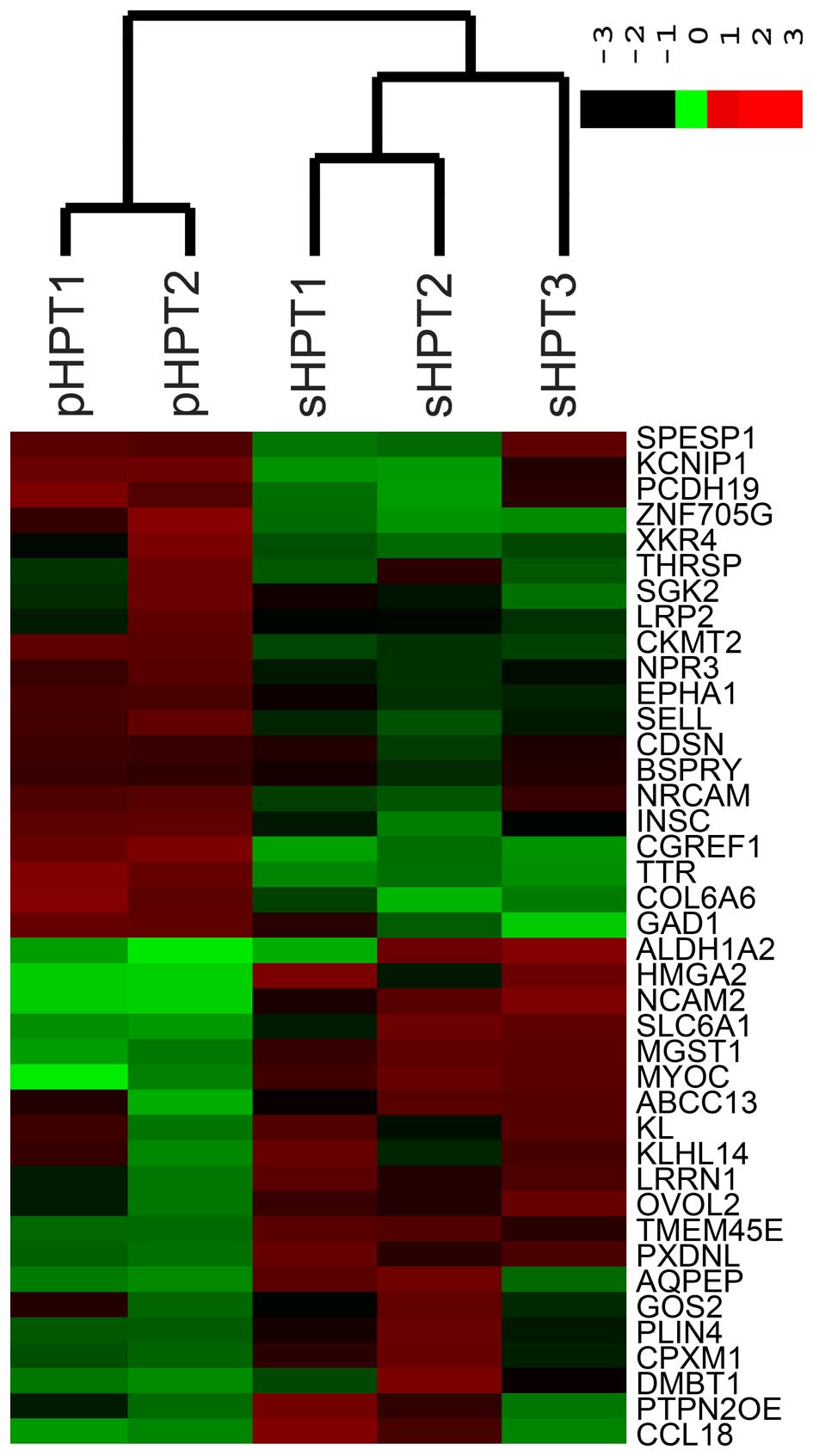
hyperplasia. Unsupervised hierarchical clustering analysis for the
expression of all genes revealed two natural subgroups containing
pHPT and sHPT, respectively.
A meta-signature was constructed to represent an
intersection of two sets of differential expression profile. Based
on predefined criteria, 339 genes were upregulated and 261 genes
were downregulated in pHPT. The ten most common leading-edge genes
are summarized in Tables I and
II. A total of 218 genes were
upregulated and 367 genes were downregulated in sHPT. The top
upregulated and downregulated genes are shown in Tables III and IV, respectively. A heat map generated from
the most differently expressed genes is presented in Fig. 1.
 | Table I.Upregulated genes in primary
hyperparathyroidism. |
Table I.
Upregulated genes in primary
hyperparathyroidism.
| Probe | Gene | Accession # | Description |
|---|
| A_23_P130333 | TTR | NM_000371 | Homo sapiens
transthyretin |
| A_33_P3814721 | INSC | NM_001031853 | Homo sapiens
inscuteable homolog (Drosophila), transcript variant 1 |
| A_32_P224525 | COL6A6 | NM_001102608 | Homo sapiens
collagen, type VI, alpha 6 |
| A_33_P3400273 | SELL | NM_000655 | Homo sapiens
selectin L, transcript variant 1 |
| A_24_P252364 | NRCAM | NM_001037132 | Homo sapiens
neuronal cell adhesion molecule, transcript variant 1 |
| A_23_P157333 | EPHA1 | NM_005232 | Homo sapiens
EPH receptor A1 |
| A_23_P350396 | CDSN | NM_001264 | Homo sapiens
corneodesmosin |
| A_33_P3244728 | LRP2 | NM_004525 | Homo sapiens
low density lipoprotein receptor-related protein 2 |
| A_23_P374689 | GAD1 | NM_000817 | Homo sapiens
glutamate decarboxylase 1 (brain, 67kDa), transcript variant
GAD67 |
| A_23_P71946 | BSPRY | NM_017688 | Homo sapiens
B-box and SPRY domain containing |
| Table II.Downregulated genes in primary
hyperparathyroidism. |
Table II.
Downregulated genes in primary
hyperparathyroidism.
| Probe | Gene | Accession # | Description |
|---|
| A_23_P36658 | MGST1 | NM_145791 | Homo sapiens
microsomal glutathione S-transferase 1, transcript variant 3 |
| A_33_P3300253 | PTPN20B | NM_001042357 | Homo sapiens
protein tyrosine phosphatase, non-receptor type 20B, transcript
variant 1 |
| A_23_P74609 | G0S2 | NM_015714 | Homo sapiens
G0/G1 switch 2 |
| A_33_P3251522 | AQPEP | NM_173800 | Homo sapiens
laeverin |
| A_33_P3400763 | PLIN4 | NM_001080400 | Homo sapiens
perilipin 4 |
| A_23_P23783 | MYOC | NM_000261 | Homo sapiens
myocilin, trabecular meshwork inducible glucocorticoid
response |
| A_21_P0000096 | CPXM1 | NM_019609 | Homo sapiens
carboxypeptidase X (M14 family), member 1, transcript variant
1 |
| A_23_P258310 | PXDNL | NM_144651 | Homo sapiens
peroxidasin homolog (Drosophila)-like |
| A_23_P55270 | CCL18 | NM_002988 | Homo sapiens
chemokine (C-C motif) ligand 18 (pulmonary and
activation-regulated) |
| A_23_P86599 | DMBT1 | NM_007329 | Homo sapiens
deleted in malignant brain tumors 1, transcript variant 2 |
| Table III.Upregulated genes in secondary
hyperparathyroidism. |
Table III.
Upregulated genes in secondary
hyperparathyroidism.
| Probe | Gene | Accession # | Description |
|---|
| A_24_P268685 | SLC6A1 | NM_003042 | Homo sapiens
solute carrier family 6 (neurotransmitter transporter), member
1 |
| A_23_P503064 | KL | NM_004795 | Homo sapiens
klotho |
| A_24_P73577 | ALDH1A2 | NM_170697 | Homo sapiens
aldehyde dehydrogenase 1 family, member A2, transcript variant
3 |
| A_23_P1682 | TMEM45B | NM_138788 | Homo sapiens
transmembrane protein 45B |
| A_23_P95930 | HMGA2 | NM_003483 | Homo sapiens
high mobility group AT-hook 2, transcript variant 1 |
| A_24_P240187 | LRRN1 | NM_020873 | Homo sapiens
leucine rich repeat neuronal 1 |
| A_23_P143348 | OVOL2 | NM_021220 | Homo sapiens
ovo-like zinc finger 2 |
| A_32_P199429 | NCAM2 | NM_004540 | Homo sapiens
neural cell adhesion molecule 2 |
| A_23_P370830 | KLHL14 | NM_020805 | Homo sapiens
kelch-like family member 14 |
| A_23_P99253 | LIN7A | NM_004664 | Homo sapiens
lin-7 homolog A (C. elegans) |
| Table IV.Downregulated genes in secondary
hyperparathyroidism. |
Table IV.
Downregulated genes in secondary
hyperparathyroidism.
| Probe | Gene | Accession # | Description |
|---|
| A_23_P144778 | CKMT2 | NM_001825 | Homo sapiens
creatine kinase, mitochondrial 2 (sarcomeric), transcript variant
1 |
| A_33_P3319248 | ZNF705G | NM_001164457 | Homo sapiens
zinc finger protein 705G |
| A_23_P131801 | SGK2 | NM_170693 | Homo sapiens
serum/glucocorticoid regulated kinase 2, transcript variant 1 |
| A_23_P30554 | KCNIP1 | NM_001034837 | Homo sapiens
Kv channel interacting protein 1, transcript variant 1 |
| A_23_P129085 | SPESP1 | NM_145658 | Homo sapiens
sperm equatorial segment protein 1 |
| A_23_P363954 | THRSP | NM_003251 | Homo sapiens
thyroid hormone responsive |
| A_32_P134007 | XKR4 | NM_052898 | Homo sapiens
XK, Kell blood group complex subunit-related family, member 4 |
| A_33_P3423230 | PCDH19 | NM_001184880 | Homo sapiens
protocadherin 19, transcript variant 3 |
| A_33_P3227793 | CGREF1 | NM_006569 | Homo sapiens
cell growth regulator with EF-hand domain 1, transcript variant
1 |
| A_23_P58676 | NPR3 | NM_001204375 | Homo sapiens
natriuretic peptide receptor C/guanylate cyclase C
(atrionatriuretic peptide receptor C), transcript variant 1 |
The gene function annotations were evaluated
according to the GO and KEGG pathway databases. For genes with
differential expression in pHPT, involved molecular functions in
order were: Fatty-acid ligase activity, calcium ion binding, alkali
metal ion binding, ligase activity forming carbon-sulfur bonds, and
symporter activity. Involved biological processes were: Ion
transport, cell adhesion, biological adhesion, cation transport,
and metal ion transport. Dysregulated pathways in pHPT are
presented in Table V.
| Table V.Pathway analysis of genes with
differential expression in primary hyperparathyroidism. |
Table V.
Pathway analysis of genes with
differential expression in primary hyperparathyroidism.
| Pathway | Count | P-value | Genes |
|---|
| Cell adhesion
molecules | 9 | 0.022 | HLA-DQB1, NRCAM,
ALCAM, SDC1, NRXN3, CD40LG, SELL, ITGA8, ITGA4 |
| PPAR signaling
pathway | 6 | 0.034 | HMGCS2, APOA5,
APOC3, SLC27A6, ACSL3, SLC27A2 |
| Neuroactive
ligand-receptor interaction | 13 | 0.035 | CSH1, S1PR3,
GABRG2, PTGER3, PRLR, RXFP1, HTR7, GRIN2A, TAAR1, ADRA1A, GABBR2,
PTGFR, GCGR |
Molecular interaction and networks contributing to
sHPT were identified. Molecular functions involved in sHPT were:
Retinal dehydrogenase activity, steroid dehydrogenase activity
acting on the CH-OH group of donors NAD or NADP as acceptor,
carbohydrate binding, sulfotransferase activity, and
acetylgalactosaminyltransferase activity. Involved biological
processes were: Cell adhesion, biological adhesion, secondary
metabolic process, skeletal system development, and regulation of
nucleotide metabolic process. Dysregulated pathways in sHPT are
listed in Table VI.
| Table VI.Pathway analysis of genes with
differential expression in secondary hyperparathyroidism. |
Table VI.
Pathway analysis of genes with
differential expression in secondary hyperparathyroidism.
| Pathway | Count | P-value | Genes |
|---|
| Tryptophan
metabolism | 6 | 0.003 | TDO2, CYP1B1, MAOA,
AOX1, ALDH2, INMT |
| Tight junction | 10 | 0.007 | PRKCQ, INADL,
MYH11, ACTN1, CLDN10, MYH7, CLDN11, CTNNA3, CTNNA2, MYL9 |
| Renin-angiotensin
system | 4 | 0.009 | ACE2, MAS1, ANPEP,
CTSG |
| Steroid hormone
biosynthesis | 5 | 0.030 | AKR1C3, CYP3A5,
CYP1B1, AKR1C4, CYP19A1 |
| O-glycan
biosynthesis | 4 | 0.041 | GALNT3, GCNT1,
GALNT13, GALNT14 |
Discussion
The exact mechanism underlying the development of
pHPT remains poorly understood (15).
In patients with multiglandular pHPT, independent genetic events
may be present in separate glands within the same individual
(16). Previous studies have
indicated that parathyroid adenomas typically harbor few somatic
variants (17,18). Mutations in the MEN1 tumor
suppressor gene and alterations in the CCND1 (cyclin
D1/PRAD1) oncogene represent the major driver in sporadic
parathyroid tumorigenesis. Cell cycle regulators (including
CDC73 aka HRPT2), growth factors, apoptosis-inducing
ligands, death receptors, and other transmitter substances have
also been implicated in the pathogenesis (19,20).
Although there is an abundance of data examining the gene
expression profiles of parathyroid adenoma, few studies have been
performed in comparing gene expressions of pHPT with sHPT
tissues.
In a previous study examining clonality in pHPT and
sHPT, 7/8 pHPT glands (6 adenomas and 2 hyperplasias) exhibited
monoclonal proliferation (21). One
parathyroid adenoma demonstrated a polyclonal pattern. In an sHPT
patient, one of the 3 hyperplastic lesions was monoclonal and the
other 2 lesions were polyclonal. This finding underscores the
complexity of pHPT pathogenesis. The results from later studies
also attest that pHPT can arise by clonal and polyclonal mechanisms
(22). This is different from sHPT in
which polyclonal growth transforms to monoclonal proliferation
during disease progression (23).
Nonetheless, there remains the possibility that similar mechanisms
regulate parathyroid cell growth in both entities. The current
study represents the first effort to compare the transcriptional
profiles between pHPT and sHPT using modern microarray
technology.
One of the limitations of the study design is the
lack of transcriptional data from normal parathyroid controls. To
overcome this ethical constraint, additional data were extracted
from a publicly available database and combined to form a
meta-signature. Regardless, the aberrantly regulated pathways that
the present study identified in different types of
hyperparathyroidism provide critical insights into the differences
in pathophysiology. For instance, cell adhesion molecules were
upregulated in pHPT but downregulated in sHPT. It is well known
that nodular hyperplasia in patients with chronic kidney disease is
associated with progressive downregulation of calcium-sensing
receptor and vitamin D receptor (6).
Activation of calcium-sensing receptor may potentiate cell adhesion
by promoting integrin binding to a fibronectin-rich matrix
(24). It is therefore reasonable
that downregulated tight junction and cell adhesion were among the
dysregulated pathways in sHPT. Conversely, expression of some
adhesion molecules including selectin L and neuronal cell adhesion
molecule were significantly increased in pHPT (Table I).
It was also noted that renin-angiotensin system was
among the dysregulated pathways in sHPT. Increasing evidence links
the renin-angiotensin-aldosterone system to calcium regulatory
systems (25). A high calcium diet
was demonstrated to downregulate angiotensin-converting enzyme of
the kidney in experimental renal failure (26). In the present study, it was
demonstrated that the expression of angiotensin I converting enzyme
2 (ACE2) was downregulated in sHPT. Recently, it has been
shown that angiotensin II infusion acutely stimulated the secretion
of parathyroid hormone in a dose-dependent manner (27). It is possible that downregulation of
angiotensin-converting enzymes in sHPT results from a negative
feedback mechanism to reduce further stimulation from angiotensin
II. Renin-angiotensin system may be a potential target for
therapeutic intervention in hyperparathyroidism.
The result of the present study point to alterations
in peroxisome proliferator-activated receptor pathway in addition
to fatty acid and amino acid metabolism in pHPT and sHPT. Metabolic
aberrations have already been established as serving essential
roles in imaging studies of hyperparathyroidism. Uptake and
accumulation of technetium-99m sestamibi in mitochondria-rich
oxyphil cells are employed as the basis of scintigraphic detection
of hyperfunctional parathyroids (28). In addition, various tracers for
positron emission tomographic scan have been exploited in
localization of abnormal parathyroid glands (29,30).
Metabolic reprogramming in neoplasms has recently been indicated as
another general hallmark of cancer. Nonetheless, metabolic
rearrangements in hyperparathyroidism remain a virtually untapped
area of investigation. Elucidating the complex interplay between
calcium homeostasis and parathyroid metabolic activity is an
exciting challenge for future research.
In conclusion, the present study demonstrates that
different pathophysiology led to differential gene profiling in
hyperparathyroidism. Systemic analysis and annotated pathway
resources were used to identify several pathways that are
dysregulated in hyperparathyroidism which may be targets of
interest.
Acknowledgements
The present study was supported by a grant (grant
no. MOST-103-2314-B-195-015-MY3) from the Ministry of Science and
Technology of Taiwan. The funder had no role in study design, data
collection and analysis, decision to publish, or preparation of the
manuscript. Parts of this paper were presented at the 17th European
Congress of Endocrinology (Dublin, Ireland, May 2015).
References
|
1
|
Fraser WD: Hyperparathyroidism. Lancet.
374:145–158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bilezikian JP, Brandi ML, Eastell R,
Silverberg SJ, Udelsman R, Marcocci C and Potts JT Jr: Guidelines
for the management of asymptomatic primary hyperparathyroidism:
Summary statement from the fourth international workshop. J Clin
Endocrinol Metab. 99:3561–3569. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheng SP, Lee JJ, Liu TP, Yang PS, Liu SC,
Hsu YC and Liu CL: Quality of life after surgery or surveillance
for asymptomatic primary hyperparathyroidism: A meta-analysis of
randomized controlled trials. Medicine (Baltimore). 94:e9312015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheng SP, Lee JJ, Liu TP, Yang TL, Chen
HH, Wu CJ and Liu CL: Parathyroidectomy improves symptomatology and
quality of life in patients with secondary hyperparathyroidism.
Surgery. 155:320–328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Elliott DD, Monroe DP and Perrier ND:
Parathyroid histopathology: Is it of any value today? J Am Coll
Surg. 203:758–765. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Drueke T, Martin D and Rodriguez M: Can
calcimimetics inhibit parathyroid hyperplasia? Evidence from
preclinical studies. Nephrol Dial Transplant. 22:1828–1839. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bingham EL, Cheng SP, Woods Ignatoski KM
and Doherty GM: Differentiation of human embryonic stem cells to a
parathyroid-like phenotype. Stem Cells Dev. 18:1071–1080. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thomopoulou GE, Tseleni-Balafouta S,
Lazaris AC, Koutselini H, Kavantzas N and Davaris PS:
Immunohistochemical detection of cell cycle regulators, Fhit
protein and apoptotic cells in parathyroid lesions. Eur J
Endocrinol. 148:81–87. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shan L, Nakamura Y, Nakamura M, Yokoi T
and Kakudo K: Genetic alterations in primary and secondary
hyperparathyroidism. Pathol Int. 48:569–574. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Westin G, Björklund P and Akerström G:
Molecular genetics of parathyroid disease. World J Surg.
33:2224–2233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheng SP, Lee JJ, Liu TP, Chen HH, Wu CJ
and Liu CL: Aluminum overload hampers symptom improvement following
parathyroidectomy for secondary hyperparathyroidism. World J Surg.
38:2838–2844. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cheng SP, Yin PH, Chang YC, Lee CH, Huang
SY and Chi CW: Differential roles of leptin in regulating cell
migration in thyroid cancer cells. Oncol Rep. 23:1721–1727.
2010.PubMed/NCBI
|
|
13
|
Au AY, McDonald K, Gill A, Sywak M,
Diamond T, Conigrave AD and Clifton-Bligh RJ: PTH mutation with
primary hyperparathyroidism and undetectable intact PTH. N Engl J
Med. 359:1184–1186. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Benjamini Y and Yekutieli D: False
discovery rate-adjusted multiple confidence intervals for selected
parameters. J Am Stat Assoc. 100:71–81. 2005. View Article : Google Scholar
|
|
15
|
Cheng SP, Doherty GM, Chang YC and Liu CL:
Leptin: The link between overweight and primary
hyperparathyroidism? Med Hypotheses. 76:94–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dwight T, Nelson AE, Theodosopoulos G,
Richardson AL, Learoyd DL, Philips J, Delbridge L, Zedenius J, Teh
BT, Larsson C, et al: Independent genetic events associated with
the development of multiple parathyroid tumors in patients with
primary hyperparathyroidism. Am J Pathol. 161:1299–1306. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Newey PJ, Nesbit MA, Rimmer AJ, Attar M,
Head RT, Christie PT, Gorvin CM, Stechman M, Gregory L, Mihai R, et
al: Whole-exome sequencing studies of nonhereditary (sporadic)
parathyroid adenomas. J Clin Endocrinol Metab. 97:E1995–E2005.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Costa-Guda J and Arnold A: Genetic and
epigenetic changes in sporadic endocrine tumors: Parathyroid
tumors. Mol Cell Endocrinol. 386:46–54. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee JY, Kim SY, Mo EY, Kim ES, Han JH,
Maeng LS, Lee AH, Eun JW, Nam SW and Moon SD: Upregulation of FGFR1
expression is associated with parathyroid carcinogenesis in HPT-JT
syndrome due to an HRPT2 splicing mutation. Int J Oncol.
45:641–650. 2014.PubMed/NCBI
|
|
20
|
Segiet OA, Deska M, Michalski M,
Gawrychowski J and Wojnicz R: Molecular profiling in primary
hyperparathyroidism. Head Neck. 37:299–307. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shan L, Nakamura M, Nakamura Y, Inoue D,
Morimoto S, Yokoi T and Kakudo K: Comparative analysis of clonality
and pathology in primary and secondary hyperparathyroidism.
Virchows Arch. 430:247–251. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi Y, Hogue J, Dixit D, Koh J and Olson
JA Jr: Functional and genetic studies of isolated cells from
parathyroid tumors reveal the complex pathogenesis of parathyroid
neoplasia. Proc Natl Acad Sci USA. 111:3092–3097. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tominaga Y, Kohara S, Namii Y, Nagasaka T,
Haba T, Uchida K, Numano M, Tanaka Y and Takagi H: Clonal analysis
of nodular parathyroid hyperplasia in renal hyperparathyroidism.
World J Surg. 20:744–750; discussion 750–752. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tharmalingam S, Daulat AM, Antflick JE,
Ahmed SM, Nemeth EF, Angers S, Conigrave AD and Hampson DR:
Calcium-sensing receptor modulates cell adhesion and migration via
integrins. J Biol Chem. 286:40922–40933. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tomaschitz A, Ritz E, Pieske B, Rus-Machan
J, Kienreich K, Verheyen N, Gaksch M, Grübler M, Fahrleitner-Pammer
A, Mrak P, et al: Aldosterone and parathyroid hormone interactions
as mediators of metabolic and cardiovascular disease. Metabolism.
63:20–31. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Porsti I, Fan M, Kööbi P, Jolma P,
Kalliovalkama J, Vehmas TI, Helin H, Holthofer H, Mervaala E, Nyman
T and Tikkanen I: High calcium diet down-regulates kidney
angiotensin-converting enzyme in experimental renal failure. Kidney
Int. 66:2155–2166. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brown JM, Williams JS, Luther JM, Garg R,
Garza AE, Pojoga LH, Ruan DT, Williams GH, Adler GK and Vaidya A:
Human interventions to characterize novel relationships between the
renin-angiotensin-aldosterone system and parathyroid hormone.
Hypertension. 63:273–280. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pons F, Torregrosa JV and Fuster D:
Biological factors influencing parathyroid localization. Nucl Med
Commun. 24:121–124. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang TS, Lee JJ, Lin YC and Cheng SP:
Fluorodeoxyglucose-avid parathyroid adenoma mimicking thyroid
incidentaloma. ANZ J Surg. 80:763–764. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hindié E, Zanotti-Fregonara P, Tabarin A,
Rubello D, Morelec I, Wagner T, Henry JF and Taïeb D: The role of
radionuclide imaging in the surgical management of primary
hyperparathyroidism. J Nucl Med. 56:737–744. 2015. View Article : Google Scholar : PubMed/NCBI
|