Introduction
Phosphatase and tensin homolog (PTEN), a lipid
phosphatase, is one of the frequently mutated or deleted tumor
suppressor genes during cancer progression (1). In particular, it is deleted in >50%
of prostate cancer cases. PTEN is a well-known tumor suppressor
protein that regulates a number of cellular processes, including
cell death and proliferation, through the phosphoinositide
3-kinase/protein kinase B/mammalian target of rapamycin
(PI3K/AKT/mTOR) pathway (2,3). However, PTEN function is
phosphatase-independent, as well as being dependent upon events in
the nucleus (4,5). It has been found that inactivated PTEN
can switch oncogenes to oncosuppressors in certain mouse models,
which suggests a dependence on the genetic context. In other words,
PTEN inactivation may change the progression of disease from an
aggressive state to safe and vice versa. In the present review,
examples of altered function in the p53, enhancer of zeste homolog
2 (EZH2), alternative reading frame 2 (ARF), zinc finger and BTB
domain-containing 7A (ZBTB7A), p27 and breast cancer 1, early onset
(BRCA1) proteins by PTEN loss/AKT activation signaling will be
discussed.
PTEN loss switches MDM2-dependent p53
downregulation through ARF
The first and classical example of PTEN inactivation
as a switch is the modulation of tumor suppressor p53 (6). p53 and PTEN control cell death and
proliferation, and they are often expressed simultaneously in
various types of tumor (7,8). Due to its short half-life, the
suppressor function of p53 heavily relies on this stabilization
(9). In normal cells, p53 levels are
kept at insignificant levels by ubiquitin-mediated proteolysis.
PTEN and p53 may form a positive feedback loop. In detail, p53 can
upregulate PTEN by binding to the PTEN promoter, thereby activating
PTEN transcription (8).
Mechanistically, PTEN inhibits AKT-mediated MDM2 phosphorylation to
prevent MDM2 from translocation into the nucleus to degrade p53
(10). Thus, wild-type PTEN can
stabilize p53. It can be stated that AKT activation with PTEN
deletion may result in the rapid degradation of p53, leading to
further PTEN-dependent tumorigenesis (11).
However, in mouse embryonic fibroblasts (MEFs) and a
mouse model of prostate cancer, it has been found that PTEN loss
does not induce p53 degradation or instability (12). On the contrary, Pten loss can also
upregulate p53, which leads to Pten loss-induced cellular
senescence through p53, and to a certain degree, restricted tumor
growth (13). Mechanistically, PTEN
loss induces ARF elevation and elevated ARF may degrade MDM2,
thereby stabilizing p53 (14,15). Indeed, in MEFs, acute loss of Pten did
not decrease p53 stability, but actually increased stability, with
co-upregulation of p19Arf (ARF in mouse) (13). Thus, PTEN loss can also induce
elevation of p53 as a molecular switch by regulating p53 stability
through the ARF-MDM2 pathway. However, the mechanism via which PTEN
can induce ARF upregulation remains unclear. Moreover, the method
by which ARF and AKT compete for the regulation of MDM2 for the
stability of p53 also requires further investigation.
PTEN loss/AKT activation switches EZH2 from
a tumor suppressor to an oncogene
Epigenetic regulators are a relatively new class of
therapeutic target for cancer treatment. The enhancer of zeste
homolog (EZH2), a catalyst of polycomb repressive complex 2 (PRC2),
is a well-known epigenetic regulator that plays oncosuppressive
roles by silencing gene expression via its histone
methyltransferase activity (16).
Moreover, it silences transcription by trimethylating H3 histone on
lysine 27 (16). Nevertheless, EZH2
expression correlates to the progression of prostate cancer,
particularly to that of castration-resistant prostate cancer (CRPC)
(17).
A recent study has reported that EZH2 can act as an
oncogenic protein. The oncogenic function of EZH2 is independent of
its role as a transcriptional repressor in cases of CRPC (17). This functional change requires
phosphorylation of EZH2 by AKT, as well as an intact
methyltransferase domain. Study results have shown that EZH2
phosphorylation at serine 21 can easily change its function from a
PRC2 catalyst to an androgen receptor transcriptional co-activator,
as it is mediated by the PI3K/AKT pathway directly and indirectly
(17). These findings indicate that
the oncogenic activity of EZH2 in CRPC is not dependent on its
polycomb-related oncosuppressive function. Furthermore, the
findings indicate that it is possible to develop inhibitors that
may specifically target the activation of EZH2 without altering its
PRC2 repressive function. Recent studies have suggested that PTEN
loss correlates with EZH2 elevation in the invasion and metastasis
of gallbladder adenocarcinoma (18).
Moreover, PTEN loss downregulates p16 expression to decrease
cellular senescence, thereby increasing cell proliferation, an
oncogenic process. This process is induced through the EZH2
elevation-mediated methylation change of the p16 promoter. In other
words, PTEN loss may upregulate EZH2 for the inhibition of cell
cycle arrest (19). Thus, PTEN
loss/AKT activation may switch EZH2 function.
Pten loss switches ZbTb7a from an oncogene
to a tumor suppressor
ZBTB7A (also known as Pokemon, LRF, OCZF and FBI-1)
is a part of the POK (Kruppel and POZ/BTB) transcription factor
family, and plays a major role during oncogenesis and cell
differentiation (20). ZBTB7A was
previously believed to be a proto-oncogene in various cancer types,
including prostate cancer (21,22).
ZBTB7A is highly expressed in Hodgkin's lymphoma, as it represses
the expression of AFR, a tumor suppressor (23). Nevertheless, a recent study conducted
by Wang et al showed that it may have oncosuppressive
functions in the case of prostate cancer upon PTEN loss (24). The prostate-specific transgene of
Zbtb7a has no association with tumorigenesis, which is
contrary to the previous hypothesis that it is an oncogene.
Unexpectedly, the inactivation of Zbtb7a accelerates Pten
loss-induced prostate cancer progression. Mechanistically, the
downregulation of retinoblastoma-associated protein by
hyper-elevated Sox9 was found to occur in Pten/Zbtb7a double-null
prostate tumors (24); this resulted
in Pten loss-induced cellular senescence being overcome (24). Therefore, Zbtb7a can also be
identified as a PTEN context-dependent cancer gene that may have
oncosuppressive and oncogenic functions in PTEN-null tumors.
PTEN loss switches ARF function through
sumoylation
ARF is another transcript of the ARF-INK4a
locus (CDKN2a) on human chromosome 9p21 (25). The CDKN2a locus encodes
p14ARF and p16INK4a, which are two cyclin-dependent
kinase inhibitors (26).
CDKN2a deficiency may lead to susceptibility to
carcinogen-induced tumors via antagonism of Mdm2-mediated p53
degradation (27). The canonical
pathway of ARF induces senescence through degradation of MDM2,
thereby stabilizing p53 (27).
Multiple studies have suggested that ARF has
oncosuppressive and oncogenic functions. For instance, it has been
shown that p19Arf deficient mice develop various types
of cancers, suggesting that ARF has oncosuppressive roles (26). On the other hand, a study by Chen
et al (28) showed that
p19Arf inactivation decreases tumorigenesis in
PTEN-deficient prostate cancers, indicating the oncogenic role of
ARF. Moreover, a number of previous studies have shown the
oncogenic role of ARF in a genetic context-dependent manner
(29,30). However, the manner by which ARF
function is switched from tumor suppressor to oncogene is unclear.
In a previous study, we found that PTEN loss may be the switch
during Zinc finger protein SNAI2 (SLUG)-mediated
epithelial-mesenchymal transition (EMT). SLUG as a transcription
factor can represses E-cadherin transcription to promote EMT in
various cancers, including prostate cancer (31). The mechanisms underlying these
pathways are not described well, particularly when it comes to
in vivo tumorigenesis during PTEN loss. In another previous
study, we showed that p14ARF (ARF in humans) stabilizes
SLUG through sumoylation at lysine residue 192 (32). This stabilization results in the
inhibition of E-cadherin in prostate cancer mouse models. On the
other hand, p19Arf inactivation leads to the reduction
of Slug levels, resulting in high E-cadherin expression (32). This inactivation delays the onset and
progression of prostate cancer in Pten/Trp53 double null mice
(32). This study suggested that PTEN
loss may be the switch for ARF function from a p53-dependent tumor
suppressor to an SLUG-EMT-dependent oncogenic protein. These novel
findings may have implications for clinical research.
Chemotherapeutic compounds that target ubiquitination to control
cancer progression are currently in clinical trial. A similar
approach can be used to develop inhibitors that will prevent cancer
metastasis by blocking SLUG sumoylation. Moreover, it is possible
to target ARF and make the treatment of prostate cancer more
efficient.
PTEN loss/AKT activation switches p27 from a
tumor suppressor to an oncogene
p27 is a tumor suppressor that represses cell
cycle progression as an inhibitor of cyclin-dependent kinase (CDK)
in the nucleus. However, the cytoplasmic localization of p27 has
been found in a number of cancers and correlates with PTEN loss in
prostate cancer (33). The
cytoplasmic localization of p27 has been also found to be
correlated with the activation of AKT in primary breast cancer
samples (34). Mechanically, the
AKT-mediated phosphorylation of p27 at threonine 157 determines
cellular p27 translocation from the nucleus to the cytoplasm
(34–36). As a consequence, cytoplasmic localized
p27 inhibits G1 cell cycle arrest (34,35). Thus,
the PTEN loss/AKT activation pathway may switch p27 from a tumor
suppressor to an oncogenic protein through phosphorylation mediated
nuclear-cytoplasmic translocation.
PTEN loss/AKT activation switches BRCA1 from
a tumor suppressor to an oncogenic resistant gene in radiation
therapy
BRCA1 is believed to act in the nucleus as a tumor
suppressor that can repair DNA (37).
The cytoplasmic localization of BRCA1 has been found in numerous
cancers, where it results in the sensitization of DNA damage and
increased apoptosis (38). Wild-type
p53 promotes BRCA1 nuclear export upon irradiation (38). Consistently, dysregulated p53 induces
the nuclear retention of BRCA1, which results in the resistance to
radiation therapy in breast cancer cells (39). While p53 is regulated indirectly or
directly by PTEN, PTEN loss may affect BRCA1 cellular localization
and thereby switch the BRCA1 from a tumor suppressor to an
oncogenic resistant gene in radiation therapy. On the other hand,
AKT has been found to phosphorylate BRCA1 to enhance its nuclear
localization and stability, which results in the inhibition of
radiation sensitivity (40). Thus,
the PTEN loss/AKT activation pathway is most likely able to convert
BRCA1 function.
In summary, with PTEN loss/AKT activation,
functional switches of targets from oncogenes to suppressors and
vice versa have been observed. These switches are associated with a
subset of previously believed oncogenes or tumor suppressors
(Fig. 1). It can be concluded that
PTEN may be a powerful switcher, playing an
oncosuppressive/oncogenic role in a context-dependent manner
through multiple pathways.
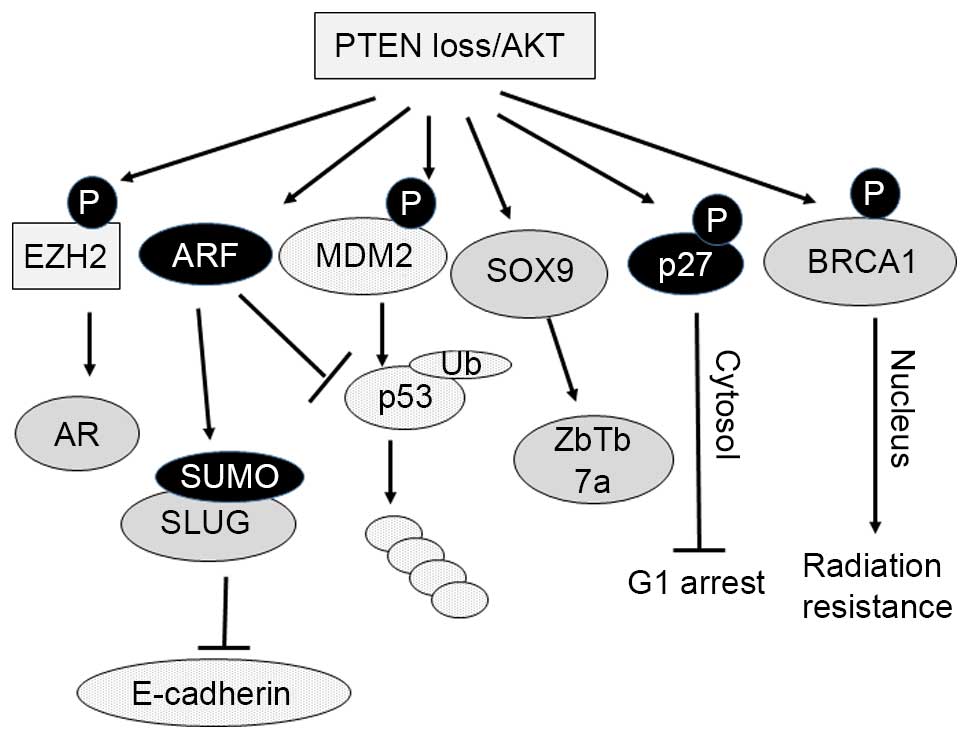 | Figure 1.PTEN loss/AKT activation converts a
subset of tumor suppressor proteins to oncogenic proteins or vice
versa through multiple pathways. PTEN, phosphatase and tensin
homolog; Akt, protein kinase B; EZH2, enhancer of zeste homolog 2;
AR, androgen receptor; ARF, alternative reading frame 2; SUMO,
small ubiquitin-like modifier; SLUG, ces-1-related zinc finger
transcription factor; E-cadherin, epithelial cadherin; MDM2, mouse
double minute 2; Ub, ubiquitin; SOX9, SRY-related HMG-box 9;
ZbTb7a, zinc finger and BTB domain-containing 7A; BRCA1, breast
cancer 1, early onset; P, phosphorylation. |
Conclusion and prospective outlook
The results of this review suggest that PTEN
inactivation may impact prostate cancer development significantly.
This issue becomes particularly important in case of cancer
treatment. For example, drugs targeting ZBTB7A may facilitate tumor
growth if PTEN is inactivated. Therefore, a personalized medicine
treatment approach should be taken into consideration, as gene
deletions or mutations induce switches of oncogenic/tumor
suppressive signaling, such as PTEN loss or mutations among
individual patients.
Finally, with the development of methods for the
transcriptional and genomic analysis of cancer cells, PTEN
inactivation can potentially be used as a prognostic and diagnostic
marker for various types of cancer, including, but not limited to,
prostate cancer. As the understanding of the role of PTEN in the
progression and initiation of cancer increases, this accumulating
knowledge may be used for the design of novel therapeutics methods
that will target PTEN-dependent pathways to treat different stages
of prostate cancer.
Acknowledgements
This manuscript was supported in part by the
Kazakhstan-China Collaboration Grant of Diagnosis and Precision
Cancer Therapy. The authors would like to thank Nazarbayev
University's grant support for the project, including the Social
Policy Grant (to Dr Yingqiu Xie), SST Professional Development
grant, and other under-review grants (IASANU Proposal grant and
ORAU grant).
References
|
1
|
Gustafson S, Zbuk KM, Scacheri C and Eng
C: Cowden syndrome: Semin Oncol. 34:428–434. 2007.
|
|
2
|
Blumenthal GM and Dennis PA: PTEN
hamartoma tumor syndromes. Eur J Hum Genet. 16:1289–1300. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Feilotter HE, Nagai MA, Boag AH, Eng C and
Mulligan LM: Analysis of PTEN and the 10q23 region in primary
prostate carcinomas. Oncogene. 16:1743–1748. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cairns P, Okami K, Halachmi S, Halachmi N,
Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS and Sidransky D:
Frequent inactivation of PTEN/MMAC1 in primary prostate cancer.
Cancer Res. 57:4997–5000. 1997.PubMed/NCBI
|
|
5
|
Song MS, Salmena L and Pandolfi PP: The
functions and regulation of the PTEN tumour suppressor. Nat Rev Mol
Cell Biol. 13:283–296. 2012.PubMed/NCBI
|
|
6
|
Ackler S, Ahmad S, Tobias C, Johnson MD
and Glazer RI: Delayed mammary gland involution in MMTV-AKT1
transgenic mice. Oncogene. 21:198–206. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bargonetti J and Manfredi JJ: Multiple
roles of the tumor suppressor p53. Curr Opin Oncol. 14:86–91. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Di Cristofano A and Pandolfi PP: The
multiple roles of PTEN in tumor suppression. Cell. 100:387–390.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Di Cristofano A, Pesce B, Cordon-Cardo C
and Pandolfi PP: Pten is essential for embryonic development and
tumor suppression. Nat Genet. 19:348–355. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou BP, Liao Y, Xia W, Zou Y, Spohn B and
Hung MC: HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2
phosphorylation. Nat Cell Biol. 3:973–982. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Downward J: Mechanisms and consequences of
activation of protein kinase B/Akt. Curr Opin Cell Biol.
10:262–267. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mayo LD, Dixon JE, Durden DL, Tonks NK and
Donner DB: PTEN protects p53 from Mdm2 and sensitizes cancer cells
to chemotherapy. J Biol Chem. 277:5484–5489. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen Z, Trotman LC, Shaffer D, Lin HK,
Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al:
Crucial role of p53-dependent cellular senescence in suppression of
Pten-deficient tumorigenesis. Nature. 436:725–730. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Freeman DJ, Li AG, Wei G, Li HH, Kertesz
N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, et
al: PTEN tumor suppressor regulates p53 protein levels and activity
through phosphatase-dependent and -independent mechanisms. Cancer
Cell. 3:117–130. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lowe SW, Cepero E and Evan G: Intrinsic
tumor suppression. Nature. 432:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cao R, Wang L, Wang H, Xia L,
Erdjument-Bromage H, Tempst P, Jones RS and Zhang Y: Role of
histone H3 lysine 27 methylation in Polycomb-group silencing.
Science. 298:1039–43. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis
RT, Wu X, Stack EC, Loda M, Liu T, et al: EZH2 oncogenic activity
in castration-resistant prostate cancer cells is
polycomb-independent. Science. 338:1465–1469. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu DC and Yang ZL: Overexpression of EZH2
and loss of expression of PTEN is associated with invasion,
metastasis and poor progression of gallbladder adenocarcinoma.
Pathol Res Pract. 207:472–478. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zeng N, Yang KT, Bayan JA, He L, Aggarwal
R, Stiles JW, Hou X, Medina V, Abad D, Palian BM, et al: PTEN
controls β-cell regeneration in aged mice by regulating cell cycle
inhibitor p16ink4a. Aging Cell. 12:1000–1011. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vredeveld LC, Rowland BD, Douma S,
Bernards R and Peeper DS: Functional identification of LRF as an
oncogene that bypasses RASV12-induced senescence via upregulation
of CYCLIN E. Carcinogenesis. 31:201–207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qu H, Qu D, Chen F, Zhang Z, Liu B and Liu
H: ZBTB7 overexpression contributes to malignancy in breast cancer.
Cancer Invest. 28:672–678. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao ZH, Wang SF, Yu L, Wang J, Chang H,
Yan WL, Zhang J and Fu K: Overexpression of Pokemon in non-small
cell lung cancer and foreshowing tumor biological behavior as well
as clinical results. Lung Cancer. 62:113–119. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aggarwal A, Hunter WJ III, Aggarwal H,
Silva ED, Davey MS, Murphy RF and Agrawal DK: Expression of
leukemia/lymphoma-related factor (LRF/POKEMON) in human breast
carcinoma and other cancers. Exp Mol Pathol. 89:140–148. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang G, Lunardi A, Zhang J, Chen Z, Ala U,
Webster KA, Tay Y, Gonzalez-Billalabeitia E, Egia A, Shaffer DR, et
al: Zbtb7a suppresses prostate cancer through repression of a
Sox9-dependent pathway for cellular senescence bypass and tumor
invasion. Nat Genet. 45:739–746. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
den Besten W, Kuo ML, Tago K, Williams RT
and Sherr CJ: Ubiquitination of and sumoylation by, the Arf tumor
suppressor. Isr Med Assoc J. 8:249–251. 2006.PubMed/NCBI
|
|
26
|
Bardeesy N, Aguirre AJ, Chu GC, Cheng KH,
Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et
al: Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain
progression of pancreatic adenocarcinoma in the mouse. Proc Natl
Acad Sci USA. 103:5947–5952. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ha L, Ichikawa T, Anver M, Dickins R, Lowe
S, Sharpless NE, Krimpenfort P, Depinho RA, Bennett DC, Sviderskaya
EV and Merlino G: ARF functions as a melanoma tumor suppressor by
inducing p53-independent senescence. Proc Natl Acad Sci USA.
104:10968–10973. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen Z, Carracedo A, Lin HK, Koutcher JA,
Behrendt N, Egia A, Alimonti A, Carver BS, Gerald W,
Teruya-Feldstein J, et al: Differential p53-independent outcomes of
p19(Arf) loss in oncogenesis. Sci Signal. 2:ra442009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Humbey O, Pimkina J, Zilfou JT, Jarnik M,
Dominguez-Brauer C, Burgess DJ, Eischen CM and Murphy ME: The ARF
tumor suppressor can promote the progression of some tumors. Cancer
Res. 68:9608–9613. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Khoo CM, Carrasco DR, Bosenberg MW, Paik
JH and Depinho RA: Ink4a/Arf tumor suppressor does not modulate the
degenerative conditions or tumor spectrum of the
telomerase-deficient mouse. Proc Natl Acad Sci USA. 104:3931–3936.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hajra KM, Chen DY and Fearon ER: The SLUG
zinc-finger protein represses E-cadherin in breast cancer. Cancer
Res. 62:1613–1618. 2002.PubMed/NCBI
|
|
32
|
Xie Y, Liu S, Lu W, Yang Q, Williams KD,
Binhazim AA, Carver BS, Matusik RJ and Chen Z: Slug regulates
E-cadherin repression via p19Arf in prostate tumorigenesis. Mol
Oncol. 8:1355–3564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Halvorsen OJ, Haukaas SA and Akslen LA:
Combined loss of PTEN and p27 expression is associated with tumor
cell proliferation by Ki-67 and increased risk of recurrent disease
in localized prostate cancer. Clin Cancer Res. 9:1474–1479.
2003.PubMed/NCBI
|
|
34
|
Liang J, Zubovitz J, Petrocelli T,
Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C,
Beniston R, et al: PKB/Akt phosphorylates p27, impairs nuclear
import of p27 and opposes p27-mediated G1 arrest. Nat Med.
8:1153–1160. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shin I, Yakes FM, Rojo F, Shin NY, Bakin
AV, Baselga J and Arteaga CL: PKB/Akt mediates cell-cycle
progression by phosphorylation of p27(Kip1) at threonine 157 and
modulation of its cellular localization. Nat Med. 8:1145–1152.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Viglietto G, Motti ML, Bruni P, Melillo
RM, D'Alessio A, Califano D, Vinci F, Chiappetta G, Tsichlis P,
Bellacosa A, et al: Cytoplasmic relocalization and inhibition of
the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated
phosphorylation in breast cancer. Nat Med. 8:1136–1144. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mullan PB, Quinn JE and Harkin DP: The
role of BRCA1 in transcriptional regulation and cell cycle control.
Oncogene. 25:5854–5863. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang H, Yang ES, Jiang J, Nowsheen S and
Xia F: DNA damage-induced cytotoxicity is dissociated from BRCA1′s
DNA repair function but is dependent on its cytosolic accumulation.
Cancer Res. 70:6258–6267. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang J, Yang ES, Jiang G, Nowsheen S,
Wang H, Wang T, Wang Y, Billheimer D, Chakravarthy AB, Brown M, et
al: p53-dependent BRCA1 nuclear export controls cellular
susceptibility to DNA damage. Cancer Res. 71:5546–5557. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nelson AC, Lyons TR, Young CD, Hansen KC,
Anderson SM and Holt JT: AKT regulates BRCA1 stability in response
to hormone signaling. Mol Cell Endocrinol. 319:129–142. 2010.
View Article : Google Scholar : PubMed/NCBI
|














