Introduction
Colorectal cancer (CRC) is one of the most common
human cancers worldwide, accounting for 1.2 million novel cases and
600,000 mortalities per year (1).
Despite the great progress in current therapeutic options,
including surgery and chemoradiotherapy, the incidence and
mortality of CRC remains high (2).
Novel strategies require investigation not only to enhance the
efficacy of currently available agents, but also to identify novel
therapeutic targets for CRC patients. Therefore, there are
concerted efforts to identify gene expression patterns that may be
used for early detection and improved prognosis prediction for
CRC.
Spleen tyrosine kinase (SYK) is a 72 kDa
non-receptor tyrosine kinase that contains two tandem Src homology
2 domains at the NH2 terminus and a kinase domain at the
COOH terminus (3). It is widely
expressed in hematopoietic cells and was hypothesized to be a
hematopoietic cell-specific signaling molecule (4). Currently, SYK is considered to be a
potential tumor suppressor in breast carcinoma (5), and there are a growing number of studies
on SYK in non-hematopoietic tumors (6–8). Several
clinical studies have indicated that patients with negative SYK
expression have a significantly lower overall survival rate
compared with patients with positive SYK expression (9–12).
Functional studies have demonstrated that dysregulation of SYK is
associated with cancer proliferation and metastasis (13–15). These
data implicate SYK as a putative tumor suppressor in cancer.
However, Luangdilok et al (16) reported that high expression of SYK was
significantly associated with recurrence and poorer survival in
squamous cell carcinomas of the head and neck, and these results
are consistent with a study concerning nasopharyngeal carcinoma
(17). Overall, the present study
hypothesizes that SYK has a complex role in multiple cancer
types.
SYK has two alternatively spliced isoforms:
Full-length [SYK(L)] and short form [SYK(S)] SYK, which lacks a
69-nucleotide exon (Fig. 1A). A
previous study by the present authors revealed that SYK(L) was
present in the cytoplasm and nucleus of breast cancer cells, and
suppressed breast cancer cell invasiveness, whereas SYK(S) was
located exclusively in the cytoplasm and did not affect breast
cancer cell invasion (18).
Consistent with these results, recent evidence revealed that
differential expression of SYK(L) and SYK(S) may contribute to
tumor biology in different ways, and may be clear indicators of
prognosis in patients with hepatocellular cancer (19). In addition, Prinos et al
(20) have reported that changing the
SYK alternative splicing pattern alters cancer cell survival and
mitotic progression. On the basis of these data, the present study
hypothesizes that SYK alternative splicing isoforms have different
functional effects in cancer and act as modulators of cancer.
Hypermethylation of the SYK gene promoter was
demonstrated to be associated with a loss of SYK gene expression in
a variety of malignant cancers, including breast cancer (21), gastric cancer, nasopharyngeal
carcinoma (22) and hepatocellular
cancer (23). In CRC, the present
authors previously demonstrated that global SYK methylation was an
independent prognostic factor for overall survival (12), but the expression and biological
functions of alternative splicing SYK isoforms in CRC remain
unclear. The present study aimed to investigate the functional
impact of SYK(L) and SYK(S) in CRC. The present study evaluated the
effect of SYK(L) and SYK(S) on proliferation, metastasis and
5-fluorouracil (5-FU) resistance in CRC cells by overexpressing
SYK(L) and SYK(S). In addition, the expression pattern of SYK
isoforms was also confirmed in CRC tissues.
Materials and methods
Clinical samples and cell lines
In total, 26 CRC samples and matched adjacent normal
samples were obtained from the Tissue Bank of The Sixth Affiliated
Hospital, Sun Yat-sen University (Guangzhou, China) between March
2010 and July 2010. All the samples were obtained with the written
informed consent of the patients and were histologically confirmed.
The Institutional Review Board of Sun Yat-sen University approved
the study.
Seven human CRC cell lines (HCT 116, SW480, RKO,
HCT-8, LoVo, HCT-15 and Caco-2) were obtained from Shanghai Cell
Collection, Chinese Academy of Science (Shanghai, China). HCT 116,
SW480, HCT-8 and HCT-15 were maintained in RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), whereas RKO,
LoVo and Caco-2 were maintained in Dulbecco's Modified Eagle's
Medium (DMEM; Thermo Fisher Scientific, Inc.). All cells were
cultured in recommended medium containing 10% fetal bovine serum
(FBS; Thermo Fisher Scientific, Inc.), and maintained at 37°C with
5% CO2 in a humidified incubator. The cell lines were
routinely authenticated (once every year) using DNA-fingerprinting
analysis, identity verification using short tandem repeat profiling
analysis, cell morphology monitoring, growth curve analysis and
mycoplasma contamination checks.
Lentiviral vector construction and
stable cell lines
The lentiviral plasmid pLVX-EGFP-3FLAG alone
(negative control) or with SYK(L) or SYK(S) cDNA were constructed
by Sunbio (Shanghai, China). The SYK(L) and SYK(S) gene cDNA were
verified by DNA sequencing and polymerase chain reaction (PCR). The
lentiviral vectors expressed green fluorescent protein (GFP);
therefore, viral titer was determined by the method of end point
dilution by counting the numbers of infected green cells at
magnification ×100 under a fluorescence microscope (Leica
Microsystems GmbH, Wetzlar, Germany). HCT 116 cells, which
expressed low detectable SYK(L) and SYK(S), were used to generate
stable cells by lentiviral infection, according to the
manufacturer's protocol. The same amount of negative control vector
was also infected in HCT 116 cells. A total of 12 h later, the
virus-containing medium was replaced with fresh complete medium.
Following a 72 h infection, 1 µg/ml puromycin was added to the
culture medium for selection of stable cells. The fluorescence of
GFP in stable cell lines was observed by fluorescence microscopy to
achieve over 100% transfection. The expression levels of SYK(L) and
SYK(S) in stable cells were evaluated by quantitative (q)PCR.
Reverse transcription-qPCR
Total RNA was extracted from tissues and transfected
cells using TRIzol Reagent (Invitrogen™; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. Reverse
transcription was performed using 0.5 µg RNA and the ReverTra
Ace® qPCR RT kit (cat. no. FSQ-301; Toyobo, Co., Ltd.,
Osaka, Japan) containing DNase I, according to the manufacturer's
protocol. qPCR was conducted on an ABI 7500 system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) using the SYBR Green
Real-Time PCR Master Mix (Thermo Fisher Scientific, Inc.), with the
following cycling conditions: 95°C for 10 min; and 40 cycles of
95°C for 15 sec; 60°C for 1 min. qPCR was performed in triplicate,
including no-template controls. In each qPCR assay, amplification
of the reference gene glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) was performed as the internal control. The PCR primers were
as follows: SYK(L), forward 5′-GCTCTGGCAGCTAGTCGA-3′ and reverse
5′-GCTTTGGGAAGGAGTATGA-3′; SYK(S), forward
5′-AAAGCAGATGGTTTGTTAAGAGTTC-3′ and reverse
5′-CTTGGGCAGGGGAGGACGCAGGATG-3′; GAPDH, forward
5′-GACAGTCAGCCGCATCTTCTT-3′ and reverse
5′-AATCCGTTGACTCCGACCTTC-3′. The relative amount of SYK(L) and
SYK(S) to GAPDH were calculated using the quantitative cycle
threshold (2−ΔΔCq) method (24).
Western blot
The cells were lysed in RIPA lysis buffer (DingGuo
Biotechnology, Co., Ltd., Beijing, China) supplemented with
phenylmethanesulfonyl fluoride (DingGuo Biotechnology, Co., Ltd.)
and centrifuged at 10,000 × g for 5 min at 4°C. The protein samples
were separated by 8% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and the products were electrotransferred to
nitrocellulose filter membrane (GE Healthcare Life Scientific,
Chalfont, UK). Rabbit anti-SYK polyclonal antibody (cat. no.
sc-1077; dilution, 1:700; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) and mouse anti-GAPDH monoclonal antibody (cat. no.
60004-1-lg; dilution, 1:4,000; Proteintech, Rosemont, IL, USA) were
used for immunodetection. Following incubations with primary
antibodies at 4°C overnight, IRDye® 800CW-conjugated
goat anti-rabbit and IRDye® 680RD-conjugated goat
anti-mouse secondary antibodies (cat. nos. P/N 925-32211 and P/N
925-68070, respectively; dilution, 1:10,000; LI-COR Biosciences,
Lincoln, NE, USA) were incubated with the membranes for 1 h at room
temperature. Proteins were detected using the Odyssey®
CLx Imaging System (LI-COR Biosciences), and the band intensities
were analyzed using the Odyssey Application 3.0 software (LI-COR
Biosciences).
Cell proliferation assays
Cell proliferation was measured using the
xCELLigence Real Time Cell Analyzer (RTCA) (ACEA Biosciences, San
Diego, CA, USA). Briefly, cells were seeded in E-plates at a
density of 6,000 cells per well and incubated at 37°C and 5%
CO2. The cell growth curves were automatically monitored
and recorded every 15 min for a total of 120 h. Cells adhered to
the bottom of each well and cell index values were measured by
monitoring cells that covered the surface of the sensor.
Experiments were repeated independently three times.
Cell proliferation was also measured by
5-ethynyl-2-deoxyuridine (EdU) assay using an EdU Assay kit
(Guangzhou Ribobio, Co., Ltd., Guangzhou, China), according to the
manufacturer's protocol. Briefly, cells were cultured in 96-well
plates and then exposed to 50 mM EdU for 2 h at 37°C. Subsequently,
the cells were fixed with 4% formaldehyde for 30 min at room
temperature. Cells were washed with phosphate-buffered saline (PBS)
and incubated with 100 µl 1X Apollo® reaction cocktail
for 30 min. The DNA contents of cells were stained with 100 µl
Hoechst 33342 for 30 min and viewed under a fluorescent
microscope.
Cell cycle assay
Cells were stained using Cell Cycle Staining kit
(Lianke Bio, Hangzhou, China) and analyzed by flow cytometry. In
brief, a pellet of ~2×105 cells was formed by
centrifugation and the cells were washed with PBS. Subsequently, 1
ml cold 75% ethanol was added to the cells at −20°C overnight.
Subsequently, the cells were rehydrated with PBS for 15 min and
incubated with 1 ml DNA staining solution at room temperature. The
cells were examined using a flow cytometer (BD Biosciences,
Franklin Lakes, NJ, USA). The results were analyzed using the
ModFit version 3.0 software (Verity Software House, Topsham, ME,
USA).
Cell migration and invasion
assays
Cell invasion assays were performed using a
24-multiwell insert plate with an 8.0 µm pore size membrane chamber
(BD Biosciences) containing a Matrigel-coated membrane. Cells were
suspended in serum-free DMEM and added to apical chambers. Cells at
a density of 40,000 cells/100 µl of DMEM media were placed into the
upper chamber of the Transwell plate and 700 µl DMEM medium
containing 20% FBS was added to the basal chambers. Following a 48
h incubation, the cells in the basal chamber were stained with
4′,6-diamidino-2-phenylindole, dihydrochloride (Roche Diagnostics
GmbH, Mannheim, Germany) and counted using a fluorescence
microscope. The cell migration assay was performed in the same way
without Matrigel. Three independent experiments were conducted.
Cell viability assay
5-FU resistance was measured by a
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay (Promega, Madison, WI, USA), which assesses the
reduction of MTS to formazan in viable cells. Briefly, cells were
seeded in 96-well plates at a density of 5,000 cells/100 µl/well.
Subsequent to 24 h, cells were treated with various concentrations
of 5-FU (0, 0.1, 1.0, 10.0, 20.0, 50.0, 100.0 and 200.0 µM).
Following a 72 h incubation, 20 µl of the combined MTS/phenazine
methosulfate solution was added to the medium and the mixture was
incubated at 37°C for 1.5 h. The absorbance was measured at 490 nm
using Varioskan™ Flash multimode reader (Thermo Fisher Scientific,
Inc.).
Statistical analysis
Statistical analysis was performed with SPSS version
16.0 software (SPSS, Inc., Chicago, IL, USA). Data are presented as
the mean ± standard deviation. Differences between groups were
compared using paired or unpaired Student's t-tests or the
Mann-Whitney U test. P≤0.05 was considered to indicate a
statistically significant difference.
Results
Stably transfected cells overexpress
SYK isoforms
To examine the biological impact of SYK splice
isoforms in CRC cells, the present study first identified the
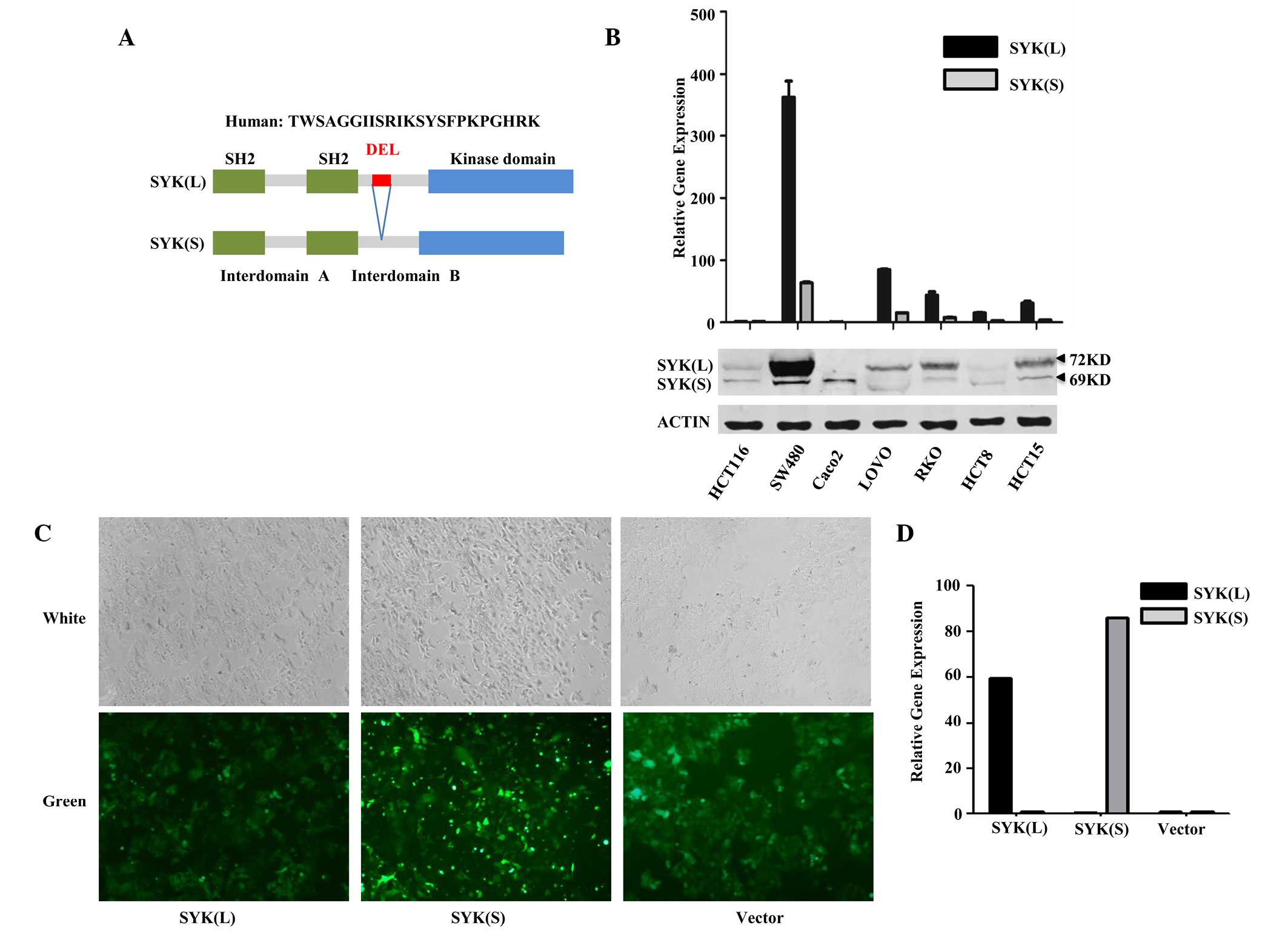
expression of SYK splice isoforms SYK(L) and SYK(S) in 7 CRC cell
lines (HCT 116, SW480, Caco2, LoVo, RKO, HCT-8 and
HCT-15) using western blot analysis and qPCR (Fig. 1B). Western blot analysis revealed that
SYK(L) was highly expressed in SW480, LoVo, HCT-15 and RKO cell
lines, with low or no expression in HCT 116, Caco2 and
HCT-8. SYK(S) protein was detected in SW480 and Caco2
cells, and was almost undetectable in other cell lines.
Subsequently, mRNA levels of SYK(L) and SYK(S) in the 7 cell lines
were identified by qPCR. The alterations in the mRNA expression of
SYK isoforms between cell lines was consistent with the protein
levels in these cells.
HCT 116 cells were selected as the target cells for
transfection with the pLVX-EGFP-3FLAG-SYK(L)/SYK(S) recombinant
lentiviral vector or empty vector. Although the expression of
SYK(L) and SYK(S) was negligible in both HCT 116 cells and HCT-8
cells, the biological functions of these isoforms of SYK were more
obvious and stable in HCT 116 cells, as compared with HCT-8 cells;
thus HCT 116 cells were selected for further studies. Results of
this transfection revealed that the transfected HCT 116 cells
expressed GFP following transfection and green fluorescence was
detected using a fluorescent microscope (Fig. 1C). qPCR demonstrated that the relative
expression levels of SYK(L) and SYK(S) were markedly increased in
the SYK(L) and SYK(S) overexpression cells compared with those in
the negative control group (Fig.
1D).
SYK(L), but not SYK(S), suppresses the
proliferation of CRC cells via cell cycle arrest
To investigate the potential effects of SYK isoforms
in CRC cells in vitro, transfected HCT 116 cells underwent
RTCA and EdU assay. As shown in Fig.
2A, the RTCA revealed that the SYK(L) overexpression group
clearly suppressed cell proliferation compared with the negative
control group (P=0.004), whereas the SYK(S) overexpression group
did not significantly affect HCT 116 cell proliferation (P=0.063).
These findings were further confirmed by an EdU assay (Fig. 2B); the percentage of EdU-positive
cells was significantly lower in cells expressing SYK(L) (P=0.020).
However, there was no significant difference between SYK(S)
overexpression group and the negative control group (P=0.054).
Overall, these findings suggest that SYK(L), and not SYK(S),
suppress the cell proliferation capability of HCT 116 cells.
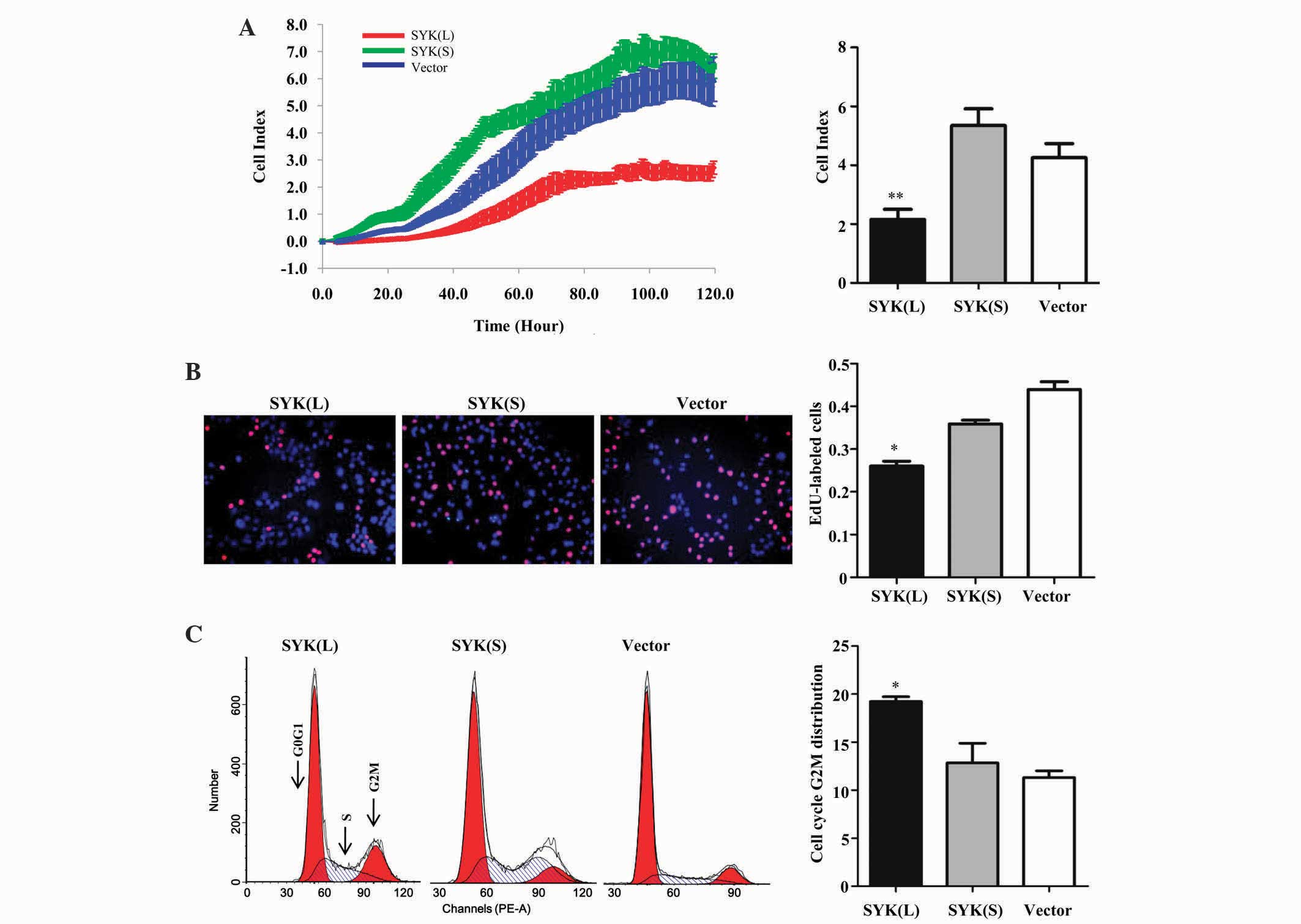 | Figure 2.Effects of ectopic SYK(L) and SYK(S)
expression on CRC tumor growth. (A) Proliferation curves of human
CRC HCT 116 cells analyzed by xCelligence real time cellular
analysis following transfection with recombinant lentivirus vector
with SYK(L) or SYK(S). Red, green and blue indicate the SYK(L)
overexpression, SYK(S) overexpression and negative control groups,
respectively. (B) Cells transfected with recombinant lentivirus
vector with SYK(L) or SYK(S) were EdU-labeled and compared with
control cells. Red and blue indicate EdU- and Hoechst 33342-labeled
cells, respectively. (C) Cell cycle progression analysis in HCT 116
cells following transfection with recombinant lentivirus vector
with SYK(L) or SYK(S). Data are presented as the mean ± standard
deviation. *P<0.05, **P<0.01 vs. empty vector transfected
cells. SYK, spleen tyrosine kinase; SYK(L), SYK full length;
SYK(S), SYK short form; EdU, 5-ethynyl-2-deoxyuridine; CRC,
colorectal cancer. |
Fluorescence-activated cell sorting analysis was
conducted to analyze the effect of the SYK splice isoforms on cell
cycle progression. As shown in Fig.
2C, the percentage of cells in the G2M-phase of the SYK(L)
overexpression group was significantly increased compared with
those of the negative control group (P=0.018), whereas SYK(S)
overexpression did not significantly alter the percentage of cells
in the G2M phase (P=0.617). Collectively, these results suggest
that SYK(L) inhibits the proliferation of HCT 116 cells via
inducing G2M phase arrest.
SYK(L) not SYK(S) suppress metastasis
in CRC cells
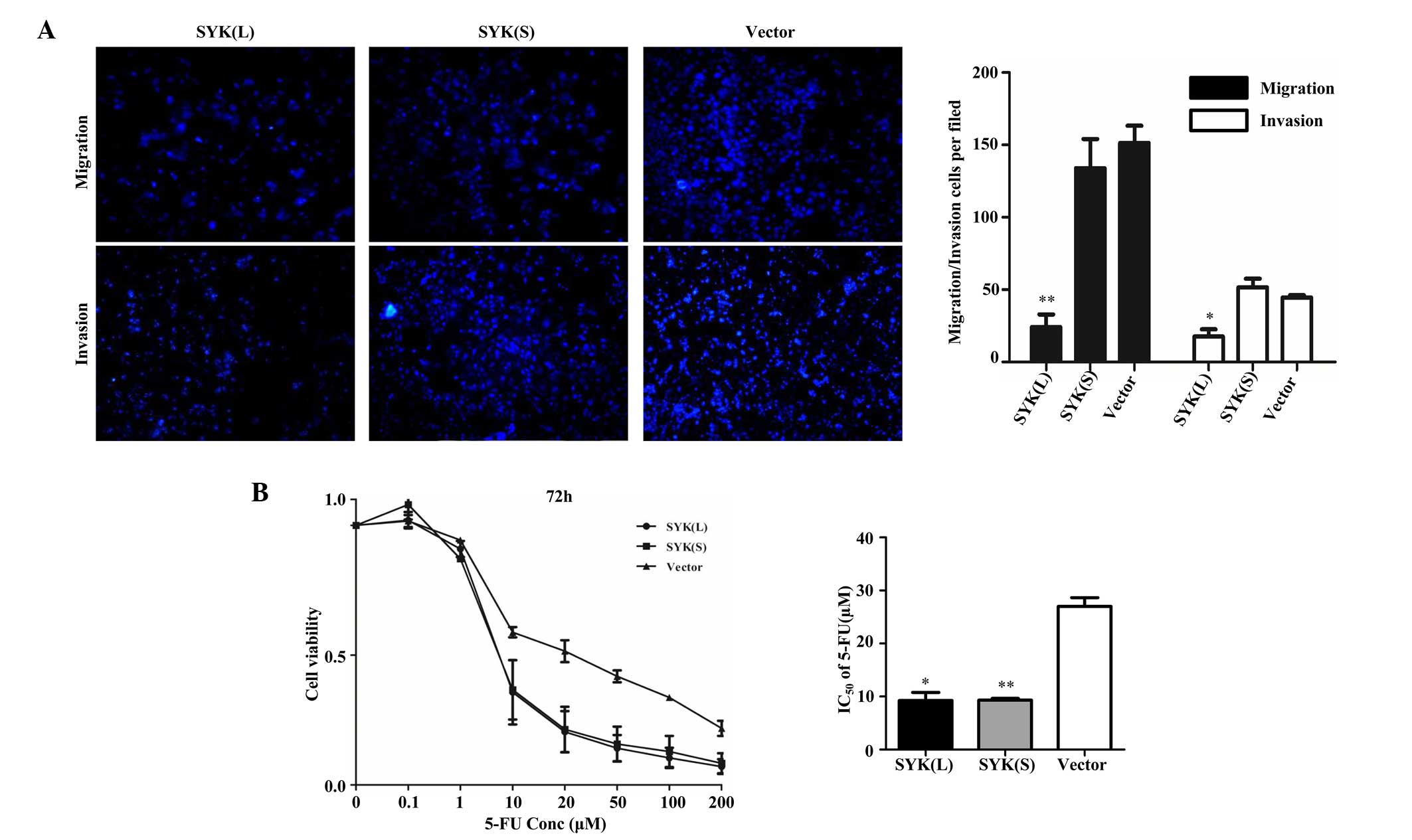
Subsequently, whether SYK(L) and SYK(S) had an
effect on the migration and invasion ability of HCT 116 cells was
analyzed by Transwell assay. Cells on the lower side of the
Transwell membrane were the migrating/invading cells.
Representative photomicrographs of Transwell assays were obtained
at magnification ×100 (Fig. 3A).
Subsequently, the number of cells were counted in nine independent
visual fields under the microscope (magnification, ×200) from three
independent experiments. Overexpression of SYK(L) significantly
decreased the migration and invasion capacity by 84% (P=0.003) and
61% (P=0.044), respectively, in HCT 116 cells compared with
negative control cells (Fig. 3A).
However, overexpression of SYK(S) did not significantly affect the
motility (P=0.325) or invasive ability (P=0.271) of the cells.
Therefore, the results revealed that SYK(L), and not SYK(S), is
involved in CRC cell migration and invasion.
SYK(L) and SYK(S) suppress the 5-FU
resistance of CRC cells
Since 5-FU is one of the most commonly used
anticancer drugs in CRC (25,26), cell viability following 5-FU treatment
of cells overexpressing SYK(L) or SYK(S), or negative control
cells, was assessed by the present study in vitro. HCT 116
cells were treated with 0, 0.1, 1.0, 10.0, 20.0, 50.0, 100.0 and
200.0 µM 5-FU and cell viability was measured by MTS assay. The
IC50 values for the SYK(L) and SYK(S) overexpression
groups and the negative control group were 9.241±1.552, 9.305±0.341
and 26.996±1.639 µM, respectively (Fig.
3B). The overexpression of SYK(L) cells exhibited a significant
reduction in cell viability compared to the negative control group
(P=0.026). Notably, the overexpression of SYK(S) in HCT 116 cells
exhibited a higher sensitivity to 5-FU compared with the negative
control group (P=0.005). These results suggest that overexpression
of SYK(L) exerts a strong synergistic effect with 5-FU on the
growth of HCT 116 cells, and overexpression of SYK(S) has an effect
on modulating CRC 5-FU sensitivity.
Expression of SYK splice isoforms in
CRC tissues
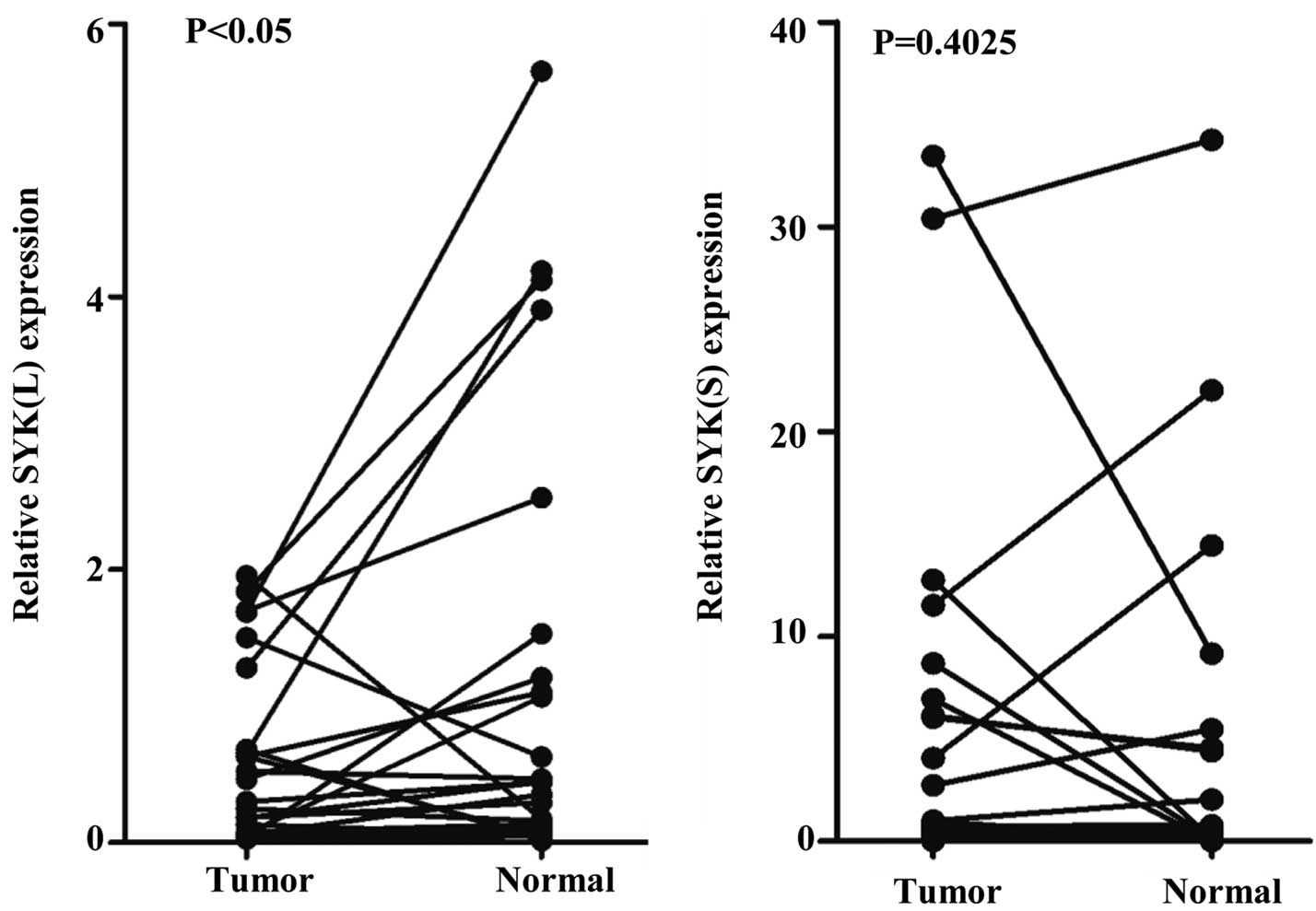
To investigate the expression pattern of SYK splice
isoforms SYK(L) and SYK(S) in CRC patients, the mRNA levels of
SYK(L) and SYK(S) were assessed in 26 tumor tissues and paired
normal tissues using qPCR. As shown in Fig. 4, the levels of SYK(L) were
significantly lower in tumor tissues compared with normal tissues
(P=0.049). However, the levels of SYK(S) had no significant
differences between tumor tissues and normal tissues (P=0.403).
Therefore, SYK(L) may be recognized as a potential tumor suppressor
in CRC.
Discussion
Alternative splicing allows pre-mRNA to be spliced
into multiple mRNAs and protein variants, which exhibit distinct
functions (27). Splice variants have
been identified for a series of functionally associated cancer
genes (28–30), suggesting that alterations in
alternative splicing isoforms may contribution to cancer biology or
carcinogenesis. A previous study performed a high-throughput survey
of 600 alternative splicing cancer-associated genes in breast
cancer, and revealed that 41 ductal breast cancer-specific markers
may be functionally associated with tumor biology (31). Miura et al (32) reported that systematic dysregulation
of alternative spliced isoforms, including vascular endothelial
growth factor A and uridine-5′-diphosphate glucuronosyltransferase
family 1, are crucial for the development of novel therapeutic
targets for CRC. Splice isoforms often have distinct functions and
even exhibit antagonistic properties of oncogenes and tumor
suppressors. Thus, it is important to understand how alternative
splicing isoforms contribute to the progression of cancer.
SYK is indispensable in inflammatory responses
(33) and has been identified as an
oncogenic driver in a broad spectrum of hematological malignancies
(34). Accumulating evidence revealed
that SYK is also required for non-hematopoietic tumors (35–39),
including colorectal, breast and hepatocellular cancer. In contrast
to the possible tumor-promoting functions for SYK in hematological
tumors (4), global SYK inversely
inhibits tumor progression in the majority of non-hematopoietic
tumors (6,8). The present authors previously
demonstrated that an aberrant expression of SYK(L) and SYK(S)
exhibits a differential phenotypic response, due to their different
subcellular localization in breast cancer (18). Hong et al (9,19) reported
that phosphorylation of Ser295 in SYK(L) was regulated by
checkpoint kinase 1, and observed that SYK(L) and SYK(S) have
opposing functions in hepatocellular carcinoma. The present study
investigated the biological functions of the two isoforms SYK(L)
and SYK(S) in CRC. In agreement with the results in breast cancer
and hepatocellular cancer, the present study demonstrated that
overexpression of SYK(L) could suppress the proliferation and
invasion of CRC cells. However, unlike the results with
hepatocellular cancer, the present study revealed that
overexpression of SYK(S) resulted in no difference in the
capability of cells to proliferate and metastasize compared with
negative control in CRC. Furthermore, the present results revealed
that SYK(L) was downregulated in 69% of 26 pairs of CRC tissues,
and the expression of SYK(S) had no significant difference between
CRC tissues and adjacent normal tissues. The differences in SYK
behavior in various cancer types has been reported to be due to the
differences in the contribution of SYK to tumor biology (19,20). The
present results indicate that splice isoforms of the SYK gene have
complex mechanisms in cancer.
5-FU is a commonly used chemotherapeutic agent in
CRC; however, drug resistance is a major reason for the failure of
this drug in the treatment of CRC patients. The present study
revealed that SYK(L) and SYK(S) increased the sensitivity of CRC
cells to 5-FU. Although overexpression of SYK(S) did not alter cell
proliferation and metastasis under normal growth conditions, unlike
SYK(L), it decreased cell viability following treatment of CRC
cells with 5-FU. These findings suggest that SYK(L) and SYK(S) have
clinical value in 5-FU cancer therapeutics. Further studies are
required to investigate the potential mechanism of SYK in 5-FU
resistance in CRC cells.
In conclusion, the present results demonstrate that
SYK(L) and SYK(S) have different expression and biological
functions in CRC. SYK(L) not only acted as a tumor suppressor in
CRC, but also had effects on chemotherapy resistance. Similarly,
SYK(S) is important in chemotherapeutic treatment of CRC. The
present study concerning the impact of alternative splicing in
cancer biology provides important insights in identifying potential
biomarkers and therapeutic targets for CRC.
Acknowledgements
The present study was supported by the National Key
Clinical Discipline, National Natural Scientific Foundation of
China (Beijing, China; grant no., 81372566), Science and Technology
Plan Project of Guangzhou (Guangzhou, China; grant no.,
2013J4500045) and Doctoral Fund of Ministry of Education of China
(Beijing, China; grant no., 20120171110095).
Glossary
Abbreviations
Abbreviations:
|
CRC
|
colorectal cancer
|
|
SYK
|
spleen tyrosine kinase
|
|
qPCR
|
quantitative polymerase chain
reaction
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
RTCA
|
real-time cellular analysis
|
References
|
1
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Guo P, Huang ZL, Yu P and Li K: Trends in
cancer mortality in China: An update. Ann Oncol. 23:2755–2762.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grädler U, Schwarz D, Dresing V, Musil D,
Bomke J, Frech M, Greiner H, Jäkel S, Rysiok T, Müller-Pompalla D
and Wegener A: Structural and biophysical characterization of the
Syk activation switch. J Mol Biol. 425:309–333. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coopman PJ and Mueller SC: The Syk
tyrosine kinase: A new negative regulator in tumor growth and
progression. Cancer Lett. 241:159–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coopman PJ, Do MT, Barth M, Bowden ET,
Hayes AJ, Basyuk E, Blancato JK, Vezza PR, McLeskey SW, Mangeat PH
and Mueller SC: The Syk tyrosine kinase suppresses malignant growth
of human breast cancer cells. Nature. 406:742–747. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang L, Devarajan E, He J, Reddy SP and
Dai JL: Transcription repressor activity of spleen tyrosine kinase
mediates breast tumor suppression. Cancer Res. 65:10289–10297.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang X, Shrikhande U, Alicie BM, Zhou Q
and Geahlen RL: Role of the protein tyrosine kinase Syk in
regulating cell-cell adhesion and motility in breast cancer cells.
Mol Cancer Res. 7:634–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ogane S, Onda T, Takano N, Yajima T,
Uchiyama T and Shibahara T: Spleen tyrosine kinase as a novel
candidate tumor suppressor gene for human oral squamous cell
carcinoma. Int J Cancer. 124:2651–2657. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hong J, Hu K, Yuan Y, Sang Y, Bu Q, Chen
G, Yang L, Li B, Huang P, Chen D, et al: CHK1 targets spleen
tyrosine kinase (L) for proteolysis in hepatocellular carcinoma. J
Clin Invest. 122:2165–2175. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nakashima H, Natsugoe S, Ishigami S,
Okumura H, Matsumoto M, Hokita S and Aikou T: Clinical significance
of nuclear expression of spleen tyrosine kinase (Syk) in gastric
cancer. Cancer Lett. 236:89–94. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peng C, Sun Q, Hao Y, Cong B, Zhao Y and
Zhao X: Syk is low-expressed in non-small-cell lung cancer and
inversely correlates with patient's survival. Acta Bioch Biophys
Sin (Shanghai). 45:149–151. 2013. View Article : Google Scholar
|
|
12
|
Yang Z, Huo L, Chen H, Ni B, Xiang J, Kang
L, Wang L, Peng J, Yuan Y and Wang J: Hypermethylation and
prognostic implication of Syk gene in human colorectal cancer. Med
Oncol. 30:2013. View Article : Google Scholar
|
|
13
|
Layton T, Stalens C, Gunderson F, Goodison
S and Sillett S: Syk tyrosine kinase acts as a pancreatic
adenocarcinoma tumor suppressor by regulating cellular growth and
invasion. Am J Pathol. 175:2625–2636. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yuan YF, Wang JP, Li JP, Wang L, Li M,
Yang Z, Zhang C and Dai JL: Frequent epigenetic inactivation of
spleen tyrosine kinase gene in human hepatocellular carcinoma. Clin
Cancer Res. 12:6687–6695. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Peng CL, Zhang Y, Sun QF, Zhao YP, Hao YT,
Zhao XG and Cong B: Inhibitory effects of Syk transfection on lung
cancer cell invasion. Asian Pac J Cancer Prev. 14:3001–3003. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luangdilok S, Box C, Patterson L, Court W,
Harrington K, Pitkin L, Rhŷs-Evans P, O-charoenrat P and Eccles S:
Syk tyrosine kinase is linked to cell motility and progression in
squamous cell carcinomas of the head and neck. Cancer Res.
67:7907–7916. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Du ZM, Kou CW, Wang HY, Huang MY, Liao DZ,
Hu CF, Chen J, Yan LX, Hu LF, Ernberg I, et al: Clinical
significance of elevated spleen tyrosine kinase expression in
nasopharyngeal carcinoma. Head Neck. 34:1456–1464. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang L, Duke L, Zhang PS, Arlinghaus RB,
Symmans WF, Sahin A, Mendez R and Dai JL: Alternative splicing
disrupts a nuclear localization signal in spleen tyrosine kinase
that is required for invasion suppression in breast cancer. Cancer
Res. 63:4724–4730. 2003.PubMed/NCBI
|
|
19
|
Hong J, Yuan YF, Wang JP, Liao Y, Zou R,
Zhu C, Li B, Liang Y, Huang P, Wang Z, et al: Expression of variant
isoforms of the tyrosine kinase SYK determines the prognosis of
hepatocellular carcinoma. Cancer Res. 74:1845–1856. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Prinos P, Garneau D, Lucier JF, Gendron D,
Couture S, Boivin M, Brosseau JP, Lapointe E, Thibault P, et al:
Alternative splicing of SYK regulates mitosis and cell survival.
Nat Struct Mol Biol. 18:673–679. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yuan Y, Mendez R, Sahin A and Dai JL:
Hypermethylation leads to silencing of the SYK gene in human breast
cancer. Cancer Res. 61:5558–5561. 2001.PubMed/NCBI
|
|
22
|
Yan C, Liu C, Jin Q, Li Z, Tao B and Cai
Z: The promoter methylation of the Syk gene in nasopharyngeal
carcinoma cell lines. Oncol Lett. 4:505–508. 2012.PubMed/NCBI
|
|
23
|
Shin SH, Lee KH, Kim BH, Lee S, Lee HS,
Jang JJ and Kang GH: Downregulation of spleen tyrosine kinase in
hepatocellular carcinoma by promoter CpG island hypermethylation
and its potential role in carcinogenesis. Lab Invest. 94:1396–1405.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Davies JM and Goldberg RM: Treatment of
metastatic colorectal cancer. Semin Oncol. 38:552–560. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shen L, Yu M, Xu X, Gao L, Ni J, Luo Z and
Wu S: Knockdown of β3GnT8 reverses 5-fluorouracil resistance in
human colorectal cancer cells via inhibition the biosynthesis of
polylactosamine-type N-glycans. Int J Oncol. 45:2560–2568.
2014.PubMed/NCBI
|
|
27
|
Ladomery M: Aberrant alternative splicing
is another hallmark of cancer. Int J Cell Biol. 2013:4637862013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hatakeyama K, Wakabayashi-Nakao K, Ohshima
K, Sakura N, Yamaguchi K and Mochizuki T: Novel protein isoforms of
carcinoembryonic antigen are secreted from pancreatic, gastric and
colorectal cancer cells. BMC Res Notes. 6:3812013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kahkhaie KR, Moaven O, Abbaszadegan MR,
Montazer M and Gholamin M: Specific MUC1 splice variants are
correlated with tumor progression in esophageal cancer. World J
Surg. 38:2052–2057. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jacob AG, O'Brien D, Singh RK, Comiskey DF
Jr, Littleton RM, Mohammad F, Gladman JT, Widmann MC, Jeyaraj SC,
Bolinger C, et al: Stress-induced isoforms of MDM2 and MDM4
correlate with high-grade disease and an altered splicing network
in pediatric rhabdomyosarcoma. Neoplasia. 15:1049–1063. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Venables JP, Klinck R, Bramard A, Inkel L,
Dufresne-Martin G, Koh C, Gervais-Bird J, Lapointe E, Froehlich U,
Durand M, et al: Identification of alternative splicing markers for
breast cancer. Cancer Res. 68:9525–9531. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Miura K, Fujibuchi W and Unno M: Splice
isoforms as therapeutic targets for colorectal cancer.
Carcinogenesis. 33:2311–2319. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Geahlen RL: Getting Syk: Spleen tyrosine
kinase as a therapeutic target. Trends Pharmacol Sci. 35:414–422.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Puissant A, Fenouille N, Alexe G, Pikman
Y, Bassil CF, Mehta S, Du J, Kazi JU, Luciano F, Rönnstrand L, et
al: SYK is a critical regulator of FLT3 in acute myeloid leukemia.
Cancer Cell. 25:226–242. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shakeel S, Mahjabeen I, Kayani MA and
Faryal R: Association of SYK genetic variations with breast cancer
pathogenesis. Asian Pac J Cancer Prev. 14:3309–3314. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hoellenriegel J, Coffey GP, Sinha U,
Pandey A, Sivina M, Ferrajoli A, Ravandi F, Wierda WG, O'Brien S,
Keating MJ and Burger JA: Selective, novel spleen tyrosine kinase
(Syk) inhibitors suppress chronic lymphocytic leukemia B-cell
activation and migration. Leukemia. 26:1576–1583. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bailet O, Fenouille N, Abbe P, Robert G,
Rocchi S, Gonthier N, Denoyelle C, Ticchioni M, Ortonne JP,
Ballotti R, et al: Spleen tyrosine kinase functions as a tumor
suppressor in melanoma cells by inducing senescence-like growth
arrest. Cancer Res. 69:2748–2756. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sung YM, Xu X, Sun J, Mueller D, Sentissi
K, Johnson P, Urbach E, Seillier-Moiseiwitsch F, Johnson MD and
Mueller SC: Tumor suppressor function of Syk in human MCF10A in
vitro and normal mouse mammary epithelium in vivo. PLoS One.
4:e74452009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Larive RM, Urbach S, Poncet J, Jouin P,
Mascré G, Sahuquet A, Mangeat PH, Coopman PJ and Bettache N:
Phosphoproteomic analysis of Syk kinase signaling in human cancer
cells reveals its role in cell-cell adhesion. Oncogene.
28:2337–2347. 2009. View Article : Google Scholar : PubMed/NCBI
|