Introduction
Neuroblastoma arises from neural crest precursors
that do not differentiate into terminal neurons. The hallmark of
these tumors are the numerous different clinical variables, ranging
from highly metastatic with rapid progression and resistance to
therapy to spontaneous regression or development into benign
ganglioneuromas (1). Neuroblastoma
accounts for >7% of malignancies in pediatric patients (<15
years) (1). Approximately 65% of
primary tumors occur in the adrenal glands. Tumors also arise in
the neck, chest and pelvis. The symptoms of neuroblastoma are
dependent on tumor location, as well as the presence or absence of
metastasis and paraneoplastic syndromes (1). At diagnosis, 40% of cases are classified
as high-risk according to age of onset (<18 months) and the
presence of genetic mutations (2).
Despite advances in chemotherapy, 60% of high-risk neuroblastomas
recur and thus mortality rates are extremely high (3). For low-risk patients, surgery alone is
curative in the majority of cases, while intermediate-risk patients
(without MYCN amplification) are administered low doses of
cyclophosphamide and vincristine until resection is possible
(4). Treatments for high-risk
patients with stage III and IV neuroblastoma include
intensification of conventional chemotherapy agents with the
addition of 13-cis-retinoic acid. Clinical trials with viral agents
able to target tumor cells or immune therapies with exposure of
neuroblastoma cells to interferon γ, prior to conventional
chemotherapy, are returning encouraging results (5).
The different clinical variables correspond to
genetic mutations in crucial genes, including oncogenes,
oncosuppressor genes and transcription factors. Furthermore,
scientists are concentrating their efforts on determining the
network of regulatory microRNAs (miRs/miRNAs) and genes in
neuroblastoma (6,7). A possible therapeutic approach would be
to enhance the expression of miRNAs that induce differentiation
while suppressing those associated with tumor progression. As well
as regulating the expression of oncogenes and transcription
factors, miRNAs are associated with epigenetic mechanisms in
neuroblastoma, such as DNA methylation and the induction of
autophagy (8–10).
The key prognostic factor in neuroblastoma is MYCN
amplification (11), which occurs in
~20% of cases and is associated with poor outcome. MYCN
amplification is correlated to the transcription of proteins that
regulate differentiation, such as nestin (12), or HMG proteins, which suppress
apoptosis, induce autophagy and amplify AKT signaling (13,14). A
recent study demonstrated that high mobility group box 1 (HMGB1) is
upregulated in neuroblastoma (15),
inducing cell proliferation and autophagy in Schwann cells. In our
recent study, the HMGB1/receptor for advanced glycation end
products (RAGE)/miR-221/222 signaling pathway, which is associated
with cellular proliferation and phosphatase and tensin homolog
(PTEN) inhibition, was identified in a model of thyroid cancer
(16). In vitro models of
neuroblastoma derive from different tumors, and maintain their
heterogeneous genetic variances and, thus, tumorigenic properties.
It is known that each neuroblastoma and its deriving cell line has
at least three different phenotypic patterns that include stem,
neuroblastic and non-neuronal cells. The different phenotypes show
different tumorigenicity. Tumorigenicity in neuroblastoma is
directly associated with MYCN amplification and differently
expressed miRNAs (17). Furthermore,
numerous miRNAs induce neuroblastoma cell differentiation and
interact with endogenous substances. The understanding of these
interactions may provide novel strategies for the treatment of
neuroblastoma.
The present study reports the role of HMGB1 in an
MYCN-amplified neuroblastoma cell line [SK-N-BE(2)] and a neuroblastoma cell line without
MYCN amplification (SH-SY5Y). The results show that HMGB1 exerts
different effects on the two cell lines and that its interaction
with miR-221/222 influences important pathways associated with cell
growth.
Materials and methods
Reagents
Anti-human PTEN monoclonal antibody (cat. no. M3627;
1:1,000 dilution) was obtained from Dako (Carpinteria, CA, USA).
Monoclonal anti-mouse IgG horseradish peroxidase conjugate (cat.
no. NXA931; 1:2,000 dilution) was purchased from GE Healthcare
(Little Chalfont, UK). Monoclonal anti-human HMGB1 (cat. no. H9537;
1:2,000) and β-actin (cat. no. A5316; 1:10,000) antibodies were
purchased from Sigma-Aldrich (St. Louis, MO, USA). Human
recombinant HMGB-1 expressed in Escherichia coli was from
Sigma-Aldrich (cat no. H4652; St. Louis, MO, USA). miRIDIAN Hairpin
Inhibitor hsa-miRNA-221 (cat. no. IH300578-07-005) and
hsa-miRNA-222 (cat. no. IH301176-02 −005) and miRIDIAN microRNA
Hairpin Inhibitor Transfection Control with Dy547 (cat. no.
FE5IP0045000105) (control oligonucleotide) and DharmaFECT 1
Transfection Reagent (cat. no. FE5T200103) were purchased from GE
Healthcare Dharmacon Inc. (Lafayette, CO, USA). The mature
miRNA-221 sequence was 5′-AGCUACAUUGUCUGCUGGGUUUC-3′ and the mature
miRNA-222 sequence was 5′-CUCAGUAGCCAGUGUAGAUCCU-3′. Reverse
transcription (RT) primers and TaqMan probes were obtained from
TaqMan miRNA assays (Applied Biosystems, Foster City, CA, USA).
Cell lines
SK-N-BE(2) (DSMZ no.
ACC632) and SH-SY5Y (DSMZ no. ACC209) cells were obtained from DSMZ
(Braunschweig, Germany) in October 2012 and maintained in RPMI-1640
(Gibco; Thermo Fisher Scientific, Inc.), supplemented with
heat-inactivated 10% fetal calf serum (FCS) containing 2 mM
L-glutamine. Where required, 10 nM HMGB1 was added to the cultures
for 24, 48 or 72 h. Cell viability was determined by a trypan blue
exclusion test. Cells were frozen in nitrogen liquid tanks
(2×106 cells/ml FCS, 10% dimethylsulfoxide) until use
between the 4th and 8th passage after revival.
Cell viability
Cell viability was determined using the Trypan blue
exclusion test. SK-N-BE(2) or SH-SY5Y
cells (5×104/ml) were suspended in a 10-µl suspension
containing trypan blue (0.4% w/v; Sigma-Aldrich) in
phosphate-buffered saline (PBS) and incubated at room temperature
for 5 min, followed by examination under a light microscope to
determine the percentage of cells with clear (viable cells) and
blue cytoplasm (non-viable cells).
Knockdown of miRNA
AntagomiRs against miR-221 and −222, and control
oligonucleotide with Dy547 were transiently transfected into
SK-N-BE(2) and SH-SY5Y cells using
DharmaFECT 1 Transfection Reagent, prior to treatment with HMGB1.
Briefly, cells were seeded in 12-well plates at a density of
2×105 cells/well and incubated at 37°C for 18 h prior to
transfection. Subsequently, culture medium was replaced with 1 ml
antibiotic-free medium containing DharmaFECT 1 Transfection Reagent
(2 µl/ml) and antagomiRs at a final concentration of 25 nM for 24
h, according to manufacturer's instructions. Controls with Dy547
and DharmaFECT 1 Transfection Reagent alone were included in the
experiments as indicators of transfection efficiency and as
negative controls, respectively. Transfection efficiency was
determined by cytofluorimetric readings in the red light spectrum
(575 nm; Cytofluorimetric Coulter Epics XL; Beckman Coulter, Brea,
CA, USA) and reached 69–76% in Dy547-positive cells.
Relative quantification of miRNA by
RT-quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from SK-N-BE(2) and SH-SY5Y cells with TRIzol reagent
(Life Technologies; Thermo Fisher Scientific, Inc.), according to
the manufacturer's instructions. Briefly, 1 ml TRIzol and 0.2 ml
chloroform (Sigma-Aldrich) were added to pellets and centrifuged at
12,000 × g at 4°C for 15 min. For the detection of mature miR-221
and −222, 50 ng total RNA was reverse transcribed using the TaqMan
MicroRNA Reverse Transcription kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.). qPCR was performed using an
miRNA-specific TaqMan assay (cat. no. PN4427975; Applied
Biosystems; Thermo Fisher Scientific, Inc.) and hsa-miR221 and −222
and an ABI Prism 7900HT Sequence Detection System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). PCR conditions were as
follows: 50°C for 2 min and 95°C for 10 min, followed by 40 cycles
of 95°C for 15 sec and 60°C for 1 min. Amplifications were
performed in triplicate and repeated twice. The ubiquitously
expressed U6b small nuclear RNA was used for normalization. An
experimental negative control (no cDNA) was used, which confirmed
the absence of amplification. Relative quantification of miRNA
expression was performed using the ∆∆Cq method (18).
Western blot analysis
Briefly, whole cell lysates were heat denatured for
5 min, loaded on a standard Tris-HCl 12.5% SDS-PAGE gel, and run on
ice at 40 V for the stacking gel and 80 V for the running gel to
separate the proteins, as previously described (12). Proteins were transferred onto a
previously activated PVDF membrane (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Membranes were then placed in Tris-buffered
saline (Sigma-Aldrich) with Tween 20 and 5% bovine serum albumin
(Sigma-Aldrich) for 1 h, and probed overnight with the specific
antibody at 4°C. At the end of incubation time, membranes were
washed with Tris-buffered saline and incubated with anti-mouse IgG
peroxidase conjugated secondary antibody (1:2,000 dilution) for 1 h
at room temperature. Membranes were stripped with stripping buffer
(β-mercaptoethanol, SDS and Tris-HCl) and incubated with β-actin
monoclonal antibody as a loading control. Signals were detected by
autoradiography (Kodak Biomax; Kodak, Rochester, NY, USA) using a
chemiluminescent peroxidase substrate kit (Sigma-Aldrich), then
quantified by densitometric analysis using Quantity-One software
(version 4.6.5; Bio-Rad Laboratories, Inc.).
Cytofluorimetric determination of cell
cycle
Parental control cells and transfectant cells
(105) were seeded into a 6-well plate with and without
10 nM HMGB1. After 24 h, cells were fixed with cold 70% ethanol in
PBS for 1 h at 4°C. After centrifugation at 200 × g for 10 min at
4°C, cells were washed once in PBS. The pellet was resuspended in a
solution of 0.5 ml propidium iodide (PI; 0.1 mg/1 ml in PBS;
Sigma-Aldrich) and 50 µl RNAse (Type I-A; 10 mg/ml in PBS;
Sigma-Aldrich). PI-stained nuclei were incubated in the dark at
room temperature for 15 min and maintained at 4°C until the nuclear
DNA content of the cells was evaluated by flow cytometry
(Cytofluorimetric Coulter Epics XL; Beckman Coulter).
Statistical analysis
All statistical analyses were performed using
KaleidaGraph version 4.5.1 (Synergy Software Inc., Reading, PA,
USA). All determinations were performed three times. Data are
expressed as means + standard deviation. The differences between
cell populations [SK-N-BE(2) cells
and SH-SY5Y cells, untransfected or transfected with antagomiRs;
untreated or treated with HMGB1] were analyzed for miR-221/222,
HMGB1 and PTEN expression, using Student's t-test. P-values were
evaluated in the differently treated cell populations. P<0.05
was considered to indicate a statistically significant
difference.
Results
Effect of HMGB1 on miR-221 and −222
expression in SK-N-BE(2) and SH-SY5Y
cells
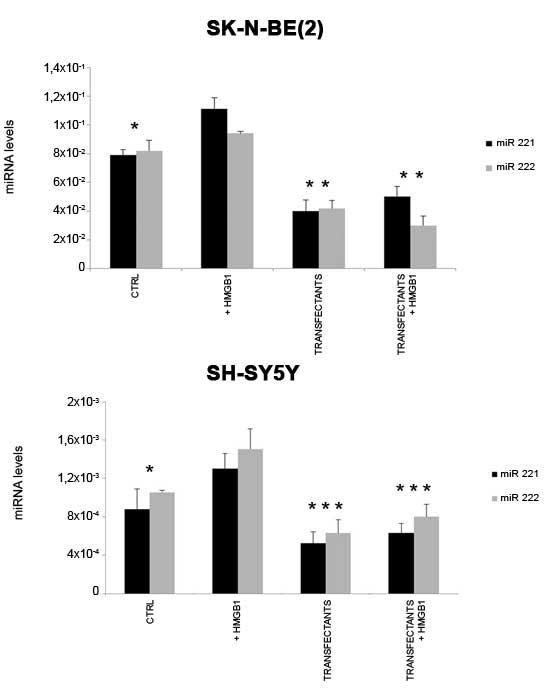
Fig. 1 shows that the
two cell lines express different amounts of miR-221 and −222.
SK-N-BE(2) cells express markedly
higher levels of miR-221 (100-fold; P=0.004) and miR-222 (80-fold;
P=0.007) than SH-SY5Y cells.
Treatment with 10 nM HMGB1 for 24 h increased
expression of miR-221 and −222 in both SK-N-BE(2) and SH-SY5Y cells. By contrast,
transfection of SK-N-BE2 cells with miR-221 and −222 antagomiRs
reduced their expression by 49% each compared with the controls. In
transfected SH-SY5Y cells, expression was reduced by 41% for
miR-221 and 40% for miR-222, compared with the controls.
The treatment of transfected cells (transfectants)
with HMGB1 for 24 h did not markedly increase or decrease the
expression of miR-221/222 in either cell line.
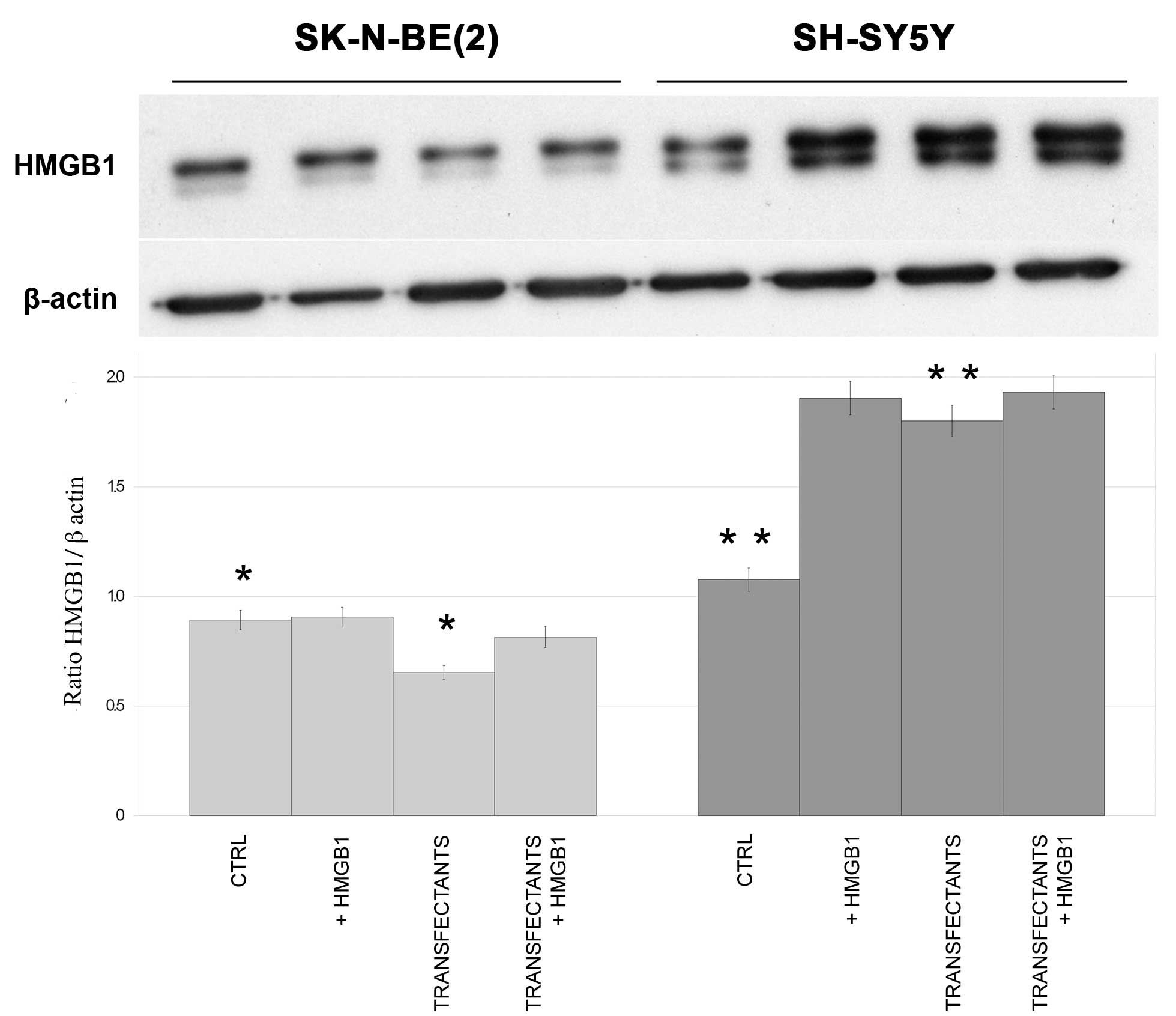
Expression of HMGB1 in parental
control and transfectant SK-N-BE(2)
and SH-SY5Y cells
Endogenous HMGB1 protein was expressed in both cell
lines (Fig. 2). While endogenous
HMGB1 levels decreased in SK-N-BE(2)
transfectants compared with the controls (P=0.04), the opposite
effect was observed in SH-SY5Y transfectants, which expressed
markedly higher levels of HMGB1 than the control SH-SY5Y cells
(P=0.003).
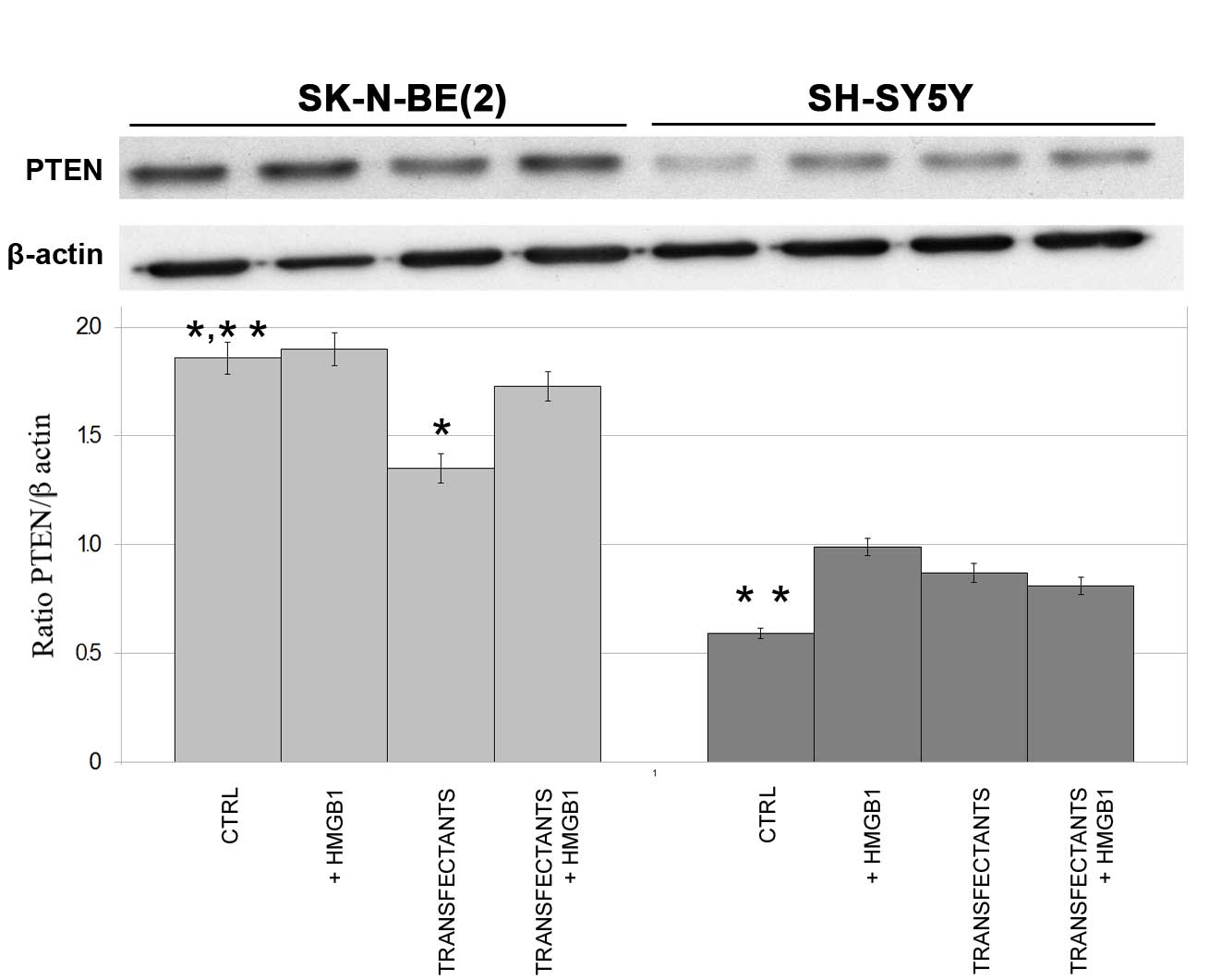
Expression of PTEN in miR-221 and −222
in parental control and transfectant SK-N-BE(2) and SH-SY5Y cells
To show that the action of exogenous HMGB1 on miRNAs
is functional, the expression of PTEN, a known target of
miR-221/222, was analyzed in control and transfectant cells.
Fig. 3 shows that PTEN expression is
markedly decreased in SK-N-BE(2)
transfectants (miR-221/222) compared with controls, while the
addition of HMGB1 increases PTEN expression to the same levels as
the controls (P=0.008). The decreased PTEN expression in
SK-N-BE(2) transfectant cells may be
explained by the different roles of miRNAs in neuroblastoma, as the
same miRNAs may induce proliferation or survival of certain cells
and concurrent differentiation of others.
In SH-SY5Y cells, HMGB1 appeared to increase PTEN
expression compared with the control cells, while it did not show
any effect in the transfectant cells. It is of note that PTEN
expression in control SH-SY5Y cells was markedly lower than in
control SK-N-BE(2) cells (P=0.001),
in agreement with a previous study (19).
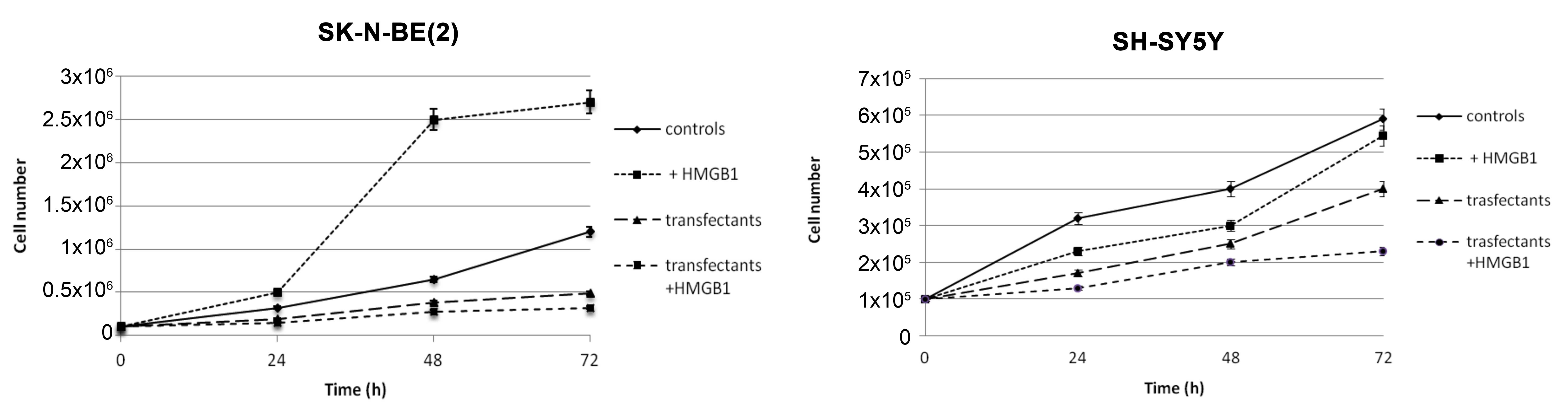
HMGB1 has opposite effects on the
growth of SK-N-BE(2) and SH-SY5Y
cells
Fig. 4 shows growth
curves of parental control and transfectant SK-N-BE(2) and SH-SY5Y cells. The addition of 10 nM
HMGB1 to SK-N-BE(2) cells for 24, 48
and 72 h induced an increase in growth of 56, 284 and 125%.
Notably, the opposite result was obtained in SH-SY5Y cells cultured
in the presence of HMGB1, with growth increasing to 28 and 75% at
24 and 48 h, but decreasing to 0.9% at 72 h. SK-N-BE(2) cells transfected with miR-221 and −222
antagomiRs demonstrated a decrease in growth compared with control
cells. HMGB1 added to transfectant SK-N-BE(2) and SH-SY5Y cells further reduced growth
compared with untreated transfectants.
Analysis of DNA content in parental control cells
and transfectants of both cell lines treated with or without HMGB1
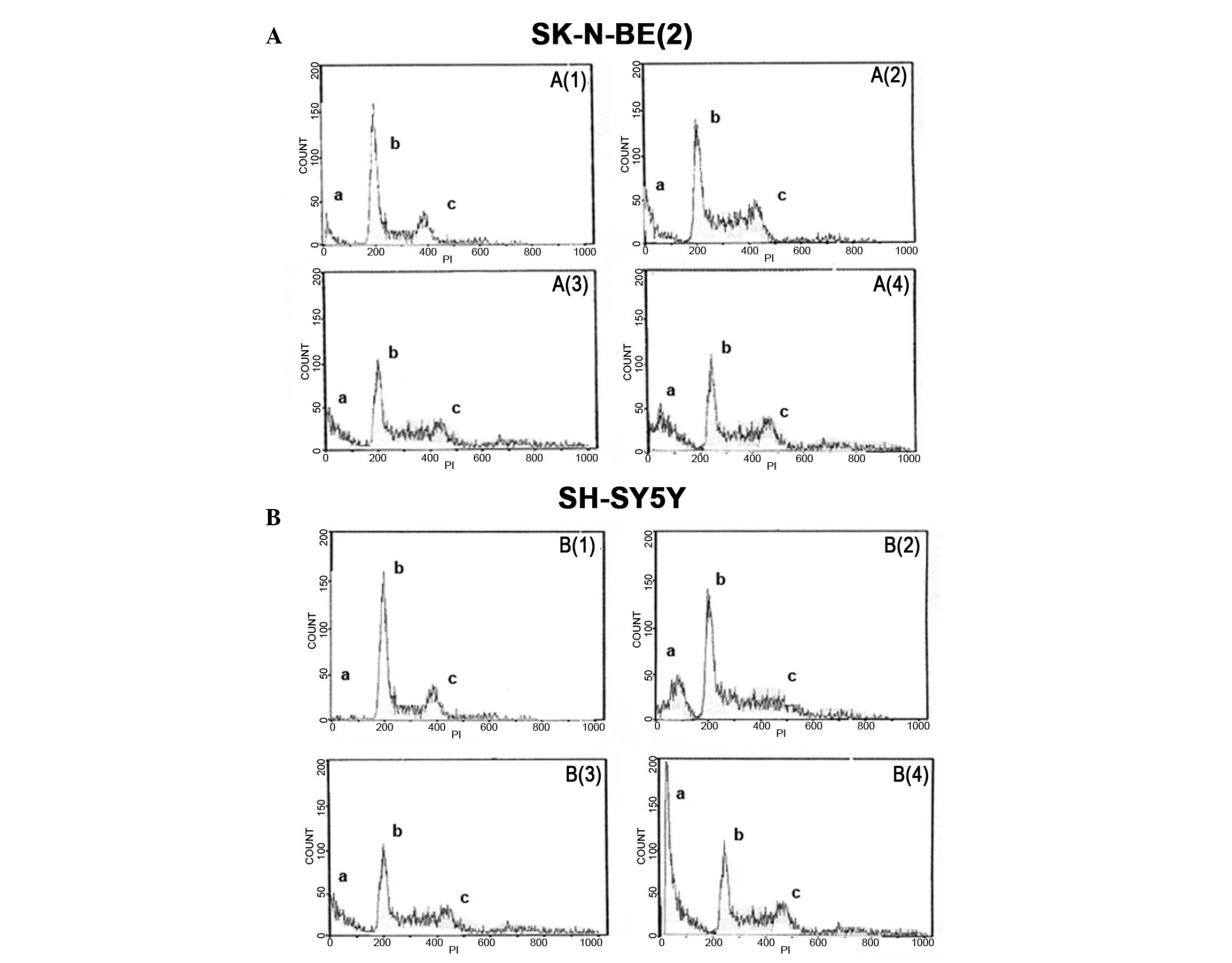
for 24 h confirmed the growth curves results. As shown in Fig. 5, a high tetraploid peak was observed
in SK-N-BE(2) cells following
treatment with HMGB1, while the tetraploid peak was reduced in
transfectant cells (A3) and there was a high hypodiploid peak in
SK-N-BE(2) transfectant cells (A4),
indicating cellular apoptosis. Regarding SH-SY5Y cells, the
reduction in growth following treatment with HMGB1 was confirmed by
the high hypodiploid peak (B2) and low tetraploidy (B4) peaks
observed in both transfectant and control cells following treatment
with HMGB1.
Discussion
Transcription of miR-221 is induced by N-Myc
transcription factor (20) in
MYCN-amplified SK-N-BE(2) cells. The
present study demonstrated that miR-221 and −222 are more highly
expressed in SK-N-BE(2) cells than in
SH-SY5Y cells. It is known that the miR-221/222 cluster targets
PTEN (21), an oncosuppressor gene
that is considered to be one of the primary regulators of
AKT-activating pathways. The PTEN gene is frequently lost on
chromosome 10q2317 in various types of human cancer. Notably,
aberrant methylation of the PTEN gene has been observed in
neuroblastoma tissues, resulting in PTEN inactivation and AKT
hyperactivation. Our previous study reported that HMGB1 decreases
PTEN expression in thyroid cancer cell lines by cooperating with
the miR-221/222 cluster. Similarly, the present study reports that
HMGB1 has an effect on neuroblastoma cell lines by enhancing
miR-221 and −222 expression in both MYCN-amplified and
non-amplified cell lines. The growth increase induced by HMGB1 in
SK-N-BE(2) cells may be explained by
the known association between N-Myc transcription factor and HMGB1,
which acts on AKT-activating pathways (22). Furthermore, the present study showed
that HMGB1 enhances miR-221/222 expression and induces growth in
SK-N-BE(2) cells. When miR-221/222
expression was reduced by antisense oligonucleotides, PTEN
expression also decreased; however, the addition of HMGB1 for 24 h
was able to re-establish PTEN function and, consequently, reduce
the cell growth rate. We hypothesize that HMGB1 exerts its action
on miR-221/222 and PTEN expression as a consequence of their
interaction. Furthermore, the decreased PTEN expression observed in
SK-N-BE(2) cells may be explained by
the different roles of miRNAs in neuroblastoma, as the same miRNAs
may induce both inhibition and potentiation of certain mRNAs in the
different neuroblastoma lines. It is known that exogenous HMGB1
binds RAGE, which in turn activates intracellular pathways that
lead to AKT activation. The present study identified a possible
pathway through which HMGB1 increases the growth of
SK-N-BE(2) cells, triggered by the
action of miR-221/222 on PTEN. This is supported by the observation
that when miR-221 and −222 function are reduced by antagomiRs,
HMGB1 increases PTEN expression. In SH-SY5Y cells, which express
markedly less miR-221 and −222 than SK-N-BE(2) cells, HMGB1 reduces growth. The
differential expression of miR-221/222 in the two cell lines
reported in the present study is consistent with a previous finding
that expression of miR-21, −221 and −335 are associated with
non-tumorigenic and neuroblastoma cell differentiation (23). By contrast, RAGE (24), which interacts with HMGB1, has a
prominent role in neuritic extension in neurons (25). During early development, neurons
undergoing differentiation express higher levels of RAGE and HMGB1
(26). A previous study (27) reported that the interaction between
RAGE and HMGB1 potentiates retinoic acid-induced differentiation of
SH-SY5Y cells. Furthermore, in thyroid cancer, in which the
miR-221/222 cluster has an oncogenic effect, it has been shown that
blocking RAGE prevents the action HMGB1 on miRNAs. Therefore, it is
hypothesized that binding of RAGE with HMGB1 can regulate the
actions of miR-221/222, even in neuroblastoma cells (unpublished
data).
In conclusion, HMGB1 acts on PTEN through the
enhancement of miR-221/222 in SK-N-BE(2) and SH-SY5Y cells. In cells transfected
with miR-221/222 antagomiRs, HMGB1 was able to enhance PTEN
expression and consequently reduce cell growth. The present study
indicates that HMGB1 contributes to neuroblastoma growth and
differentiation by cooperating with miRNAs that in turn function as
oncogenes, oncosuppressor genes or differentiating factors. The
present study indicates that HMGB1 signaling bridges the
extracellular microenvironment of neuroblastoma and its
tumorigenicity via the enhancement of miR-221/222 that in turn
suppress the oncosuppressor PTEN. Accordingly, targeting HMGB1 and
miR-221/222 may provide novel strategies for therapies in
neuroblastoma as extracellular HMGB1 is an inflammatory molecule
that is implicated in the pro-tumorigenic microenvironment and
increased expression of oncogenic miRs in neuroblastoma.
Acknowledgements
The authors thank Dr Emily Bowles for English
proofreading.
References
|
1
|
Maris JM, Hogarty MD, Bagatell R and Cohn
SL: Neuroblastoma. Lancet. 369:2106–2120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thompson D, Vo KT, London WB, Fischer M,
Ambros PF, Nakagawara A, Brodeur GM, Matthay KK and DuBois SG:
Identification of patient subgroups with markedly disparate rates
of MYCN amplification in neuroblastoma: A report from the
International Neuroblastoma Risk Group project. Cancer.
122:935–945. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pinto NR, Applebaum MA, Volchenboum SL,
Matthay KK, London WB, Ambros PF, Nakagawara A, Berthold F,
Schleiermacher G, Park JR, et al: Advances in risk classification
and treatment strategies for neuroblastoma. J Clin Oncol.
33:3008–3017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rubie H, De Bernardi B, Gerrard M, et al:
Excellent outcome with reduced treatment in infants with
nonmetastatic and unresectable neuroblastoma without MYCN
amplification: results of the prospective INES 99.1. J Clin Oncol.
(29): 449–455. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wagner LM and Danks MK: New therapeutic
targets for the treatment of high-risk neuroblastoma. J Cell
Biochem. (107): 46–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang L, Che XJ, Wang N, Li J and Zhu MH:
Regulatory network analysis of microRNAs and genes in
neuroblastoma. Asian Pac J Cancer Prev. 15:7645–7652. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin RJ, Lin YC, Chen J, Kuo HH, Chen YY,
Diccianni MB, London WB, Chang CH and Yu AL: MicroRNA signature and
expression of dicer and drosha can predict prognosis and delineate
risk groups in neuroblastoma. Cancer Res. 70:7841–7850. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cui C, Yu J, Huang S, Zhu H and Huang Z:
Transcriptional regulation of gene expression by micro RNAs
endogenous decoys of transcription factors. Cell Physiol Biochem.
33:1698–1714. 2010. View Article : Google Scholar
|
|
9
|
Mei H, Lin ZY and Tong QS: The roles of
microRNAs in neuroblastoma. World J Pediatr. 10:10–16. 2010.
View Article : Google Scholar
|
|
10
|
Samaraweera L, Grandinetti KB, Huang R,
Spengler BA and Ross RA: MicroRNAs define distinct human
neuroblastoma cell phenotypes and regulate their differentiation
and tumorigenicity. BMC Cancer. 14:3092014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schulte JH, Horn S, Otto T, Samans B,
Heukamp LC, Eilers UC, Krause M, Astrahantseff K, Klein-Hitpass L,
Buettner R, et al: MYCN regulates oncogenic MicroRNAs in
neuroblastoma. Int J Cancer. 122:699–704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thomas SK, Messam CA, Spengler BA, Biedler
JL and Ross RA: Nestin is a potential mediator of malignancy in
human neuroblastoma cells. J Biol Chem. 279:27994–27999. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rothermund K, Rogulski K, Fernandes E,
Whiting A, Sedivy J, Pu L and Prochownik EV: C-Myc-independent
restoration of multiple phenotypes by two C-Myc target genes with
overlapping functions. Cancer Res. 65:2097–2107. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Armstrong MB, Mody RJ, Ellis DC, Hill AB,
Erichsen DA and Wechsler DS: N-Myc differentially regulates
expression of MXI1 isoforms in neuroblastoma. Neoplasia.
15:1363–1370. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu Y and Song L: HMGB1-induced autophagy
in Schwann cells promotes neuroblastoma proliferation. Int J Clin
Exp Pathol. 8:504–510. 2015.PubMed/NCBI
|
|
16
|
Mardente S, Mari E, Massimi I, Fico F,
Faggioni A, Pulcinelli F, Antonaci A and Zicari A: HMGB1-induced
cross talk between PTEN and miRs 221/222 in thyroid cancer. Biomed
Res Int 2015:: 512027. 2015. View Article : Google Scholar
|
|
17
|
Ayers D, Mestdagh P, Van Maerken T and
Vandesompele J: Identification of miRNAs contributing to
neuroblastoma chemoresistance. Comput Struct Biotechnol J.
13:307–319. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qiao L, Paul P, Lee S, Qiao J, Wang Y and
Chung DH: Differential regulation of cyclin-dependent kinase
inhibitors in neuroblastoma cells. Biochem Biophys Res Commun.
435:295–299. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guglielmi L, Cinnella C, Nardella M,
Maresca G, Valentini A, Mercanti D, Felsani A and D'Agnano I: MYCN
gene expression is required for the onset of the differentiation
programme in neuroblastoma cells. Cell Death Dis. 5:e10812014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li W, Guo F, Wang P, Hong S and Zhang C:
miR-221/222 confers radioresistance in glioblastoma cells through
activating Akt independent of PTEN status. Curr Mol Med.
14:185–195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mardente S, Mari E, Consorti F, Di Gioia
C, Negri R, Etna M, Zicari A and Antonaci A: HMGB1 induces the
overexpression of miR-222 and miR-221 and increases growth and
motility in papillary thyroid cancer cells. Oncol Rep.
28:2285–2289. 2012.PubMed/NCBI
|
|
23
|
Zhao Z, Ma X, Hsiao THM, Lin G, Kosti A,
Yu X, Suresh U, Chen Y, Tomlinson GE, Pertsemlidis A and Du L: A
high-content morphological screen identifies novel microRNAs that
regulate neuroblastoma cell differentiation. Oncotarget.
15:2499–2512. 2014. View Article : Google Scholar
|
|
24
|
Schmidt AM, Yan SD, Yan SF and Stern DM:
The biology of the receptor for advanced glycation end products and
its ligands. Biochim Biophys Acta. 1498:99–111. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sajithlal G, Huttunen H, Rauvala H and
Munch G: Receptor for advanced glycation end products plays a more
important role in cellular survival than in neurite outgrowth
during retinoic acid-induced differentiation of neuroblastoma
cells. J Biol Chem. 277:6888–6897. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen J, Song M, Yu S, Gao P, Yu Y, Wang H
and Huang L: Advanced glycation endproducts alter functions and
promote apoptosis in endothelial progenitor cells through receptor
for advanced glycation endproducts mediate overpression of cell
oxidant stress. Mol Cell Biochem. 335:137–146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang L, Li S and Jungalwala FB: Receptor
for advanced glycation end products (RAGE) mediates neuronal
differentiation and neurite outgrowth. J Neuroscience Res.
86:1254–1266. 2010. View Article : Google Scholar
|