Introduction
Glioblastoma multiforme (GBM) is an astrocytoma of
malignancy grade 4, thus, it is a glioma with the highest degree of
histological abnormality (1,2). This malignant glioma grows rapidly due
to the loss of signals that inhibit cell cycle progression and the
increased signaling mediated by growth factors (3,4). The
standard treatment consists of maximum surgical resection,
radiotherapy and chemotherapy with temozolomide (TMZ), which is the
first-line therapeutic agent for GBM (3,5). TMZ is a
lipophilic substance with a low molecular weight (194.15 g/mol),
meaning it is well absorbed by the oral route and crosses the
blood-brain barrier to reach the target site (6). This alkylating agent generates cytotoxic
DNA lesions in tumor cells that results in cell cycle arrest and
apoptosis. Chemotherapy with TMZ has shown clinical benefits in
increasing mean survival time and improving patient quality of life
(7). However, survival time is only
extended by 2.5 months in comparison with radiation therapy alone.
Indeed, ~70% of patients with GBM present no benefit following TMZ
treatment, highlighting the urgent need for novel anticancer
approaches (8).
Voltage-gated potassium channels have gained
increased attention in research regarding tumor biology and
therapeutics (9). Ether à go-go 1
(Eag1) is a potassium channel that is overexpressed in various
types of tumor, which plays a role in tumorigenesis (10,11). Both
low-grade and high-grade human gliomas express Eag1 (12). To the best of our knowledge, no
previous studies have evaluated whether TMZ effects Eag1 expression
in glioblastoma cells. However, TMZ is known to lead to an increase
in p53 protein expression, which accounts for its antitumor effects
(13). In addition, technologies that
restore wild-type p53 function improve the effects of TMZ on glioma
cells (14). p53 protein negatively
regulates Eag1 (15,16); therefore, we hypothesize that TMZ, at
least indirectly, may reduce Eag1 expression, amplifying the drug
effects on glioma cells.
Eag1 is a definitive oncological target. Previous
studies have utilized different approaches to reveal that
decreasing Eag1 activity can undermine tumor progression (17). First, Eag1 blockers imipramine and
astemizole were shown to reduce tumor cell growth (18–20).
Monoclonal antibodies against Eag1 also had the ability to control
tumor development, and proved as effective as the standard agent,
cyclophosphamide, in a mouse model of breast cancer (21). Finally, RNA interference (RNAi), the
‘state of art’ tool for gene therapy, has also been applied to
determine the role of Eag1 in cancer. RNAi-based agents for cancer
therapy are already in clinical testing and will become approved
treatments in the coming years (22).
Small-interfering RNAs (siRNAs) designed to silence Eag1 may reduce
the growth of different cancer cells in culture (23). Our previous studies developed a
plasmid vector able to express short hairpin RNA (shRNA) that
targets Eag1 mRNA, termed pKv10.1-3. This expression vector also
reduced the viability of glioblastoma cells and increased the cell
damage caused by interferon-γ (IFN-γ), a therapeutic agent for
brain tumors (24,25).
In the present study, the effect of TMZ on Eag1
expression was examined in U87MG glioblastoma cells. In addition,
the study evaluated whether silencing Eag1 with shRNA would
increase the cell damage caused by TMZ, the first-line therapeutic
agent for glioblastoma.
Materials and methods
Cell culture
U87MG human glioblastoma cells were purchased from
the Bank of Cells of Rio de Janeiro (Rio de Janeiro, Brazil). Cells
were maintained with Dulbecco's modified Eagle medium (DMEM)/F12
[supplemented with 10% (v/v) heat-inactivated fetal calf serum, 1%
GlutaMAX and 1% penicillin/streptomycin solution, all obtained from
Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA)] in
25-cm3 culture flasks at 37°C in a 5% CO2
atmosphere.
Drug treatment
TMZ (Orion Corporation, Espoo, Finland) and
astemizole (Sigma-Aldrich, Madrid, Spain) were dissolved in
dimethyl sulfoxide (DMSO; Sigma-Aldrich) and stored at −20°C. The
final concentration of DMSO in culture medium did not exceed 0.01%;
therefore, it had no influence on cell viability, according to
preliminary experiments (data not shown). Details of each
experimental procedure are presented in the figure legends and
schematic representations.
Cell viability measurement by MTT
assay
Cell viability was determined by the MTT method.
Briefly, glioma cells were grown in 96 well plates in triplicates
(104 cells/well) at 37°C in a 5% CO2
atmosphere. ATZ blocker (5 µM) was added 30 minutes prior to TMZ
(250 µM) treatment for 24 h. Subsequently, culture medium was
exchanged and incubated for an additional 48 h. Mock control group
were cells without ATZ blocker and TMZ treatment. After each
experimental treatment, the cells were incubated with 15 µl MTT (5
mg/ml in DMEM) for 4 h at 37°C. Subsequently, the medium was
removed and 150 µl DMSO was added to each well to dissolve formazan
crystals. A microplate reader (SpectraMax M2 Microplate Reader;
Molecular Devices, Sunnyvale, CA, USA) calibrated to read
absorbance at 595 nm was used to quantify the formazan product. All
experiments were performed in triplicate. The number of independent
assays performed are indicated in the figure legends.
Cell transfection method
The present study used a previously described shRNA
expression vector targeting the Eag1 mRNA sequence
(5′-GTCCACTTGGTCCATGTCCAG-3′), termed pKv10.1-3, and the negative
control pScramble (24). Following
the experimental treatment, Lipofectamine 2000 and Opti-MEM
(Invitrogen; Thermo Fisher Scientific, Inc.) were used to transfect
pKv10.1-3 and pScramble, according to the manufacturer's
instructions. Both fragments were cloned into the pSilencer 3.1-H1
vector. Briefly, glioblastoma cells were grown for 24 h and then
transfected in Opti-MEM for 6 h at 37°C in a 5% CO2 atmosphere.
Subsequently, the medium was exchanged for DMEM/F12 (Thermo Fisher
Scientific, Inc.), following by addition of TMZ at 250 µM or 500
µM. Glioblastoma cells without any treatment comprised the mock
control group.
RNA isolation, cDNA synthesis and
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR)
Monolayer cell cultures were grown in 6-well plates
with a density of 6×105 cells per well. Total RNA was
extracted using an RNeasy Mini kit (Qiagen, Hilden, Germany),
according to the manufacturer's instructions, and quantified by
fluorometry (Qubit 2.0 firmware 3.11; Thermo Fisher Scientific,
Inc.). Purity was considered acceptable for RNA/protein ratios
>1.8. RNA integrity was analyzed by agarose gel electrophoresis
using ethidium bromide (Invitrogen™; Thermo Fisher Scientific,
Inc.). 1 Kb DNA Ladder (Thermo Fisher Scientific, Inc.) was used as
a molecular weight marker. Absence of DNA contamination following
RNA extraction was checked by fluorometry and qPCR with negative RT
controls. cDNA synthesis was performed from 2.5 ng total RNA using
oligo(dT) primers (SuperScript First-Strand Synthesis System for
RT-PCR; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. The qPCR reaction was conducted in a
QuantStudio 12K Flex Real-Time PCR System (Applied Biosystems™;
Thermo Fisher Scientific, Inc.). The forward and reverse primers
for Eag1 were 5′-TTGGAGATGTGTTCTGGAAGGAA-3′ and
5′-AGGGCATCCCGCTTGATC-3′, respectively (26). Poly(A) polymerase alpha (PAPOLA) was
used as the reference gene with the following primers: Forward,
5′-GCTACGAAGACCAGTCCATTG-3′ and reverse, 5′-TGTTGGTCACAGATGCTGCT-3′
(24,27). PAPOLA was selected as the reference
gene based on a previous study that evaluated the stability of
various endogenous reference genes in tumor cells (27). That study also presented four genes
that had a high gene expression stability score; therefore, the
present study analyzed these four genes: TATA-binding protein
(forward 5′-GCTGGCCCATAGTGATCTTT-3′ and reverse
5′-CTTCACACGCCAAGAAACAGT-3′); GC-rich promoter binding protein
(forward 5′-TCACTTGAGGCAGAACACAGA-3′ and reverse
5′-AGCACATGTTTCATCATTTTCAC-3′); Cullin 1 (forward
5′-GCGAGGTCCTCACTCAGC-3′ and reverse
5′-TTCTTTCTCAATTAGAATGTCAATGC-3′); and PAPOLA. Among them, PAPOLA
showed the lowest rate of Cq value variation (qPCR) in glioma cells
with Eag1 silencing compared with controls (~2%), an experimental
condition that also reduces cell viability. Amplification products
were detected via intercalation of the fluorescent dye Fast SYBR
Green Master Mix (Applied Biosystems™). Briefly, 10 µl reaction mix
contained 5.0 µl Fast SYBR Green Master mix, 2.0 µl cDNA, and 0.4
µl each forward and reverse primer (10 pmol/µl). The PCR cycling
conditions included an initial denaturation step at 95°C for 5 min,
followed by 40 cycles of amplification (95°C for 1 min, 60°C for 1
min). Each sample was analyzed in triplicate, and the assay
included non-template negative RT controls. The relative
quantification method (ΔΔCq) was used to express the RNAi effects
on Eag1 mRNA expression (28).
Samples without RNA were used as RT negative controls.
Flow cytometry apoptosis assay
An Alexa Fluor 488 Annexin V/Dead Cell Apoptosis kit
(Invitrogen; Thermo Fisher Scientific, Inc.) was used for apoptosis
analysis. Samples were prepared according to the manufacturer's
protocol with minor modifications. In brief, 1×105 cells
were plated in 12-well plates. Following treatment, cells were
washed with phosphate-buffer saline (PBS) and resuspended in a
solution containing 100 µl binding buffer, 5 µl Annexin
V-fluorescein isothiocyanate (FITC) and 10 µl propidium iodide (PI)
for 10 min in the dark at room temperature. Next, 400 µl binding
buffer was added to the cells and 10,000 events were acquired for
each sample. PI fluorescence was analyzed in a flow cytometer
(FACSVerse™; BD Biosciences, Franklin Lakes, NJ, USA) at 617 nm and
the Annexin V-FITC fluorescence was detected at 488 nm. Following
acquisition, data were analyzed using FlowJo version 7.6.5 software
(Tree Star Inc., Ashland, OR, USA).
Immunocytochemistry and image
analysis
For immunocytochemical detection of Eag1, U87MG
glioblastoma cells (1×105) were first cultured on
coverslips in 24-well plates. Then, cells were fixed with 4%
paraformaldehyde for 15 min and permeabilized with 10% Triton X-100
in PBS for 10 min. Nonspecific binding was blocked with 5% horse
serum (Gibco™; Thermo Fisher Scientific, Inc.) in PBS for 1 h. The
previously described Eag1 green fluorescence-labeled primary mouse
monoclonal antibody, anti-Eag1.62.mAb (kindly provided by Prof.
Walter Stuhmer, Max Planck Institute for Biophysical Chemistry,
Göttingen, Germany) (21), was used
at a concentration of 1 µg/ml (1:500 dilution) and incubated
overnight at 4°C. The cells were incubated with Alexa Fluor
488-labeled anti-mouse IgG antibody (catalog no., A-11001; 1:2,000
dilution; Molecular Probes; Thermo Fisher Scientific, Inc.) for 1 h
in the dark at room temperature. The cell nuclei were labeled with
Hoechst 33342 dye (Sigma-Aldrich) for 5 min at room temperature. To
evaluate the morphology of cytoskeleton, a commercial kit for
phalloidin detection (Alexa Fluor 532 Phalloidin; 200 U/ml;
Molecular Probes; Thermo Fisher Scientific, Inc.) was used,
according to the manufacturer's protocol. The coverslips were
mounted and observed with a confocal microscope (TCS SP5; Leica
Microsystems, Wetzlar, Germany).
The following formula was used to measure the
corrected total cell fluorescence (CTCF) for Eag1: CTCF =
integrated density - (area of selected cell × mean fluorescence of
background readings). Cell morphology was visualized using a binary
(mask-like) image of phalloidin detection and cell nuclei labeled
with Hoechst 33342 dye (Sigma-Aldrich). Image analysis was
conducted using ImageJ version 1.47 software (National Institutes
of Health, Bethesda, MD, USA). Each experimental condition was
performed in triplicate in three independent experiments.
Statistical analysis
All data were analyzed using SPSS software (version
20.0; IBM SPSS, Armonk, NY, USA) and expressed as mean ± standard
error of the mean. One-way analysis of variance followed by Tukey's
test was used to determine differences between groups. Differences
between pairs of experimental groups were analyzed by Student's
t-test. P<0.05 was used to indicate a statistically significant
difference.
Results
TMZ affects the viability of U87MG
glioblastoma cells
Initially, a dose-response curve was constructed to
examine the cell damage of TMZ on glioblastoma cells by performing
an MTT assay. The mock control group, without any treatment, was
taken as 100% viability. TMZ caused a decrease in cell viability in
all time periods examined (24, 48 and 72 h). As shown in Fig. 1A, the intensity of cell damage varied
according to the TMZ concentration and the duration of treatment.
At 125 µM, TMZ caused a slight but stable decrease in cell
viability compared with the mock group (100%) to 91, 87 and 89%
after 24, 48 and 72 h, respectively (Fig.
1A). However, cells exposed to 250 µM TMZ exhibited a
significant time-dependent change in viability, from 84% at 24 h to
54% at 72 h (P=0.001) and 75% at 48 h to 54% at 72 h (P=0.003)
(Fig. 1A). At the highest
concentration evaluated (500 µM) TMZ also caused a time-dependent
effect. The viability of glioblastoma cells significantly varied
between 59 and 13% between the 24 and 72 h time-points (Fig. 1A; P=0.001).
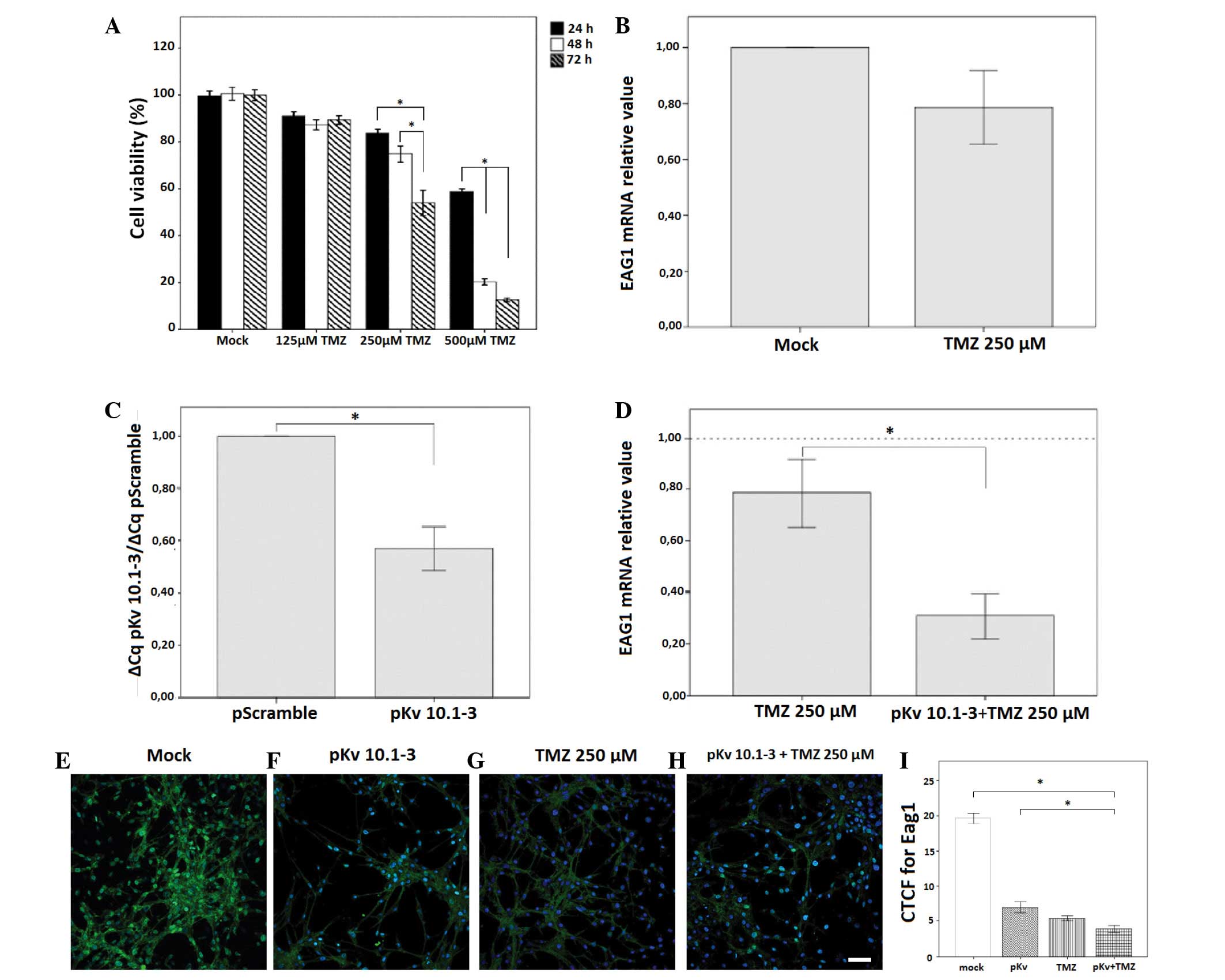 | Figure 1.Effects of TMZ and pKv10.1-3 on U87MG
cell viability and Eag1 mRNA expression. (A) Cell viability in each
TMZ-treated group (125, 250 or 500 µM) was expressed in comparison
with the mock control values at 24, 48 and 72 h, as determined by
MTT assay. Experimental groups were analyzed in triplicate in eight
independent assays. The results are represented as a percentage of
the mock control group value. (B) Eag1 mRNA expression in glioma
cells treated with 250 µM TMZ per 72 h, as revealed by reverse
transcription-quantitative polymerase chain reaction. (C) Effects
of pKv10.1-3 (0.2 µg) on Eag1 mRNA expression compared with a
control vector (pScramble). (D) pKv10.1-3 amplified the effect of
250 µM TMZ on Eag1 mRNA expression. Data were normalized to the
reference gene poly(A) polymerase alpha and compared using the
2−ΔΔCq method (n=3). (E-H) Eag1 immunocytochemistry in
U87MG cells. Representative confocal microscopy images showing the
expression of Eag1 in the (E) mock control group, and the reduced
cell filament following treatment with (F) pKv10.1-3, (G) 250 µM
TMZ or (H) pKv10.1-3 and 250 µM TMZ. Scale bar, 20 µm. (I) CTCF for
Eag1. Eag1 intensity was significantly decreased in all treated
groups in comparison with the mock. Eag1 intensity was
significantly lower in the cell group treated with pKv10.1-3 and
250 µM TMZ in comparison with pKv10.1-3 alone. *P<0.05.
Experimental groups were analyzed in triplicate in three
independent experiments. All results are expressed as the mean ±
standard error of the mean. CTCF, corrected total cell
fluorescence; TMZ, temozolomide; pKv, pKv10.1-3. |
Thus, at 72 h, glioblastoma cells presented a more
linear response to the increasing doses of TMZ (Fig. 1A), with 250 µM TMZ decreasing cell
viability to a value close to 50%. Therefore, treatment with 250 µM
TMZ for 72 h was selected for all subsequent analyses of the role
of Eag1 on the effects of TMZ.
Effects of TMZ and pKv10.1-3 on
glioblastoma cell viability and Eag1 expression
Initially, Eag1 mRNA expression was determined in
TMZ-treated glioblastoma cells by performing RT-qPCR. Treatment
with TMZ (250 µM) for 72 h tended to decrease Eag1 mRNA expression
(0.78-fold reduction; P=0.129; Fig.
1B). In addition, the shRNA expression vector, pKv10.1-3 (0.2
µg), was able to knock down Eag1 mRNA expression. Transfected cells
presented a 0.57-fold decrease in Eag1 expression compared with the
pScramble (0.2 µg) negative control group, according to
2−ΔΔCq values (P=0.043; Fig.
1C). Finally, the effect of TMZ on Eag1 expression was
determined in cells pre-transfected with pKv10.1-3. The vector
significantly enhanced the downregulation of Eag1 expression caused
by TMZ on glioblastoma cells at 72 h. Eag1 mRNA content was reduced
from 0.78-fold, for 250 µM TMZ alone to 0.31-fold for 250 µM TMZ
plus pKv10.1-3 (P=0.020; Fig.
1D).
Eag1 expression in U87MG cells was also examined by
immunocytochemistry (Fig. 1E-H). Eag1
was detected using anti-Eag1.62.mAb labeled with green fluorescence
and nuclei were counterstained with Hoechst 33342 (blue) prior to
examination by confocal microscopy. Merged squares show the
combined images of both Hoechst staining and Eag1 immunodetection
(Fig. 1E-H). Eag1 was highly
expressed in human U87MG glioblastoma cells (CTCF, 19.65±0.76;
Fig. 1E and I) while TMZ and the
pKv10.1-3 vector decreased this expression of Eag1 (Fig. 1F-H). Treatment with pKv10.1-3, 250 µM
TMZ alone or 250 µM TMZ and pKv10.1-3 significantly reduced the
CTCF values to 6.97±0.79, 5.37±0.37 and 3.86±0.49, respectively,
compared with the mock control (P=0.001; Fig. 1I). Eag1 CTCF values was significantly
lower in the cell group treated with pKv10.1 3 and 250 µM TMZ in
comparison with pKv10.1 3 alone (P=0.049; Fig. 1I).
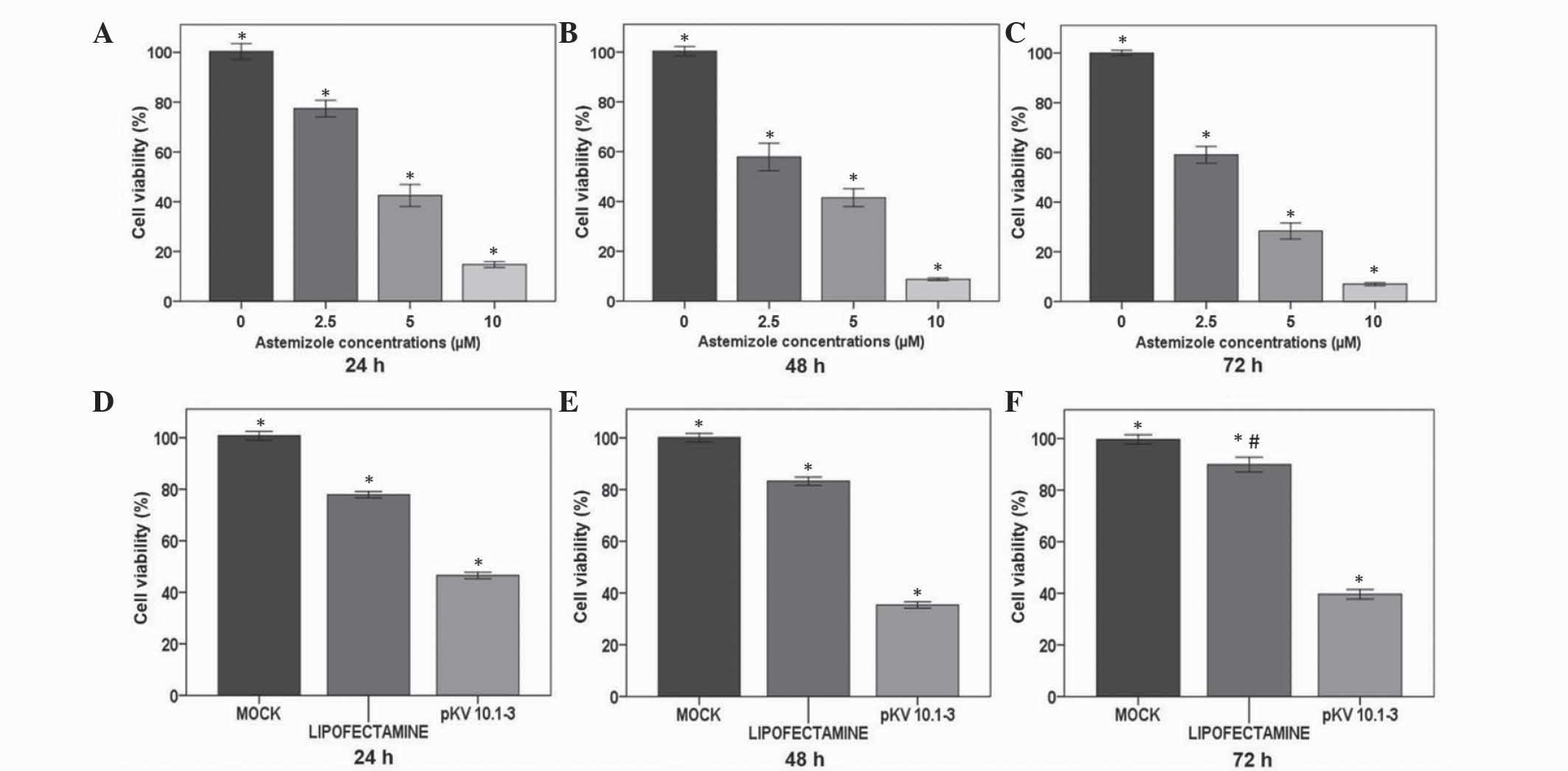
Astemizole and pKv10.1-3 reduce the
viability of glioblastoma cells
The role of Eag1 on the viability of glioblastoma
cells was examined using astemizole, a potassium channel blocker,
and pKv10.1-3, a plasmid that silences Eag1. Astemizole caused a
dose-dependent effect on the viability of glioblastoma cells
treated with this drug for 72 h, as determined by MTT assay
(Fig. 2A-C). At 24 h, cells treated
with 2.5, 5 or 10 µM astemizole exhibited significant reductions in
cell viability of 23, 57 and 85%, respectively, compared with 0 µM
astemizole (P=0.001; Fig. 2A). At the
same concentrations, 48 h of treatment with the drug also caused a
dose-response effect, with cell viability decreasing by 42 at 2.5
µM up to 93% at 10 µM, compared with 0 µM (P=0.001; Fig. 2B). Furthermore, at 72 h, astemizole
also reduced the viability of glioma cells by 41, 72 and 93% at
doses of 2.5, 5 and 10 µM, compared with 0 µM (P=0.001; Fig. 2C).
RNAi of Eag1 also affected glioblastoma cell
viability. Cells transfected with pKv10.1-3 (0.2 µg) showed a
stable decrease in viability between 24 and 72 h, as observed by
MTT assay. Values of cell viability decreased to 47, 35 and 40% at
24, 48 and 72 h, respectively, after transfection, compared with
mock control cells (P=0.001; Fig.
2D-F). Lipofectamine also caused a decrease in glioblastoma
cell viability compared with the mock control that was more intense
at 24 h (22%) than at 72 h (10%). Taken together, the results of
astemizole and pKv10.1-3 MTT assays confirm a role for Eag1 in
preserving glioblastoma cell viability.
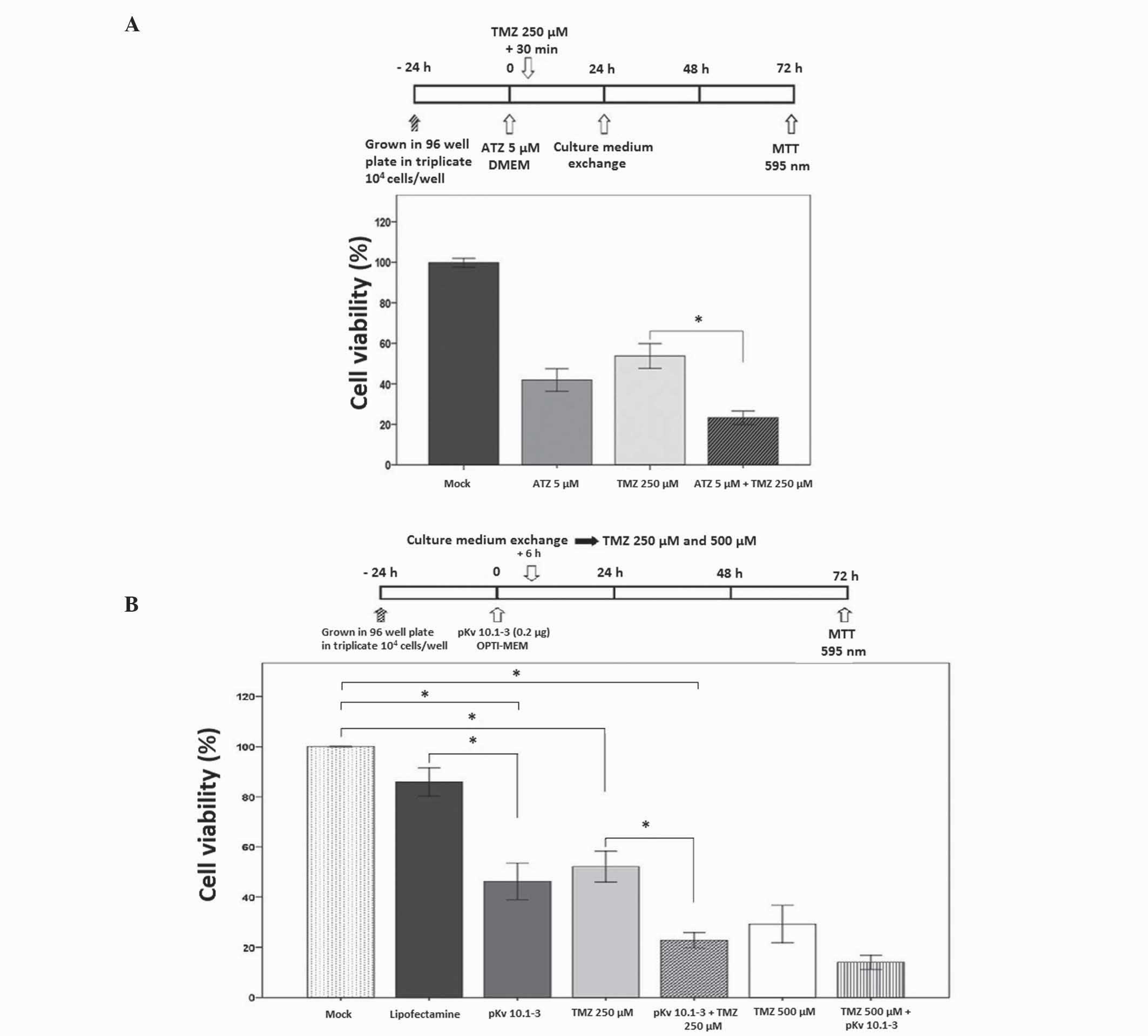
Suppression of Eag1 by astemizole or
pKv10.1-3 sensitizes cells to TMZ
The present study explored the effects of Eag1
potassium channel suppression on the injury caused to glioma cells
by TMZ. Cell preparations were treated with TMZ following Eag1
suppression, which varied according to each experimental method.
Eag1 blockade with astemizole required 30 min incubation, whereas 6
h was required for pKv10.1-3 transfection. Experimental groups
pretreated with the Eag1 channel blocker astemizole or pKv10.1-3
exhibited a stronger response to TMZ, as examined by MTT assay.
Cells incubated with astemizole (5 µM) for 30 min followed by 250
µM TMZ (Fig. 3A) exhibited a 77%
decrease in cell viability compared with the mock control, a value
significantly higher than in groups treated with astemizole (58%)
or TMZ alone (46%; P=0.001; Fig.
3A).
Furthermore, glioblastoma cells with silenced Eag1
expression using pKv10.1-3 exhibited a greater reduction in cell
viability following the addition of TMZ (Fig. 3B). In the TMZ (250 µM) plus pKv10.1-3
(0.2 µg) group, cell viability decreased by 77% compared with the
mock control (P=0.001). This effect was significantly higher in
comparison with that found in the group treated with TMZ alone
(48%; P=0.004). Groups treated with 500 µM TMZ plus pKv10.1-3 also
exhibited a greater decrease in cell viability compared with the
TMZ only group. Thus, the vector was able to enhance the effects of
TMZ on glioblastoma cells at drug doses of 250 and 500 µM,
indicating a dose-response effect.
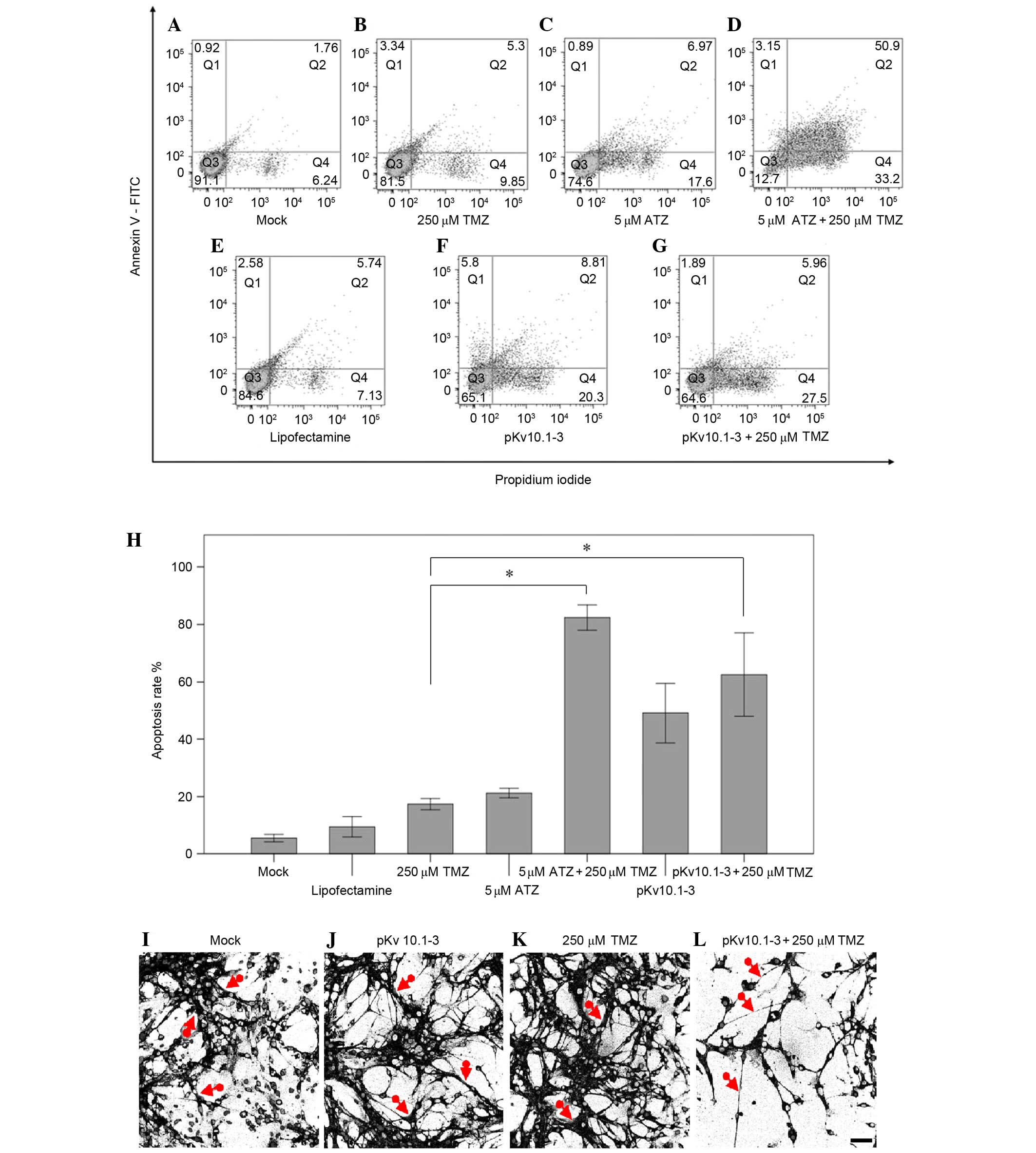
Apoptosis of GBM cells determined by
flow cytometry
The role of Eag1 suppression on the rate of
apoptosis was also examined in glioblastoma cells treated with TMZ.
Flow cytometry analysis employing an Annexin V-FITC/PI double
staining assay was performed. As shown in Fig. 4A-G, early apoptosis (Annexin V+/PI-)
and later stage apoptosis (Annexin V+/PI+) are represented in
quadrants Q4 and Q2, respectively. The mock control group
corresponds to untreated cells. Suppression of Eag1 increased the
induction of apoptosis (Q4 and Q2) caused by TMZ (Fig. 4H). For example, treating with TMZ and
blocking Eag1 with astemizole significantly increased the
glioblastoma cell apoptosis rate induced by TMZ alone by 4.7-fold,
from 17 to 82% (P=0.001; Fig. 4B vs. D
and H). In addition, treating with TMZ and silencing Eag1 with
pKv10.1-3 increased the rate of apoptosis triggered by TMZ alone by
3.6-fold (17 vs. 63%; P=0.011; Fig. 4B
vs. G and H).
Cell groups subjected to Eag1 suppression and TMZ
treatment also showed changes in cell morphology, as revealed by
confocal microscopy (Fig. 4I-L). The
cell filaments in the pKv10.1-3 plus 250 µM TMZ treatment group
were relatively thin and less dense compared with the mock controls
(Fig. 4I-L, red arrows). The mock
group (Fig. 4I) and those that
received pKv10.1-3 (Fig. 4J) or 250
µM TMZ (Fig. 4K) alone showed a more
stable morphology and survival rate, compared with the group
treated with pKv10.1-3 plus 250 µM TMZ (Fig. 4L). Furthermore, glioblastoma cells
treated with pKv10.1 3 plus 250 µM TMZ showed lower adherence with
round and floating shapes, in comparison with TMZ alone, as
determined by visual inspection.
Discussion
Ion channels have a critical role in tumorigenesis
(29). They regulate the flux of ions
across the plasma membrane, which influences cell cycle, growth and
apoptosis (30). Thus, deregulated
activity or expression of ion channels will favor the loss of
normal control of cell division, a classic hallmark of cancer
(31). Previous studies revealed that
Eag1 potassium channels have a role in cell growth, neoplastic
behavior and malignancy (10,17). Numerous tumor types display
deregulated Eag1 function, including breast cancer cells (20), tumors of head and neck (32), colon (33,34),
esophagus (35), cervix (36), and leukemia (37). Gliomas also overexpress Eag1,
irrespective of their malignancy grade (12). The present study confirmed that U87MG
glioblastoma cells in culture exhibit high expression levels of
Eag1 (Fig. 1E).
The aforementioned result confirmed a role for Eag1
in tumorigenesis, highlighting Eag1 as a promising target for
anticancer therapy. However, as Eag1 is involved in
electrophysiology, inhibiting channel function may cause adverse
effects. In a previous study, an Eag1 knock-out mouse was developed
and characterized (38). The animals
showed no changes in embryogenesis, brain development or electrical
properties of cerebellar Purkinge cells; only mild behavioral
changes occurred. Animals that lack an active channel also showed
no marked changes in physiology, thus, Eag1 appears to be a safe
and a promising target for cancer treatment. In fact, studies with
monoclonal antibodies, siRNAs, astemizole and imipramine have
confirmed that Eag1 inhibition can control cancer development
(18,21,23,39).
Expression of Eag1 was increased in the brain tumors of patients
with shorter overall survival (40).
Patients with brain metastasis exhibiting low Eag1 expression and
receiving drugs that block Eag1 (tricyclic antidepressants)
presented a longer overall survival. These data strongly suggest
that Eag1 plays a role in the growth of human brain tumors,
revealing this channel as a potential oncologic target.
Astemizole is an antihistamine that also blocks the
Eag1 channel. The drug has shown activity against different cancer
types, including hepatocellular carcinoma, breast tumors and
cervical cancer cells (41–43). The current results corroborated the
aforementioned findings in glioblastoma cells, reinforcing the role
of Eag1 in cancer cell growth. Astemizole caused a dose-dependent
decrease in glioblastoma cell viability following 24, 48 and 72 h
of treatment. The potential use of astemizole in cancer treatment,
however, is hindered by its side effects (39). Astemizole may cause ventricular
arrhythmia, a rare but potentially fatal toxic effect (44,45). For
brain tumors, a second pharmacological property of astemizole would
also undermine its efficacy: Its inability to penetrate the
blood-brain barrier. Although this pharmacokinetic property is
valuable for antihistamines in order to avoid sedation, it is
undesirable for drugs that must reach injured brain tissues, as
occurs for infiltrating gliomas (46). Thus, the blood-brain tumor barrier
represents an obstacle for antiglioma chemotherapy. In the core
area of glioblastomas this barrier is leaky, however, it remains
unchanged in large parts, blocking the access of anticancer drugs
to tumor tissues (47). Thus, a novel
therapeutic strategy to inhibit Eag1 is urgently required.
In a previous study, synthetic siRNAs showed the
ability to silence Eag1 expression in various types of cancer cells
(23). Eag1-targeted siRNAs decreased
Eag1 mRNA and protein expression, which resulted in decreased
growth in the majority of cell types investigated. Among the four
siRNAs evaluated, Kv10.1-3 caused the greatest silencing effect on
Eag1, targeting nucleotides 1793–1813 of Eag1 mRNA (http://www.ncbi.nlm.nih.gov/nuccore; NM_172362).
However, glioblastoma cells were not examined in this previous
study. Therefore, the present study employed the same Eag1 target
sequence, which was cloned in an shRNA expression vector termed
pKv10.1-3. Our previous study reported that glioblastoma cells
transfected with pKv10.1-3 were less viable than untreated controls
cells. Indeed, this Eag1 silencing vector also intensified IFN-γ
damage of glioma cells (24). The
present study revealed that pKv10.1-3 sensitized glioblastoma cells
to TMZ, the first-choice drug for the treatment of GBM. It was
observed that Eag1 has a role in the cell damage caused by TMZ on
glioblastoma cells. First, Eag1 is highly expressed in glioblastoma
cells (Fig. 1E). Second, pKv10.1-3 in
association with TMZ increased the silencing effect on Eag1 mRNA
caused by TMZ alone (0.31- vs. 0.78-fold reduction; Fig. 1D). Third, the effect displayed by
pKv10.1-3 on glioblastoma cell viability at 24, 48 and 72 h was
also observed for astemizole, an Eag1 channel blocker, on a
dose-dependent scale (Fig. 2).
Fourth, astemizole and pKv10.1-3 treatment both sensitized cells to
TMZ injury, decreasing glioblastoma cell viability (77% for
astemizole plus TMZ vs. 46% for TMZ alone, Fig. 3A; 63% for pKv10.1-3 plus TMZ vs. 34%
for TMZ alone, Fig. 3B). Fifth, these
treatments also caused changes in glioblastoma cell morphology and
caused poor adherence in culture flasks. Finally, both astemizole
and pKv10.1-3 significantly increased the rate of apoptosis caused
by TMZ, as determined by flow cytometry (Fig. 4A-G).
The use of RNAi for Eag1 silencing has previously
been reported in other cancer cell lines. A viral vector expressing
shRNA to Eag1 reduced tumor growth and angiogenesis of osteosarcoma
(48), and siRNAs to Eag1 sensitized
ovarian cancer cells to cisplatin (49). To the best of our knowledge, no
previous study has examined whether Eag1 gene silencing is able to
sensitize glioblastoma cells to TMZ.
In conclusion, the present study revealed that
suppression of Eag1 improves the action of TMZ on glioblastoma
cells. In addition to highlighting a role for Eag1 in TMZ injury,
the current findings also indicate a possible application for Eag1
silencing as an anticancer treatment strategy. Future preclinical
studies should analyze pKv10.1-3 in animal models of glioma to
confirm if Eag1 is a target for RNAi-based gene therapy.
Acknowledgements
The present study received financial support from
the following Brazilian agencies: Financiadora de Estudos e
Projetos (grant no. 2013/05179); Coordenação de Aperfeiçoamento de
Pessoal de Nível Superior (Programa Nacional de Pós-doutorado;
grant no. 3731-37/2010); Conselho Nacional de Desenvolvimento
Científico e Tecnológico (grant no. 467467/2014-5); Fundação de
Apoio à Pesquisa do Distrito Federal (grant no. 2010/00302-9). The
study has been previously published on the University of Brasília
website as the doctoral thesis of Mr. Fernando Francisco Borges
Resende at http://repositorio.unb.br/bitstream/10482/20136/1/2016_FernandoFranciscoBorgesResende_Parcial.pdf.
References
|
1
|
Maher EA, Furnari FB, Bachoo RM, Rowitch
DH, Louis DN, Cavenee WK and DePinho RA: Malignant glioma: Genetics
and biology of a grave matter. Genes Dev. 15:1311–1333. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lassman AB: Molecular biology of gliomas.
Curr Neurol Neurosci Rep. 4:228–233. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gladson CL, Prayson RA and Liu WM: The
pathobiology of glioma tumors. Annu Rev Pathol. 5:33–50. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nagasawa DT, Chow F, Yew A, Kim W, Cremer
N and Yang I: Temozolomide and other potential agents for the
treatment of glioblastoma multiforme. Neurosurg Clin N Am.
23:307–322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chamberlain MC: Temozolomide: Therapeutic
limitations in the treatment of adult high-grade gliomas. Expert
Rev Neurother. 10:1537–1544. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pardo LA and Stümer W: The roles of K (+)
channels in cancer. Nat Rev Cancer. 14:39–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pardo LA, del Camino D, Sánchez A, Alves
F, Brüggemann A, Beckh S and Stühmer W: Oncogenic potential of EAG
K(+) channels. EMBO J. 18:5540–5547. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hemmerlein B, Weseloh RM, de Queiroz F
Mello, Knötgen H, Sánchez A, Rubio ME, Martin S, Schliephacke T,
Jenke M, Heinz-Joachim-Radzun, et al: Overexpression of Eag1
potassium channels in clinical tumours. Mol Cancer. 5:412006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Patt S, Preussat K, Beetz C, Kraft R,
Schrey M, Kalff R, Schönherr K and Heinemann SH: Expression of
ether à go-go potassium channels in human gliomas. Neurosci Lett.
368:249–253. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hermisson M, Klumpp A, Wick W, Wischhusen
J, Nagel G, Roos W, Kaina B and Weller M: O6-methylguanine DNA
methyltransferase and p53 status predict temozolomide sensitivity
in human malignant glioma cells. J Neurochem. 96:766–776. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim SS, Rait A, Kim E, Pirollo KF, Nishida
M, Farkas N, Dagata JA and Chang EH: A nanoparticle carrying the
p53 gene targets tumors including cancer stem cells, sensitizes
glioblastoma to chemotherapy and improves survival. ACS Nano.
8:5494–5514. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lin H, Li Z, Chen C, Luo X, Xiao J, Dong
D, Lu Y, Yang B and Wang Z: Transcriptional and
post-transcriptional mechanisms for oncogenic overexpression of
ether à go-go K+ channel. PLoS One. 6:e203622011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Essmann F and Schulze-Osthoff K:
Translational approaches targeting the p53 pathway for anticancer
therapy. Br J Pharmacol. 165:328–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pardo LA and Stühmer W: EAG1: An emerging
oncological target. Cancer Res. 68:1611–1613. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gavrilova-Ruch O, Schönherr K, Gessner G,
Schönherr R, Klapperstück T, Wohlrab W and Heinemann SH: Effects of
imipramine on ion channels and proliferation of IGR1 melanoma
cells. J Membr Biol. 188:137–149. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
García-Ferreiro RE, Kerschensteiner D,
Major F, Monje F, Stühmer W and Pardo LA: Mechanism of block of
hEag1 K+ channels by imipramine and astemizole. J Gen Physiol.
124:301–317. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roy J, Vantol B, Cowley EA, Blay J and
Linsdell P: Pharmacological separation of hEAG and hERG K+ channel
function in the human mammary carcinoma cell line MCF-7. Oncol Rep.
19:1511–1516. 2008.PubMed/NCBI
|
|
21
|
Gómez-Varela D, Zwick-Wallasch E, Knötgen
H, Sánchez A, Hettmann T, Ossipov D, Weseloh R, Contreras-Jurado C,
Rothe M, Stühmer W and Pardo LA: Monoclonal antibody blockade of
the human Eag1 potassium channel function exerts antitumor
activity. Cancer Res. 67:7343–7349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Burnett JC and Rossi JJ: RNA-based
therapeutic: Current progress and future prospects. Chem Biol.
19:60–71. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weber C, de Queiroz F Mello, Downie BR,
Suckow A, Stühmer W and Pardo LA: Silencing the activity and
proliferative properties of the human EagI potassium channel by RNA
interference. J Biol Chem. 281:13030–13037. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cunha LC, Del Bel E, Pardo L, Stühmer W
and Titze-DE-Almeida R: RNA interference with EAG1 enhances
interferon gamma injury to glioma cells in vitro. Anticancer Res.
33:865–870. 2013.PubMed/NCBI
|
|
25
|
Kane A and Yang I: Interferon-gamma in
brain tumor immunotherapy. Neurosurg Clin N Am. 21:77–86. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mareschi K, Novara M, Rustichelli D,
Ferrero I, Guido D, Carbone E, Medico E, Madon E, Vercelli A and
Fagioli F: Neural differentiation of human mesenchymal stem cells:
Evidence for expression of neural markers and each K+ channel type.
Exp Hematol. 34:1563–1572. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kwon MJ, Oh E, Lee S, Roh MR, Kim SE, Lee
Y, Choi YL, In YH, Park T, Koh SS and Shin YK: Identification of
novel reference genes using multiplatform expression data and their
validation for quantitative gene expression analysis. PLoS One.
4:e61622009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kunzelmann K: Ion channels and cancer. J
Membr Biol. 205:159–173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kondratskyi A, Kondratska K, Skryma R and
Prevarskaya N: Ion channels in the regulation of apoptosis. Biochim
Biophys Acta. 1848:2532–2546. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lang F, Föller M, Lang KS, Lang PA, Ritter
M, Gulbins E, Vereninov A and Huber SM: Ion channels in cell
proliferation and apoptotic cell death. J Membr Biol. 205:147–157.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Menéndez ST, Villaronga MA, Rodrigo JP,
Alvarez-Teijeiro S, García-Carracedo D, Urdinguio RG, Fraga MF,
Pardo LA, Viloria CG, Suárez C and García-Pedrero JM: Frequent
aberrant expression of the human ether à go-go (hEAG1) potassium
channel in head and neck cancer: Pathobiological mechanisms and
clinical implications. J Mol Med (Berl). 90:1173–1184. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ding XW, Yan JJ, An P, Lü P and Luo HS:
Aberrant expression of ether à go-go potassium channel in
colorectal cancer patients and cell lines. World J Gastroenterol.
13:1257–1261. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ousingsawat J, Spitzner M, Puntheeranurak
S, Terracciano L, Tornillo L, Bubendorf L, Kunzelmann K and
Schreiber R: Expression of voltage-gated potassium channels in
human and mouse colonic carcinoma. Clin Cancer Res. 13:824–831.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ding XW, Luo HS, Jin X, Yan JJ and Ai YW:
Aberrant expression of Eag1 potassium channels in gastric cancer
patients and cell lines. Med Oncol. 24:345–350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ortiz CS, Montante-Montes D, Saqui-Salces
M, Hinojosa LM, Gamboa-Dominguez A, Hernández-Gallegos E,
Martínez-Benítez B, Del Rosario Solís-Pancoatl M, Garcia-Villa E,
Ramírez A, et al: Eag1 potassium channels as markers of cervical
dysplasia. Oncol Rep. 26:1377–1383. 2011.PubMed/NCBI
|
|
37
|
Agarwal JR, Griesinger F, Stühmer W and
Pardo LA: The potassium channel Ether à go-go is a novel prognostic
factor with functional relevance in acute myeloid leukemia. Mol
Cancer. 9:182010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ufartes R, Schneider T, Mortensen LS, de
Juan Romero C, Hentrich K, Knoetgen H, Beilinson V, Moebius W,
Tarabykin V, Alves F, et al: Behavioural and functional
characterization of Kv10.1 (Eag1) knockout mice. Hum Mol Genet.
22:2247–2262. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
García-Quiroz J and Camacho J: Astemizole:
An old anti-histamine as a new promising anti-cancer drug.
Anticancer Agents Med Chem. 11:307–314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Martínez R, Stühmer W, Martin S, Schell J,
Reichmann A, Rohde V and Pardo L: Analysis of the expression of
Kv10.1 potassium channel in patients with brain metastases and
glioblastoma multiforme: impact on survival. BMC Cancer.
15:8392015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
de Guadalupe Chávez-López M, Pérez-Carreón
JI, Zuñiga-García V, Díaz-Chávez J, Herrera LA, Caro-Sánchez CH,
Acuña-Macías I, Gariglio P, Hernández-Gallegos E, Chiliquinga AJ
and Camacho J: Astemizole-based anticancer therapy for
hepatocellular carcinoma (HCC), and Eag1 channels as potential
early-stage markers of HCC. Tumour Biol. 36:6149–6158. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
García-Quiroz J, García-Becerra R,
Santos-Martínez N, Barrera D, Ordaz-Rosado D, Avila E, Halhali A,
Villanueva O, Ibarra-Sánchez MJ, Esparza-López J, et al: In vivo
dual targeting of the oncogenic Ether-à-go-go-1 potassium channel
by calcitriol and astemizole results in enhanced antineoplastic
effects in breast tumors. BMC Cancer. 14:7452014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
de Guadalupe Chávez-López M,
Hernández-Gallegos E, Vázquez-Sánchez AY, Gariglio P and Camacho J:
Antiproliferative and proapoptotic effects of astemizole on
cervical cancer cells. Int J Gynecol Cancer. 24:824–828. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
de Abajo FJ and Rodríguez LA: Risk of
ventricular arrhythmias associated with nonsedating antihistamine
drugs. Br J Clin Pharmacol. 47:307–313. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Oppenheimer JJ and Casale TB: Next
generation antihistamines: Therapeutic rationale, accomplishments
and advances. Expert Opin Investig Drugs. 11:807–817. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
González MA and Estes KS: Pharmacokinetic
overview of oral second-generation H1 antihistamines. Int J Clin
Pharmacol Ther. 36:292–300. 1998.PubMed/NCBI
|
|
47
|
vanTellingen O, Yetkin-Arik B, de Gooijer
MC, Wesseling P, Wurdinger T and de Vries HE: Overcoming the
blood-brain tumor barrier for effective glioblastoma treatment.
Drug Resist Updat. 19:1–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wu J, Wu X, Zhong D, Zhai W, Ding Z and
Zhou Y: Short Hairpin RNA (shRNA) Ether à go-go 1 (Eag1) inhibition
of human osteosarcoma angiogenesis via VEGF/PI3K/AKT signaling. Int
J Mol Sci. 13:12573–12583. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hui C, Lan Z, Yue-Li L and Li-Lin H and
Li-Lin H: Knockdown of Eag1 expression by RNA interference
increases chemosensitivity to cisplatin in ovarian cancer cells.
Reprod Sci. 22:1618–1626. 2015. View Article : Google Scholar : PubMed/NCBI
|