Introduction
Colorectal carcinoma (CRC) is one of the most common
types of malignancy and has a poor prognosis (1). It is a major cause of cancer-associated
morbidity and mortality worldwide, and the incidence of this
disease has gradually increased in recent years (1,2). The
development of CRC is associated with genetic and epigenetic
factors, resulting in changes in the expression of various
oncogenes, which are tumor suppressor genes that lead to
dysregulation of intestinal epithelial cell proliferation,
survival, differentiation and apoptosis (1,3,4).
The phosphatidylinositol-3 kinase (PI3K)/v-akt
murine thymoma viral oncogene homolog (Akt) and the mitogen
activated protein kinase (MAPK), also termed Raf-MAPK kinase
(MEK)-extracellular signal-regulated kinase (ERK), signaling
pathways are commonly dysregulated and hyperactivated in cancers,
including CRC, by genetic and epigenetic aberrations (4–8). Due to
the fact that suppression of the PI3K/Akt and MAPK/ERK signaling
pathways leads to the blockade of cell proliferation, these
cascades are important during cancer development for the control of
cell cycle progression and cell growth (6–10).
Akt, a serine-threonine protein kinase, is a
downstream effector and central signaling molecule in the PI3K
pathway and plays an important role in controlling the balance
between cell survival and apoptosis (11,12).
Aberrant cell growth and survival may be promoted by the PI3K
pathway due to Akt phosphorylation (12). In addition, the PI3K/Akt signaling
pathway is frequently activated in numerous types of human cancers,
including CRC, and has been linked to cancer development (10,13,14).
The MAPK/ERK pathway is also a major signal
transduction cascade for cellular processes, such as cell
proliferation, cell cycle progression, survival, differentiation
and apoptosis (15,16). Activation of MAPK signaling results in
increased phosphorylation and activation of ERK1/2, which in turn,
phosphorylates several proteins associated with cell cycle
progression and cell motility (17).
In addition, this signaling pathway is critical for development and
progression of human cancers and results in increased cancer cell
proliferation, resistance to apoptosis, and therefore, tumor growth
(18,19).
Certain studies demonstrated that cancer development
may be stopped or slowed down by inhibition of these signal
transduction pathways, such as PI3K/Akt and MAPK/ERK (14,19–21). These
pathways have been attractive therapeutic targets in the treatment
of CRC and numerous compounds inhibit these signaling pathways at
several levels; therefore, they may be considered potentially
useful for CRC treatment (7,8,22).
Selective inhibition of Akt and ERK represents a potential approach
for the treatment of CRC. The small molecule Akt/protein kinase B
inhibitor,
4-amino-5,8-dihydro-5-oxo-8-β-D-ribofuranosyl-pyrido[2,3-d]pyrimidine-6-carboxamide
(API-1) is a novel and potent selective inhibitor of Akt signaling
that binds to the pleckstrin homology (PH) domain of Akt and blocks
its membrane translocation, which leads to inhibition of
Akt-regulated cell growth and cell survival in vitro and
in vivo (23,24). Additionally, FR180204 (FR) is a potent
and selective adenosine triphosphate (ATP)-competitive inhibitor of
ERK1 and ERK2, and inhibits the kinase activity of ERK1 and ERK2
(25).
In the present study, the role of Akt and ERK in
cell growth and apoptosis was focused on in DLD-1 and LoVo cell
lines using the specific Akt inhibitor API-1 and ERK1/2 inhibitor
FR. In addition, the present study aimed to investigate the
possible synergistic apoptotic and antiproliferative effects of a
novel combination of API-1 and FR in CRC cells and their effects on
PI3K and MAPK signaling pathways, including changes in the mRNA and
protein expression levels of these cascade components.
Materials and methods
Chemicals and antibodies
The reagents used in the present study were
purchased from the following suppliers: FR and API-1 from Tocris
Bioscience (Bristol, UK); RPMI-1640 medium, fetal bovine serum
(FBS), L-glutamine and penicillin/streptomycin from Gibco (Thermo
Fisher Scientific, Inc., Waltham, MA, USA); water soluble
tetrazolium-1 (WST-1), Cytotoxicity Detection Kit Plus, Cell
Proliferation ELISA colorimetric kit and Cell Death Detection ELISA
Plus kit from Roche Diagnostics GmbH (Mannheim, Germany); and
PathScan ® Cleaved Caspase-3 (Asp175) Sandwich ELISA kit and
monoclonal rabbit antibodies against β-actin (ACTB; catalog no.,
4970; dilution, 1:1,000), B-cell lymphoma-2 (BCL2)-associated X
protein (BAX; catalog no., 5023; dilution, 1:1,000), BCL2
antagonist/killer (BAK; catalog no., 12105; dilution, 1:1,000),
cyclin D1 (CYCD1; catalog no., 2978; dilution, 1:1,000), cMYC
(catalog no., 13987; dilution, 1:1,000), Akt (catalog no., 4685;
dilution, 1:1,000), ERK1/2 (catalog no., 4370; 1:2,000),
phosphorylated Akt (pAkt; catalog no., 4060; dilution, 1:2,000),
phosphorylated ERK1/2 (pERK1/2; catalog no., 4094; dilution,
1:1,000) and horseradish peroxidase (HRP)-conjugated anti-rabbit
IgG secondary antibody (catalog no., 7074; dilution, 1,1000) were
provided by Cell Signaling Technology (Danvers, MA, USA). All other
chemicals and reagents were obtained from Sigma-Aldrich (St. Louis,
MO, USA).
Cell culture
The human CRC DLD-1 (catalog no., CCL-221; American
Type Culture Collection, Manassas, VA, USA) and LoVo (catalog no.,
CCL-229; American Type Culture Collection) cell lines were cultured
in RPMI-1640 medium containing 10% FBS, 2 mM L-glutamine, 100 U/ml
penicillin and 100 µg/ml streptomycin. The cells were maintained in
a humidified atmosphere incubator at 37°C, with a 5% CO2
atmosphere. FR and API-1 were dissolved in dimethyl sulfoxide
(DMSO) to make 1 mM stock solutions that were kept at −20°C. The
stock solutions were freshly diluted with cell culture medium to
the required concentration immediately prior to use. The final
concentration of DMSO in culture medium during the treatment of
cells did not exceed 0.5% (v/v).
Cell viability and apoptotic
analyses
To detect the effect of FR and API-1 on cell
viability following treatment, a WST-1 cell proliferation assay was
performed. In brief, DLD-1 and LoVo cells were seeded into 96-well
plates (1×104 cells/well) containing 100 µl of the
growth medium in the absence or presence of increasing
concentrations of FR (1–150 µM) and API-1(0.1–100 µM) and then
incubated at 37°C and 5% CO2 for 24 and 48 h. At the end
of the incubation period, the medium was removed, 100 µl WST-1 was
added and the cell solution was incubated at 37°C for 4 h. Formazan
dye produced by viable cells was quantified by measuring absorbance
at a wavelength of 450 nm using a microplate reader (Spectramax M3;
Molecular Devices, Sunnyvale, CA, USA). All experiments were
performed four times and the experiment was repeated twice.
Fluorescence microscopic analysis of
cell death
Cell death was assessed using ethidium bromide (EB)
and acridine orange (AO) staining. The DLD-1 and LoVo cells were
plated in 24-well plates at a concentration of 2×105
cells/well and incubated at 37°C for 24 h. Following exposure of
the cells to FR (10 µM), API-1 (0.5 µM or 2 µM for DLD-1 cells and
0.5 µM or 1 µM for LoVo cells) or a combination of the two for 24
and 48 h, cells were harvested and cell suspensions (supernatants)
were collected and then centrifuged at 130 × g for 5 min. The
pellet was resuspended in 25 µl cold phosphate-buffered saline
(PBS) buffer and 4 µl AO/EB dye mix (100 µg/ml in PBS). The stained
cell suspensions (10 µl) were placed on a microscope slide and
covered with a coverslip. The stained cells were examined under a
fluorescence microscope (Olympus, Tokyo, Japan) and images were
captured to determine the presence of apoptotic cells. To determine
the apoptotic index, the number of apoptotic cells
(yellowish-orange cells) was divided by the total number of counted
cells and multiplied by 100 to calculate the percentage of
apoptotic cells.
Lactate dehydrogenase (LDH) release
assay
The release of LDH, a marker of cytotoxicity, from
treated DLD-1 and LoVo cells was measured using the Cytotoxicity
Detection Kit Plus, according to the manufacturer's protocol.
Briefly, cells were treated with FR (10 µM), API-1 (0.5 µM or 2 µM
for DLD-1 cells and 0.5 µM or 1 µM for LoVo cells) or a combination
of the two for 24 and 48 h. To determine the release of LDH, the
supernatant of treated cells was transferred to a 96-well plate. At
the end of the period, the absorbance of the samples was measured
at a wavelength of 490 nm using the Spectramax M3 microplate
reader. The level of released LDH from treated LoVo and DLD-1 cells
was expressed as a percentage of the positive control (2% Triton
X-100).
Cell proliferation assay
The bromodeoxyuridine (BrdU) assay is an immunoassay
for the quantification of BrdU incorporation into newly synthesized
DNA of actively proliferating cells (26,27). Cell
proliferation was measured using the BrdU cell proliferation
enzyme-linked immunosorbent assay (ELISA). DLD-1 and LoVo cells
(1×104 cells/well) were incubated in 96-well plates for
24 h and then treated with various selected concentrations of FR
(10 µM) and/or API-1 (0.5 µM or 2 µM for DLD-1 cells and 0.5 µM or
1 µM for LoVo cells) for 24 and 48 h. Culture medium (RPMI-1640)
was used as a control. Cell proliferation was quantified by the
measurement of BrdU incorporation during DNA synthesis, according
to the manufacturer's protocol, using the Spectramax M3 microplate
reader at 450 nm.
DNA fragmentation analysis
The quantitative DNA fragmentation assay, a marker
for apoptosis, was performed using the Cell Death Detection ELISA
Plus kit, according to the manufacturer's protocol. Briefly, the
cells were plated into 96-well plates at a density of
2×104 cells/well in 200 µl RPMI-1640 medium and
incubated overnight at 37°C. Subsequently, the cells were treated
with FR or API-1 for 24 and 48 h at 37°C. Following the ELISA
procedure, performed according to the manufacturer's protocol,
cytoplasmic histone-associated DNA fragments were measured at 405
nm using the Spectramax M3 microplate reader. The results are
represented as the mean enrichment factor [absorbance of the
treated (apoptotic) cells/absorbance of the untreated (control)
cells].
Caspase-3 activity assay
To verify the presence of caspase-3 activity, which
is an important sign of apoptosis, in the cells, a caspase-3
activity assay was performed using the PathScan® Cleaved Caspase-3
(Asp175) Sandwich ELISA kit, according to the manufacturer's
protocol. Briefly, following treatment with either FR (10 µM) and
API-1 (0.5 µM or 2 µM for DLD-1 cells and 0.5 µM or 1 µM for LoVo
cells) alone or in combination at the indicated concentrations for
24 and 48 h, protein content of cell lysates was determined using a
bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL,
USA). At the end of this period, the absorbance of the cleaved
caspase-3 levels was measured at 405 nm using the Spectramax M3
microplate reader.
Quantitative reverse
transcription-polymerase chain reaction (RT-PCR) analysis
Total RNA was extracted from each sample using High
Pure RNA Isolation kit (Roche Diagnostics GmbH), according to the
manufacturer's protocol. Equal amounts of RNA were then reverse
transcribed into complementary DNA (cDNA) using Transcriptor First
Strand cDNA Synthesis kit (Roche Diagnostics GmbH). Quantitative
RT-PCR was performed using a LightCycler® 480 instrument (Roche
Diagnostics GmbH) with Light Cycler 480 Probes Master mix (Roche
Diagnostics GmbH), according to the manufacturer's protocol.
Gene-specific intron spanning primers and probes were designed
using the online Universal Probe Library (UPL) Assay Design Center
(Roche Diagnostics GmbH). The sequences of the primers and UPL
numbers are shown in Table I. The
mRNA level of glyceraldehyde-3-phosphate dehydrogenase was used to
normalize the levels of the genes of interest. Each sample was
tested in triplicate. Fold change in BCL2, BCL2L1, BAX, BAK,
BCL2-like 11 (BIM), CYCD1 and cMYC gene expressions were calculated
using the 2-ΔΔCq method (28) and
significant differences were identified using a Student's t-test on
normalized Cq values.
 | Table I.Gene-specific primer sequences and
UPL probe numbers used in quantitative reverse
transcription-polymerase chain reaction. |
Table I.
Gene-specific primer sequences and
UPL probe numbers used in quantitative reverse
transcription-polymerase chain reaction.
| Gene | Primer sequence
(5′-3′) | UPL probe
number |
|---|
| GAPDH |
|
|
|
Forward |
AGCCACATCGCTCAGACAC | 60 |
|
Reverse |
GCCCAATACGACCAAATCC |
|
| BCL2 |
|
|
|
Forward |
AGTACCTGAACCGGCACCT | 75 |
|
Reverse |
GCCGTACAGTTCCACAAAGG |
|
| BCL2L1 |
|
|
|
Forward |
AGCCTTGGATCCAGGAGAA | 28 |
|
Reverse |
GCTGCATTGTTCCCATAGAGT |
|
| BAX |
|
|
|
Forward |
ATGTTTTCTGACGGCAACTTC | 57 |
|
Reverse |
ATCAGTTCCGGCACCTTG |
|
| BAK |
|
|
|
Forward |
AAACTGGGCTCCCACTCA | 66 |
|
Reverse |
CAGTGGAGGACGGGATCA |
|
| BIM |
|
|
|
Forward |
CATCGCGGTATTCGGTTC | 70 |
|
Reverse |
GCTTTGCCATTTGGTCTTTTT |
|
| CYCD1 |
|
|
|
Forward |
TGTCCTACTACCGCCTCACA | 16 |
|
Reverse |
CAGGGCTTCGATCTGCTC |
|
| cMYC |
|
|
|
Forward |
GCTGCTTAGACGCTGGATTT | 66 |
|
Reverse |
TAACGTTGAGGGGCATCG |
|
Western blot analysis
The effects of FR, API-1 and a combination of the
two on protein expression in LoVo and DLD-1 cells were determined
using western blot analysis. Proteins were isolated from the cell
lysate at the different time points and protein contents of each
cell lysate were measured using BCA Protein Assay kit, according to
the manufacturer's protocol. Western blotting was performed as
previously described (29) Bands were
visualized via an enhanced chemiluminescence (ECL) system (Kodak
Gel Logic 2200 PRO Imaging System; Carestream Health, Rochester,
NY, USA). The expression of the BAX, BAK, CYCD1, cMYC, Akt, pAkt,
ERK1/2 and pERK1/2 proteins was normalized against a loading
control, β-actin. The Kodak Gel Logic 2200 PRO Imaging System
Software semiquantitatively detects protein bands against loading
control densitometrically and provides relative band signals.
Statistical analysis
Quantitative data were represented as the mean ±
standard deviation. The differences between groups were assessed
for statistical significance using Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference. All studies were repeated in at least 3 independent
experiments. Calculations of relative mRNA expression levels of
BCL2, BCL2 like 1 (BCL2L1), BAX, BAK, BAD, BIM, CYCD1 and cMYC were
performed using Relative Expression Software Tool 2009 v2.0.13
(Qiagen, Hilden, Germany) (30).
Results
Effects of FR and API-1 alone and in
combination on the viability of LoVo cells
To evaluate the effects of FR and API-1 on the
viability of human CRC cells, DLD-1 and LoVo cells were exposed to
increasing concentrations of FR (1–150 µM) and API-1 (0.1–100 µM)
for 24 and 48 h. FR and API-1 each decreased cell proliferation in
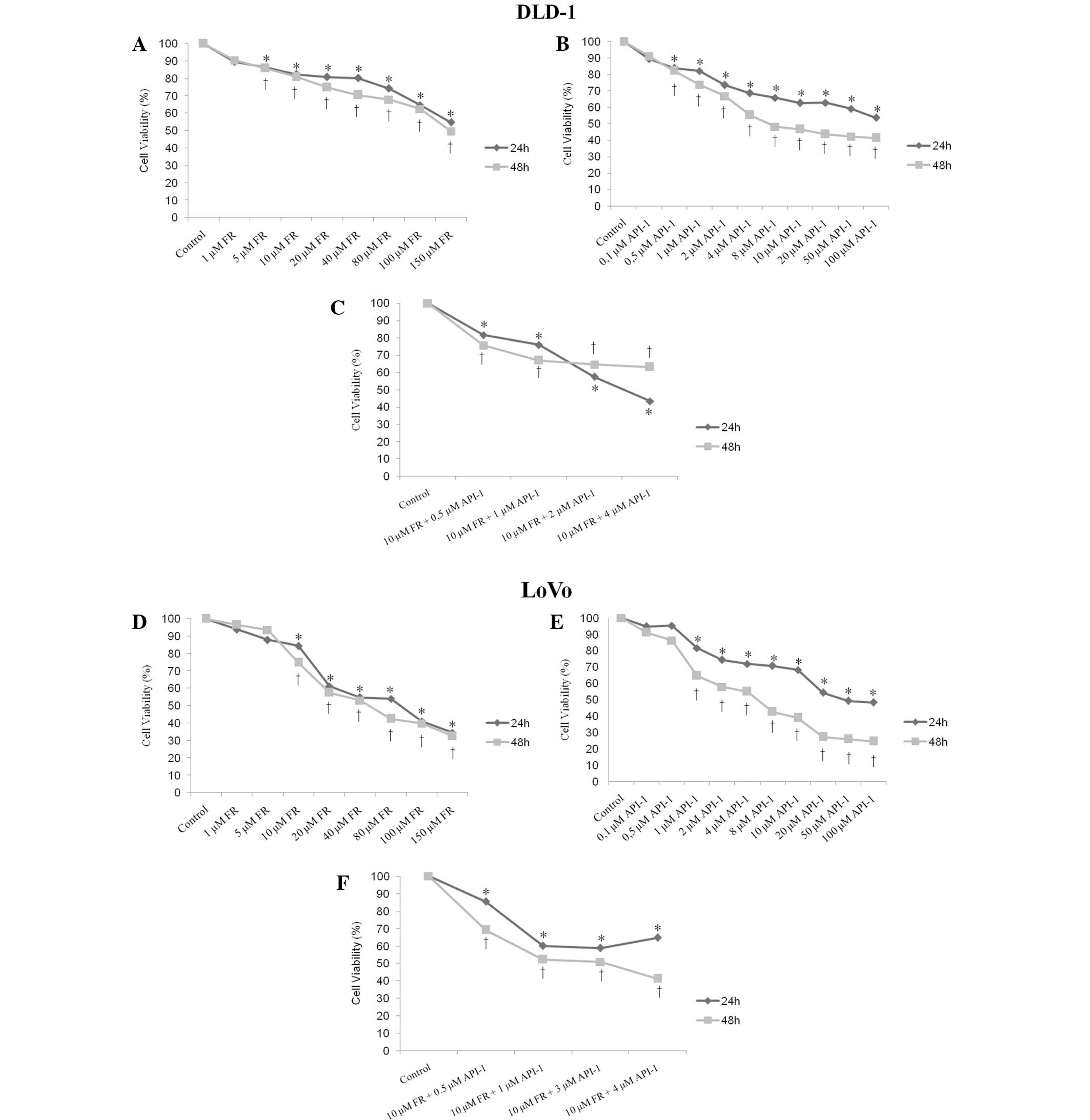
a time- and dose-dependent manner. As shown in Fig. 1A and D, while ~30% of the cells were
determined as viable in LoVo cells, viability of DLD-1 cells was
found to be almost 50% at the highest concentration of FR (150 µM)
at 24 and 48 h compared with the untreated controls. The
half-maximal inhibitory concentration (IC50) value of FR
was 150 µM for 24 and 48 h in DLD-1 cells (Fig. 1A). The IC50 value of FR was
also found to be 80 µM for 24 h and 40 µM for 48 h in LoVo cells
(Fig. 1D). The present study also
examined the effect of API-1 on DLD-1 and LoVo cells. Cell
viability of LoVo and DLD-1 cells was found to be 46 and 53% at the
highest concentration of API-1 (100 µM) at 24 h, respectively
(Fig. 1B and E). The IC50
value of API-1 was 100 µM for 24 h and 4 µM for 48 h for DLD-1
cells (Fig. 1B). The IC50
value of API-1 was also 20 µM for 24 h and 4 µM for 48 h for LoVo
cells (Fig. 1E).
To study antiproliferative effects of combined
treatment with FR and API-1, DLD-1 and LoVo cells were exposed to
10 µM FR at various concentrations of API-1 (0.5–4 µM) for 24 and
48 h. Combined treatment with FR and API-1 also resulted in
dose-dependent reduction of DLD-1 and LoVo cell viability (Fig. 1C and F). The results revealed that the
cell viability was 81, 75 and 57% in DLD-1 cells treated with 10 µM
FR and 0.5, 1 or 2 µM API-1, respectively, at 24 h. The cell
proliferation decreased 75, 66 and 64% in DLD-1 cells treated with
10 µM FR and 0.5, 1 or 2 µM API-1, respectively, compared with
untreated controls at 48 h (Fig. 1C).
In LoVo cells, cell viability was 85, 60 and 58% in cells treated
with 10 µM FR and 0.5, 1 or 3 µM API-1, respectively, at 24 h.
Following treatment for 48 h with 10 µM FR and 0.5, 1 or 3 µM
API-1, cell viability was 69, 52 and 50%, respectively, in LoVo
cells (Fig. 1F).
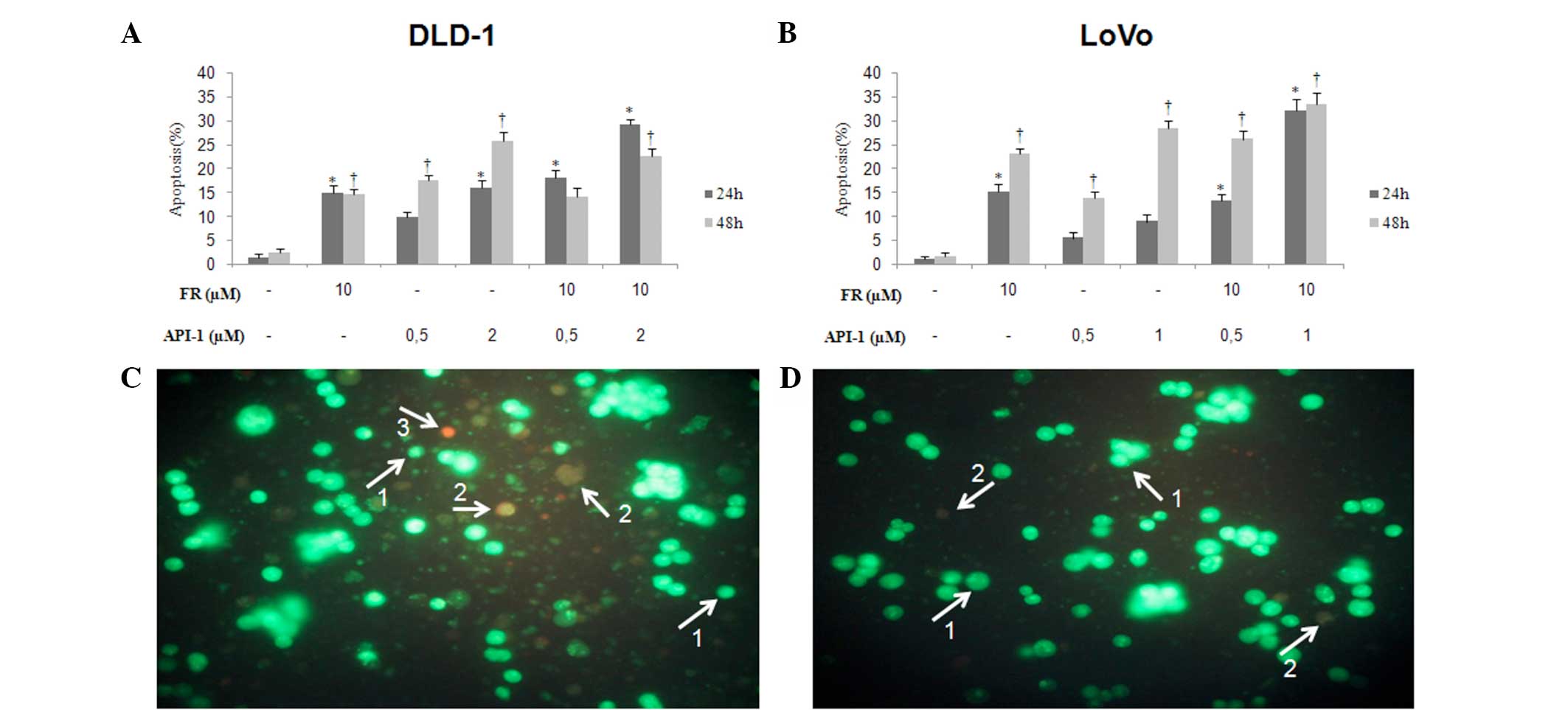
Morphological analysis by fluorescence
microscopy
To identify whether FR and/or API-1 induced
apoptosis or necrosis, double-staining using AO/EB was performed
using fluorescence microscopy. As shown in representative images
(Fig. 2), morphological findings
indicated that treatment with FR and API-1 alone induced apoptosis
in DLD-1 and LoVo cells. In LoVo cells, 10 µM FR significantly
increased the percentage of apoptotic cells in a time-dependent
manner (P=0.02). Additionally, 0.5 µM API-1 had no significant
effects in the two cell lines. However, at concentrations of 1 µM
API-1 in LoVo cells and 2 µM API-1 in DLD-1 cells, the percentages
of apoptotic cells were significantly increased at 48 h (LoVo,
P=0.005; DLD-1, P=0.016). Following treatment for 24 and 48 h, LoVo
and DLD-1 cells treated with FR + API-1 showed an increased number
of apoptotic cells in a dose- and time-dependent manner. The
maximal apoptotic death was observed in LoVo cells treated with 10
µM FR and 1 µM API-1 and DLD-1 cells treated with 10 µM FR and 2 µM
API-1 (DLD-1 at 24 h, P=0.005 and 48 h, P=0.011; LoVo at 24 h,
P=0.003 and 48 h, P=0.002).
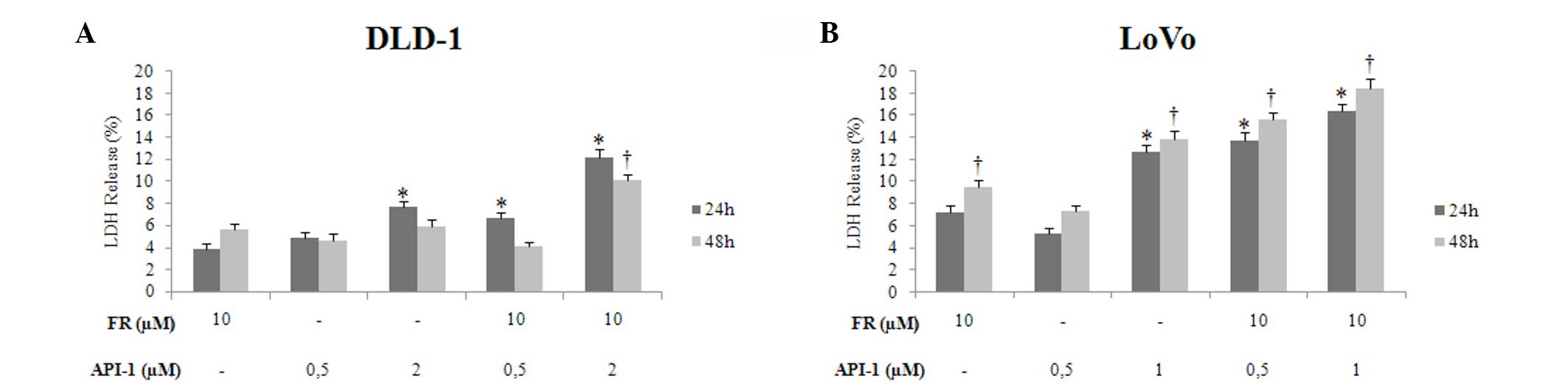
Cytotoxic effects of treatment with FR
and API-1 alone or in combination
To determine the cytotoxic effects of FR and API-1
on DLD-1 and LoVo cell lines, the cells were incubated with FR and
API-1 alone or in combination for 24 and 48 h using an LDH assay.
The results of these assays showed that there were time- and
dose-dependent increases in cytotoxicity, particularly in the LoVo
cell line. As shown in Fig. 3A,
treatment with 2 µM API-1 caused an increase in cytotoxicity at 24
h in DLD-1 cells (P=0.027), whereas treatment with 10 µM FR alone
for 48 h increased cytotoxic effects compared with control LoVo
cells. Treatment with 1 µM API-1 alone also caused a time-dependent
increase in cytotoxicity in LoVo cells (24 h, P=0.007; 48 h,
P=0.005; Fig. 3B). The results
revealed that FR + API-1 exhibited a cytotoxic effect in a
dose-dependent manner in terms of the two cell lines. As shown in
Fig. 3, the highest cytotoxicity rate
in DLD-1 cells was observed following treatment with 10 µM FR and 2
µM API-1 (24 h, P=0.005; 48 h, P=0.012), and the highest
cytotoxicity rate in LoVo cells was observed following treatment
with 10 µM FR and 1 µM API-1 for 24 h (P=0.003) and 48 h
(P=0.002).
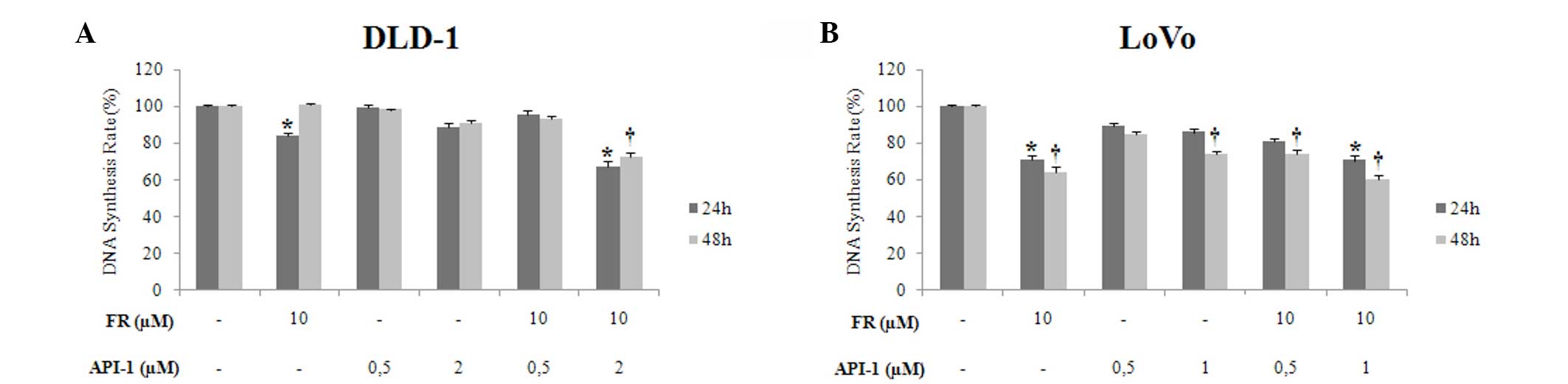
Effect of FR and API-1 alone or in
combination on DNA synthesis
To determine the antiproliferative effects of FR and
API-1 on DLD-1 and LoVo cell lines, DNA synthesis was measured by
BrdU assay. Treatment with FR and API-1 alone inhibited DNA
synthesis in a dose-dependent manner at 24 and 48 h, particularly
in LoVo cell lines. Incubation with 10 µM FR significantly reduced
the DNA synthesis rate in LoVo cells at 24 and 48 h (P=0.001), but
the DNA synthesis rate of DLD-1 cells was only reduced by treatment
with 10 µM FR for 24 h (P=0.002) (Fig. 4A
and B). Treatment with 1 µM API-1 alone also significantly
reduced the DNA synthesis rate in a time-dependent manner in LoVo
cells (P=0.001). Treatment of LoVo cells with 10 µM FR and 1 µM
API-1 and DLD-1 cells with 10 µM FR and 2 µM API-1 significantly
reduced the DNA synthesis rate subsequent to 24 and 48 h
(P=0.001).
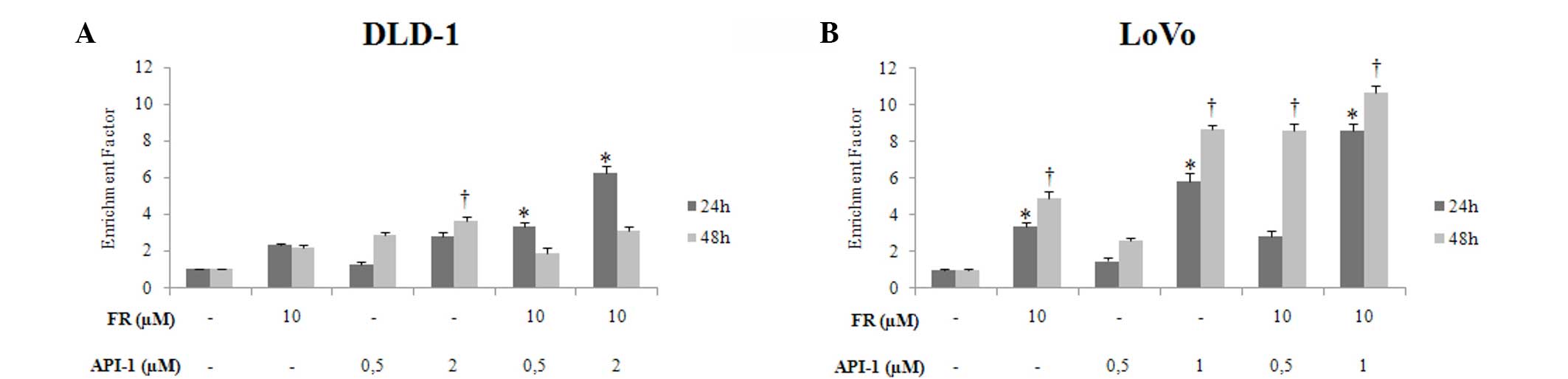
Effects of FR and API-1 alone or in
combination on DNA fragmentation
To examine the possible apoptotic effects of
treatment with FR and API-1 alone and in combination, the levels of
mono-oligo nucleosome fragments were quantified using Cell Death
Detection ELISA assay. DLD-1 and LoVo cells were treated with
different concentrations of FR (10 µM) and API-1 (0.5 µM or 2 µM
for DLD-1 cells and 0.5 µM or 1 µM for LoVo cells) alone or in
combination for 24 and 48 h prior to analyzing DNA fragmentation.
The results showed that treatment with FR alone elevated the DNA
fragmentation rate at 24 and 48 h in DLD-1 and LoVo cells when
compared with the control cells (DLD-1, P=0.001; LoVo, P=0.001). In
the DLD-1 and LoVo cell lines, a dose- and time dependent increase
in apoptotic cells was detected following treatment with API-1
alone. When LoVo cells were exposed to 10 µM FR and 1 µM API-1,
there was a 10.6-fold increase in DNA fragmentation observed at 48
h compared with untreated controls (Fig.
5A and B). Additionally, subsequent to 10 µM FR and 2 µM API-1
treatment, the DNA fragmentation rate of DLD-1 cells increased 6.26
fold at 24 h. The highest fragmentation rate was observed for the
aforementioned 10 µM FR and 1 µM API-1 concentrations for 48 h in
LoVo cells (P=0.001) and 10 µM FR and 2 µM API-1 concentrations for
24 h in DLD-1 cells (P=0.002) (Fig.
5).
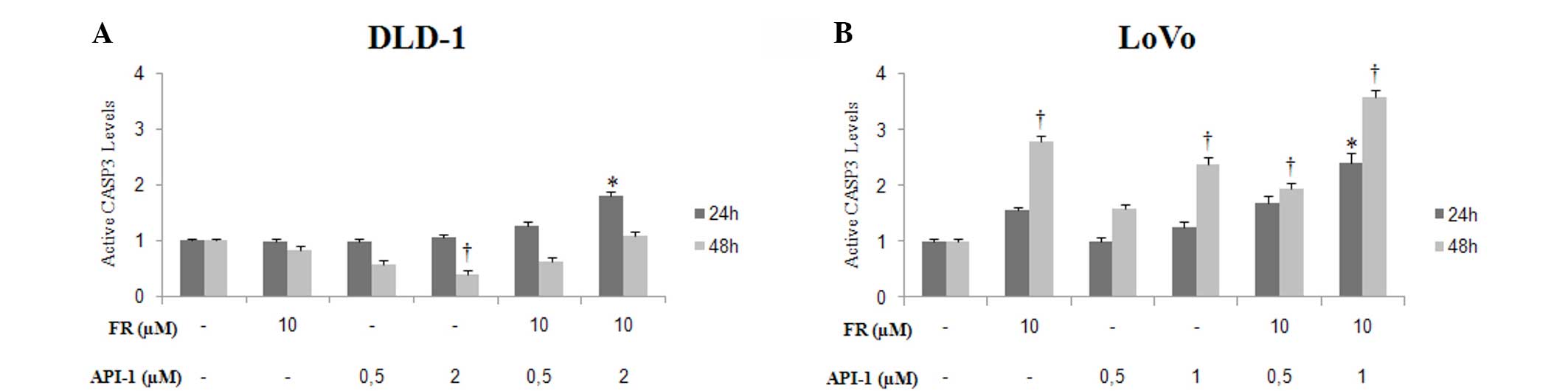
Treatment with FR and API-1 alone or
in combination induced caspase-3 cleavage
Cleaved caspase-3 protein levels were examined by
ELISA to assess caspase activity, in order to determine the nature
of apoptosis induction by treatment with FR and API-1 alone and in
combination. Following treatment with 10 µM FR, caspase-3 activity
was found to be increased in LoVo cells at 48 h and DLD-1 cells at
24 h post-treatment. Cleaved caspase-3 protein levels increased in
a dose- and time-dependent manner following API-1 treatment in LoVo
cells (Fig. 6). In addition, the
combination of FR and API-1 caused a significant increase in
cleaved caspase-3 levels in LoVo cells subsequent to 24 and 48 h of
treatment (24 h, P=0.001; 48 h, P=0.001) (Fig. 6B). However, in DLD-1 cells, caspase-3
activity was only significantly increased subsequent to 24 h of
treatment with FR and API-1 alone or in combination (FR, P=0.001;
API-1, P=0.001). Co-treatment with FR and API-1 at the highest
concentration in these cells markedly induced cleaved caspase-3
levels compared with FR and API-1 treatment alone (DLD-1 at 24 h,
P=0.001; LoVo at 24 h and 48 h, P=0.01) (Fig. 6A).
Expression of BCL2, BCL2L1, BAX, BAK,
BIM, CYCD1 and cMYC in FR and API-1-treated cells
The mRNA levels of BCL2, BCL2L1, BAX, BAK, BIM,
CYCD1 and cMYC were evaluated by quantitative RT-PCR. It was found
that treatment with FR and API-1 alone and in combination
significantly downregulated the BCL2 (FR, P=0.001; API-1, P=0.001;
FR + API-1, P=0.022) and BCL2L1 (FR, P=0.001; API-1, P=0.001; FR +
API-1, P=0.001) mRNA levels in LoVo cells. In addition, BCL2 and
BCL2L1 mRNA levels were significantly decreased in DLD-1 cells
subsequent to combined treatment with FR and API-1 (BCL2, P=0.027;
BCL2L1, P=0.019). Compared with the controls, proapoptotic BAX
(API-1, P=0.001; FR + API-1, P=0.033), BAK (API-1, P=0.001; FR +
API-1, P=0.049) and BIM (API-1, P=0.001; FR + API-1, P=0.033) mRNA
levels were also significantly upregulated following treatment with
1 µM API-1 and 10 µM FR + 1 µM API-1 in LoVo cells. In DLD-1 cells,
BAX mRNA levels were increased following treatment with API-1 alone
and FR + API-1. In addition to BAX mRNA levels, combination
treatment caused upregulation of BAK mRNA levels (Fig. 7A and B). Additionally, 10 µM FR
treatment did not significantly change BAX, BAK and BIM mRNA
expression levels (P>0.05). Compared to control cells, the CYCD1
mRNA levels were significantly decreased subsequent to combined
treatment with FR and API-1 for 24 h (P=0.001), but not 48 h, in
LoVo cells (P>0.05). Significantly downregulated CYCD1 mRNA
levels were also determined following combined treatment with FR
and API-1 at 24 and 48 h in DLD-1 cells (24 h, P=0.027; 48 h,
P=0.001). By contrast, Fig. 7
indicated that cMYC mRNA levels were significantly decreased
subsequent to treatment with FR + API-1 for 24 and 48 h in LoVo and
DLD-1 cells (DLD-1 at 24 h, P=0.041 and 48 h, P=0.001; LoVo at 24
h, P=0.031 and 48 h, P=0.021).
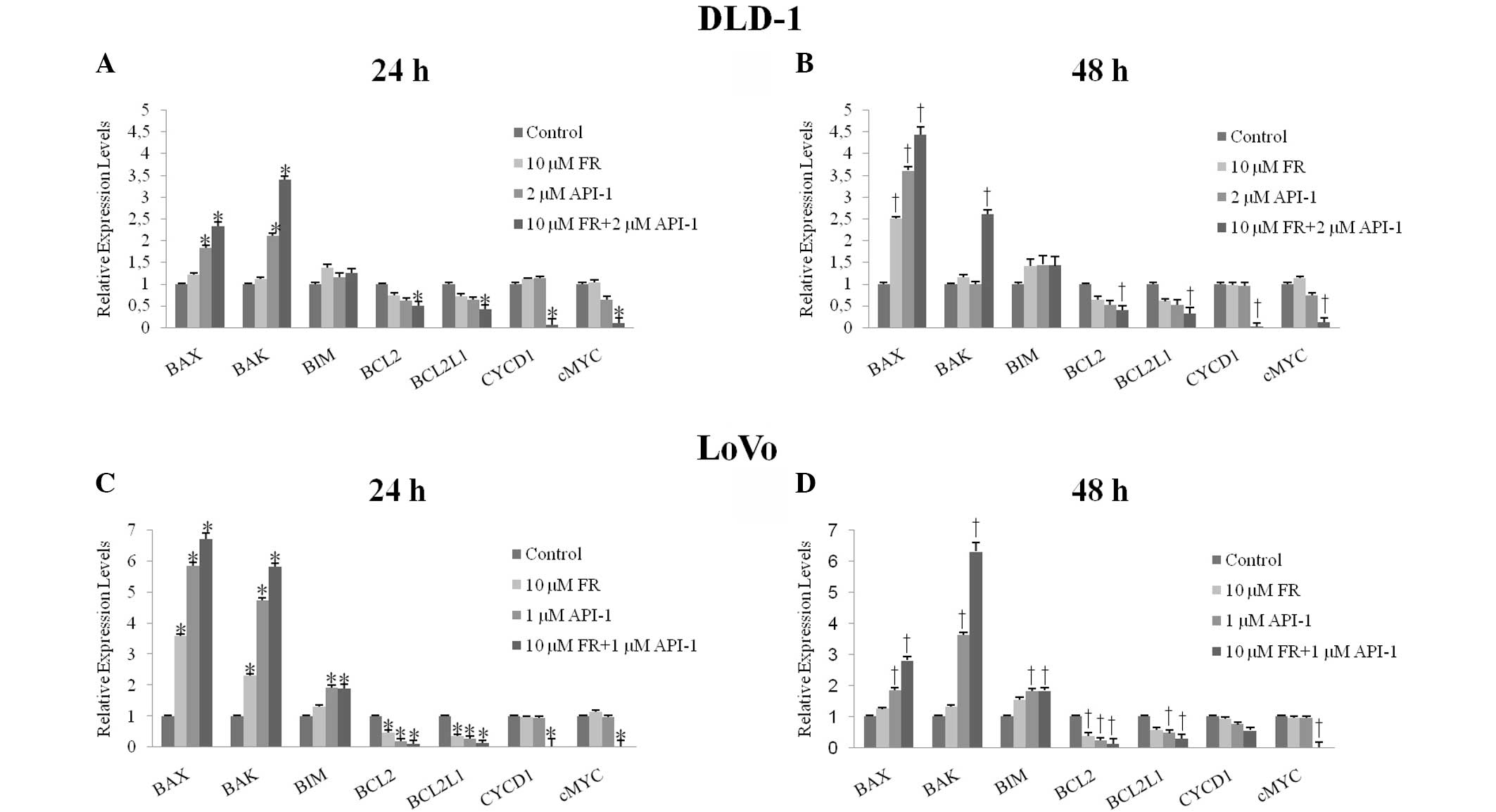 | Figure 7.Quantitative reverse
transcription-polymerase chain reaction was used to examine the
mRNA expression levels of BCL2, BCL2L1, BAX, BAK, BİM, CYCD1 and
cMYC genes subsequent to treatment with FR and API-1 alone or in
combination for (A) 24 h and (B) 48 h in DLD-1 cells and (C) 24 h
and (D) 48 h in LoVo cells. *P<0.05 vs. control cells for 24 h.
†P<0.05 vs. control cells for 48 h. mRNA, messenger RNA; FR,
FR180204; API-1,
4-amino-5,8-dihydro-5-oxo-8-β-D-ribofuranosyl-pyrido[2,3-d]pyrimidine-6-carboxamide;
BCL2, B-cell lymphoma-2; BCL2L1, BCL2 like 1; BAX, BCL2-associated
X protein; BAK, BCL2 antagonist/killer; BIM, BCL2-like 11; CYCD1,
cyclin D1. |
Effects of treatment with FR and/or
API-1 on BAX, BAK, CYCD1 and cMYC protein levels
To investigate the effects of treatment with FR and
API-1 alone and in combination on LoVo cells, the expression of
BAX, BAK, CYCD1 and cMYC proteins was assessed by western blot
analysis. The expression of CYCD1 and cMYC proteins was consistent
with results from quantitative RT-PCR analysis. As illustrated in
Fig. 8, while FR and API-1 had no
significant effects on the levels of CYCD1 and cMYC, combination
treatment of the two agents resulted in a marked reduction of CYCD1
and cMYC at 24 and 48 h in LoVo and DLD-1 cells. Additionally, in
comparison with the mRNA expression levels, there was an induction
in BAK and BAX proteins in the DLD-1 and LoVo cells. Western blot
analysis showed that treatment with API-1 alone increased the
expression of BAK protein at 24 h in DLD-1 cells (Fig. 8A). Treatment with FR and API-1 alone
for 24 and 48 h also upregulated the expression of BAK protein in
LoVo cells (Fig. 8B). Additionally,
the protein level of BAK was increased following combined treatment
with FR and API-1 in these CRC cells at 24 and 48 h. The western
blot analysis also showed a similar trend between BAX protein
levels and gene expression. As demonstrated in Fig. 8, the expression levels of BAX protein
were upregulated in the presence of API-1 alone and FR + API-1 in
these cells.
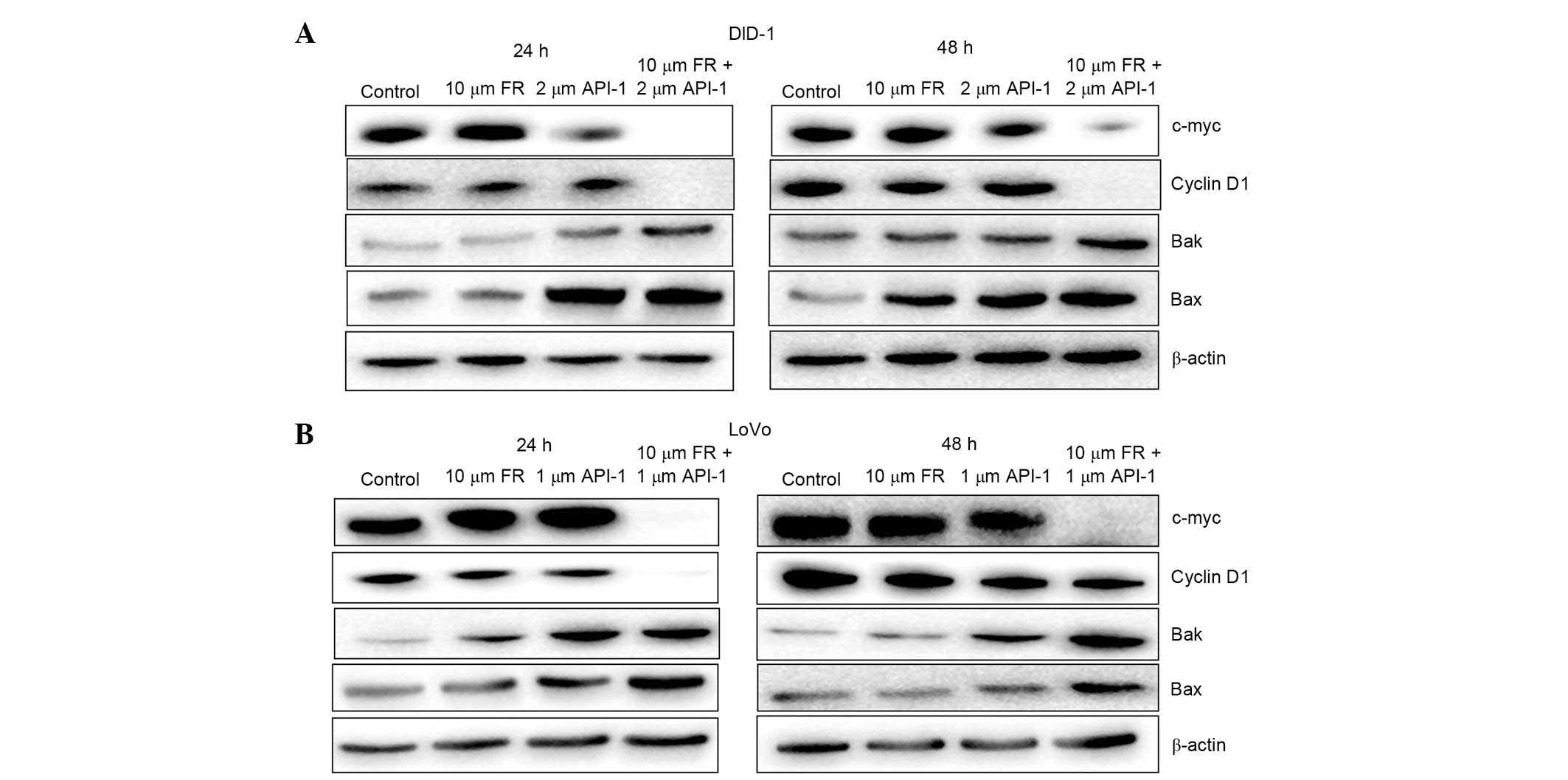 | Figure 8.Effect of FR and API-1 alone or in
combination treatment on the protein expression of BAX, BAK, cyclin
D1 and cMYC in (A) DLD-1 and (B) LoVo cells at 24 and 48 h, as
measured by western blot analysis. The reference protein β-actin
was used as an internal loading control. FR, FR180204; API-1,
4-amino-5,8-dihydro-5-oxo-8-β-D-ribofuranosyl-pyrido[2,3-d]pyrimidine-6-carboxamide;
BCL2, B-cell lymphoma-2; BAX, BCL2-associated X protein; BAK, BCL2
antagonist/killer. |
Effect of treatment with FR and API-1
alone or in combination on pAkt and pERK1/2 protein levels
The present study investigated the effect of
treatment with FR and API-1 for 24 and 48 h on the phosphorylation
of Akt and ERK1/2 in LoVo and DLD-1 cells. As shown in Fig. 9, in the presence of FR, pERK1/2
expression was downregulated, while the expression of total ERK1/2
did not change. Additionally, the phosphorylation of ERK1/2 was
also inhibited by treatment of cells with FR and API-1 in
combination for 24 and 48 h (Fig. 9).
Additionally, the total protein level of Akt did not alter
following treatment with FR and API-1 alone or in combination. In
addition, pAkt (Ser473) protein levels were significantly decreased
following treatment with API-1 in these cells for 24 and 48 h.
Additionally, combined treatment with FR and API-1 significantly
inhibited Akt activation, indicated by the decrease in the
phosphorylation of Akt at 24 and 48 h. The protein level of pAkt,
however, was markedly decreased by treatment with API-1 alone and
the combination of FR and API-1 at 48 h in DLD-1 cells.
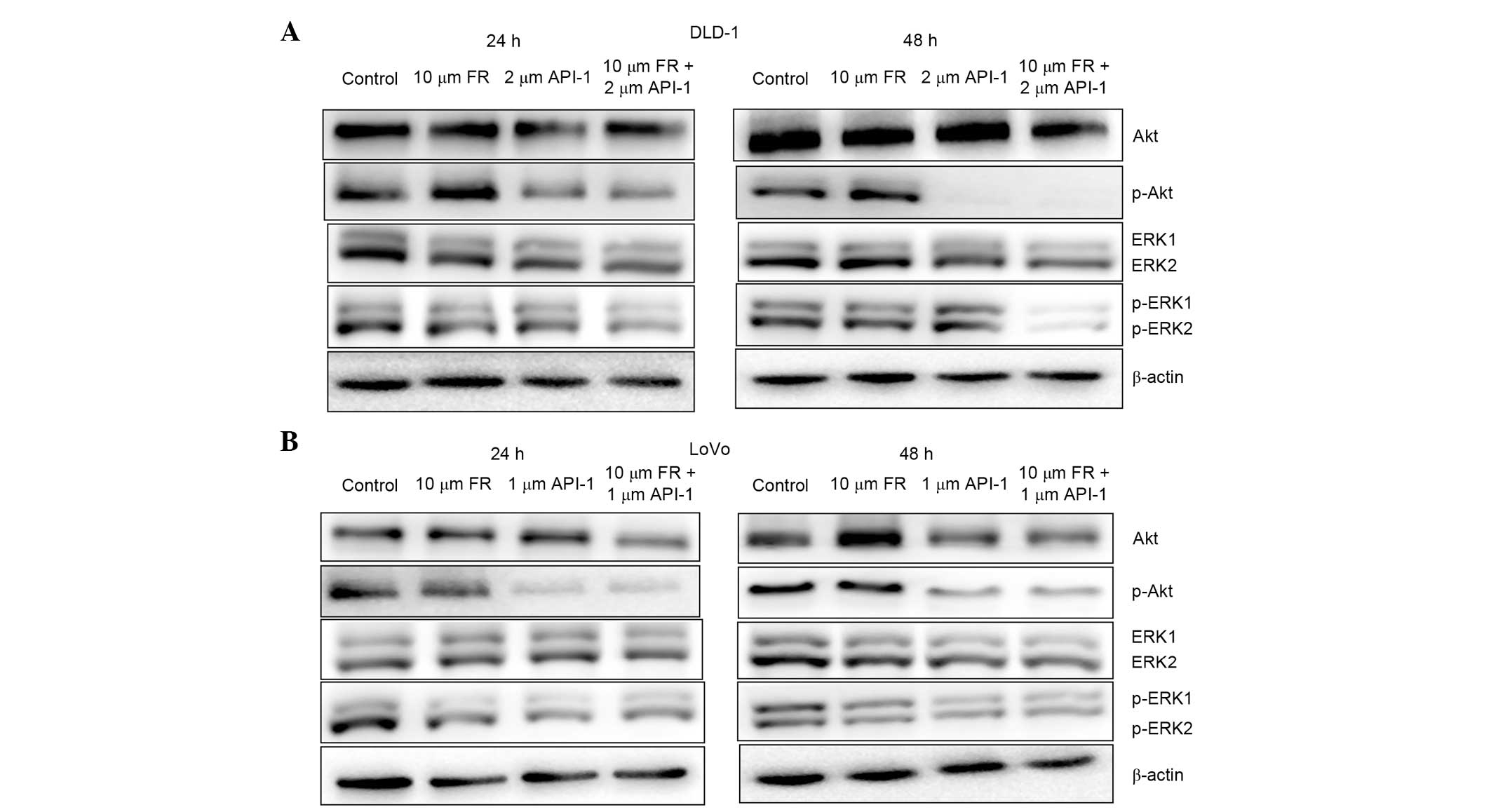 | Figure 9.Effect of treatment with FR and API-1
alone or in combination on on intracellular signaling pathways in
(A) DLD-1 and (B) LoVo cells. Cells were treated with the indicated
concentrations of these agents for 24 and 48 h and protein levels
of p-Akt, total Akt, p-ERK1/2 and total ERK1/2 were assayed by
western blot analysis. The reference protein β-actin was used as an
internal loading control. FR, FR180204; API-1,
4-amino-5,8-dihydro-5-oxo-8-β-D-ribofuranosyl-pyrido[2,3-d]pyrimidine-6-carboxamide;
Akt, v-akt murine thymoma viral oncogene homolog; p-Akt,
phosphorylated Akt; ERK1/2, extracellular signal-regulated kinase
1/2; p-ERK1/2, phosphorylated ERK1/2. |
Discussion
The PI3K/Akt and Raf/MEK/ERK pathways are critical
signal transduction pathways for regulating cell survival,
proliferation, differentiation, migration, motility, metabolism and
drug resistance (31–33). Mutationally activated or inactivated
components of these pathways give rise to oncogenic transformation
of the normal cells and cause a variety of cancers, such as CRC,
melanoma, and lung, pancreatic and breast cancers (33–35).
Therefore, numerous chemotherapeutic regimens targeting Ras
downstream effectors are widely-adopted strategy for clinical
success (35–37). Numerous studies have shown that
inhibition of the components one signaling pathway leads to
hyperactivation of another signaling pathways and addresses the
issue of resistance, positive feedback mechanisms and resulting in
increased toxicity (38,39). Therefore, combinational therapy
regimens targeting PI3K/Akt and Raf/MEK/ERK effectors appear to be
improved therapeutic approaches to inhibit cell proliferation and
induce apoptosis (19,39–41). The
present study aimed to investigate the apoptotic and
anti-proliferative effects of the selective Akt inhibitor API-1 and
selective ERK1/2 inhibitor FR in DLD-1 and LoVo CRC cells.
According to previous studies, DLD-1 cells harbor a constitutively
active mutation in PIK3CA, which evokes resistance to monotherapy
regimens (42). Briefly, the present
study mainly focused on the effects of different concentrations of
API-1 combined with a randomized concentration of FR. In the
literature, the antiproliferative effects of FR were also evaluated
at concentrations of 10–50 µM FR in various cancer cell lines,
including adrenocortical, mesothelioma, prostate, pancreatic and
CRC cell lines, via inhibiting ERK1/2 in an ATP-competitive manner
(25,43–47). In
the study by Ohori et al (25), it was noted that FR was selective for
ERK1/2 at concentrations <30 µM compared to another kinases,
such as human recombinant MAPK kinase 1, MAPK kinase 4, IκB kinase
α, protein kinase C α, Src, Syc and platelet-derived growth factor
α (25). Therefore, the lower
concentration of FR (10 µM) was chosen for single and combination
regimens to eliminate the possible non-selective kinase inhibitory
effects of FR, in the DLD-1 and LoVo cell lines.
Several studies have described the effects of API-1
as a single agent or in combination with other inhibitors of cell
proliferation and/or cell death in various cancer cells (24,48,49). In
the study by Li et al (24),
it was demonstrated that significantly decreased cell viability and
induced apoptosis in 3 different non-small cell lung carcinoma and
3 various head and neck squamous cell carcinoma cell lines
following API-1 treatment. This study found that treatment with
API-1 sensitized these 6 cells to apoptosis in a PI3K/Akt-dependent
manner. The antiproliferative response of these cells to API-1 was
determined to be dose-dependent, and increased cell death was
observed subsequent to exposure to API-1 for 72 h (24). Similarly, Toulany et al
(49) showed that treatment with
API-1 in human lung adenocarcinoma cells inhibited cell viability
and enhanced radiation-dependent apoptosis. Dirican et al
(46) investigated the possible
synergistic cytotoxic/apoptotic effects of docetaxel and
thymoquinone (TQ) in the presence of 10 µM FR in DU-145 cells and
drug-refractory prostate cancer cells. It was identified that the
ERK1/2 inhibitor FR enhanced the combination treatment of docetaxel
and TQ effects on inhibition of cell viability (46). Additionally, Doghman and Lalli
(47) showed that FR treatment alone
in 3 different adrenocortical carcinoma cell lines reduced cell
viability. This study also found that FR and BEZ-235 combination
treatment reduced the viability of LoVo cells more than
single-agent treatments (47).
Furthermore, as shown in Fig. 1C and
F, when elimination of the synergistic crosstalk mechanism
between the ERK and Akt pathways via FR was attempted, it was
observed that the resistance mechanism in DLD-1 cells appeared to
be activated in a time-dependent manner. As previously reported,
inhibiting the ERK signaling pathway leads the hyperactivation of
the Akt signaling pathway and vice versa (38,39). This
may be the cause of the increase in LoVo cell viability at 10 µM FR
+ ≥3 µM API-1 at 24 h. Concentrations of 10 µM FR + 2 µM API-1 for
DLD-1 cells and 10 µM FR + 1 µM API-1 for LoVo cells were therefore
used as optimal combination regimens at 24 h for DLD-1 cells and 48
h for LoVo cells. To support the WST assay results, cytotoxicity
analysis was performed using a commercial kit that measures the
amount of LDH released from damaged cells. According to Fig. 3, the levels of LDH leakage were
increased with the rising concentrations of treatment with single
agents at 24 h in DLD-1 cells, but at 48 h in LoVo cells.
Additionally, LoVo cells demonstrated an increased response to FR
and FR + API-1 treatments compared with DLD-1 cells at each time
interval.
Induction of apoptosis in cancer cells has already
been used as key indicator for the efficacy of a chemotherapeutic
agent (50,51). Cells undergo typical morphological
changes during apoptosis and can be detected using specific dyes,
such as AO/EB. Therefore, the present study aimed to elucidate
whether the cells undergo apoptosis or necrosis with single and/or
combination treatment. The present fluorescent microscopy results
showed that LoVo cells were much more susceptible to apoptosis than
DLD-1 cells with FR treatment. In addition, when FR treatment was
combined with API-1 treatment, the apoptotic ratio was
significantly increased in a dose-dependent manner. The
proapoptotic effects of FR and/or API-1 in CRC cells were further
confirmed using the Cell Death Detection ELISA assay, which is more
sensitive to detect apoptosis rather than necrosis. In LoVo cells,
API-1 treatment synergistically increased the DNA fragmentation
ratio of the FR treated cells, particularly at 48 h. By contrast,
treatment with 2 µM AP-1 alone appeared to induce apoptosis in more
DLD-1 cells compared with the combination regimens at 48 h.
Overall, the apoptotic cell ratio and DNA fragmentation results
suggested that when FR treated cells were administered with low
dose API-1, the ERK pathway was inhibited at first, which led to
hyperactivation of Akt pathway via crosstalk mechanisms; API-1 has
limited activity to overcome the resistance and crosstalk
mechanisms. However, increased concentrations of API-1 showed a
significant proapoptotic profile and eliminated the compensatory
mechanisms in FR-treated CRC cells, but the DNA fragmentation ratio
at 48 h in DLD-1 cells was not significantly increased. In
addition, as mentioned in the results section, when DLD-1 cells
were treated with FR and API-1, cleaved caspase-3 levels were
elevated in a dose-dependent manner at 24 h, but not at 48 h. By
contrast, LoVo cells were more susceptible to caspase-dependent
cell death at 48 h. However, when compared to the DNA fragmentation
results, DLD-1 cells appeared to have possible alternative
apoptotic mechanisms along with caspase mediated cell death. In
accordance with the present results, Hong et al showed that
DLD-1 cells may undergo caspase-independent apoptosis via
mitochondrial ROS production and JNK activation when
apoptosis-associated speck-like protein containing a caspase
recruitment domain (ASC) gene, which is epigenetically silenced in
numerous human cancers, was reactivated (52). Additionally, Nakagawa et al
demonstrated that the phytochemical α-mangostin induces apoptosis
by reducing mitochondrial membrane potential and increasing
endonuclease-G mediated nucleosomal DNA fragmentation (53). Therefore, the present study aimed to
reveal whether mitochondrial BCL2 family members were associated
with apoptotic cell death subsequent to single-agent and
combination treatment via mRNA and protein analysis.
Several studies have suggested that FR exerts
inhibitory effects against several cancer cell lines via
suppression of ERK1/2 phosphorylation (43,45,47,54).
For example, Doghman and Lalli (47)
showed that the protein level of pERK was decreased, but the level
of total ERK was not altered, in FR-treated adrenocortical cancer
cells induced by BEZ235 in vitro (47). Another study revealed that treatment
with FR blocked the phosphorylation and subsequent nuclear
translocation of ERK1/2 in response to EGF stimulation in U138
glioblastoma cells (43). In
addition, it has been found that treatment with FR reduced ERK1/2
phosphorylation in Caco-2, HCT-116 and SW-620 CRC cell lines
(45). API-1 inhibits phosphorylation
of Akt by binding to the PH domain of Akt, preventing
phosphorylation at Thr308 and Ser473 and blocking the recruitment
of Akt to the plasma membrane. In the present study, it was
observed that phosphorylated Akt (Ser473) was downregulated, but
the level of total Akt was not altered in LoVo cells treated with
API-1 alone and API-1 + FR. Similar results were also found in
several cancer cells, and it was found that treatment with API-1
decreased the level of p-Akt, but total Akt levels were not changed
(23,24). Additionally, the present study found
the proapoptotic BAX, BAK and BIM mRNA levels were significantly
increased following treatment with API-1 alone and in combination
with FR treatment. However, the levels of antiapoptotic BCL2 and
BCL2L1 were significantly decreased following combination treatment
in DLD-1 and LoVo cells at 24 and 48 h. Additionally, the western
blot results supported that treatment with API-1 alone and in
combination with FR activates mitochondrial apoptotic pathways. By
contrast, the changes in the mRNA and protein levels of BCL2 family
members following treatment with FR or API-1 alone showed that the
PI3KCA-wild-type LoVo cells were more susceptible to apoptosis
compared with DLD-1 cells. Combination treatment appeared to be an
alternative therapeutic approach for each CRC cell line.
CYCD1 is an important cell cycle regulator that
facilitates the transition between G1 and S phase and is
overexpressed in the majority of cancer cells, such as CRC
(48,55). The cMYC protooncogene, usually
implicated in cell transformation, differentiation and cell-cycle
progression, also has a central role in certain forms of apoptosis.
However, these two markers are regulated mainly by the Raf/MEK/ERK
pathways, and the expression of the markers was not significantly
changed at mRNA and protein levels subsequent to 10 µM FR
treatment. However, API-1 + FR treatment regimens significantly
decreased cMYC levels, which prevented proliferation in each cell
line. In addition, CYCD1 mRNA and protein levels were reduced
subsequent to treatment of the two cell lines with API-1 and FR,
with the exception of mRNA levels of LoVo cells at 48 h. The
reduction in CYCD1 levels made it difficult for cells to enter to
the S phase. In addition, the results of BrdU incorporation, which
is a key marker for the DNA synthesis rate at S phase, also
revealed that cell cycle arrest did not completely occur following
combination treatment (10 µM FR + 2 µM API-1 for DLD-1 cells and 10
µM FR + 1 µM API-1 for LoVo cells), as shown in the results
section. The results of BrdU incorporation, which is a key marker
for the DNA synthesis rate at S phase, revealed that cell cycle
arrest did not completely occur following combination treatment in
the cells, as shown in the results section. Flow cytometry analysis
is required to reveal the cell cycle progression subsequent to FR
and API-1 treatments in these cell lines.
The Raf/MEK/ERK and PI3K/Akt signaling pathways are
deregulated in various human cancers, including CRC. Therefore, the
components of these signaling cascades are attractive targets for
the treatment of CRC. The current study showed that combination
treatment with the selective Akt inhibitor API-1 and selective
ERK1/2 inhibitor FR prevents proliferation and induces apoptosis
compared to single treatment in vitro via deregulating the
crosstalk mechanisms. Additional analyses are required to elucidate
the effects of combination regimens on cell cycle progression, cell
proliferation and apoptosis in vitro and in vivo.
Acknowledgements
The present study was supported by The Scientific
and Technological Research Council of Turkey (grant number
111S054).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Inoue-Choi M, Lazovich D, Prizment AE and
Robien K: Adherence to the World Cancer Research Fund/American
Institute for Cancer Research recommendations for cancer prevention
is associated with better health-related quality of life among
elderly female cancer survivors. J Clin Oncol. 31:1758–1766. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Haggar FA and Boushey RP: Colorectal
cancer epidemiology: Incidence, mortality, survival, and risk
factors. Clin Colon Rectal Surg. 22:191–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bardhan K and Liu K: Epigenetics and
colorectal cancer pathogenesis. Cancers (Basel). 5:676–713. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
vanEngeland M, Derks S, Smits KM, Meijer
GA and Herman JG: Colorectal cancer epigenetics: Complex
simplicity. J Clin Oncol. 29:1382–1391. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu FZ, Yu BH, Li DL, Ke HL, Guo XZ and
Xiao XY: PI3K expression and PIK3CA mutations are related to
colorectal cancer metastases. World J Gastroenterol. 18:3745–3751.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ye Q, Cai W, Zheng Y, Evers BM and She QB:
ERK and AKT signaling cooperate to translationally regulate
survivin expression for metastatic progression of colorectal
cancer. Oncogene. 33:1828–1839. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang J, Roberts TM and Shivdasani RA:
Targeting PI3K signaling as a therapeutic approach for colorectal
cancer. Gastroenterology. 141:50–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fearon ER: Molecular genetics of
colorectal cancer. Annu Rev Pathol. 6:479–507. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu M and Grady WM: Therapeutic targeting
of the phosphatidylinositol 3-kinase signaling pathway: Novel
targeted therapies and advances in the treatment of colorectal
cancer. Therap Adv Gastroenterol. 5:319–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Franke TF, Kaplan DR and Cantley LC: PI3K:
Downstream AKTion blocks apoptosis. Cell. 88:435–437. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Itoh N, Semba S, Ito M, Takeda H, Kawata S
and Yamakawa M: Phosphorylation of Akt/PKB is required for
suppression of cancer cell apoptosis and tumor progression in human
colorectal carcinoma. Cancer. 94:3127–3134. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arcaro A and Guerreiro AS: The
phosphoinositide 3-kinase pathway in human cancer: Genetic
alterations and therapeutic implications. Curr Genomics. 8:271–306.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luo J, Manning BD and Cantley LC:
Targeting the PI3K-Akt pathway in human cancer: Rationale and
promise. Cancer Cell. 4:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Daouti S, Wang H, Li WH, Higgins B,
Kolinsky K, Packman K, Specian A Jr, Kong N, Huby N, Wen Y, et al:
Characterization of a novel mitogen-activated protein kinase kinase
1/2 inhibitor with a unique mechanism of action for cancer therapy.
Cancer Res. 69:1924–1932. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang SH, Sharrocks AD and Whitmarsh AJ:
MAP kinase signalling cascades and transcriptional regulation.
Gene. 513:1–13. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lange F, Franz B, Maletzki C, Linnebacher
M, Hühns M and Jaster R: Biological and molecular effects of small
molecule kinase inhibitors on low-passage human colorectal cancer
cell lines. Biomed Res Int. 2014:5686932014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cossa G, Gatti L, Cassinelli G, Lanzi C,
Zaffaroni N and Perego P: Modulation of sensitivity to antitumor
agents by targeting the MAPK survival pathway. Curr Pharm Des.
19:883–894. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
De Luca A, Maiello MR, D'Alessio A,
Pergameno M and Normanno N: The RAS/RAF/MEK/ERK and the PI3K/AKT
signalling pathways: Role in cancer pathogenesis and implications
for therapeutic approaches. Expert Opin Ther Targets. 16(Suppl 2):
S17–S27. 2012. View Article : Google Scholar
|
|
20
|
Gollob JA, Wilhelm S, Carter C and Kelley
SL: Role of Raf kinase in cancer: Therapeutic potential of
targeting the Raf/MEK/ERK signal transduction pathway. Semin Oncol.
33:392–406. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McCubrey JA, Milella M, Tafuri A, Martelli
AM, Lunghi P, Bonati A, Cervello M, Lee JT and Steelman LS:
Targeting the Raf/MEK/ERK pathway with small-molecule inhibitors.
Curr Opin Investig Drugs. 9:614–630. 2008.PubMed/NCBI
|
|
23
|
Kim D, Sun M, He L, Zhou QH, Chen J, Sun
XM, Bepler G, Sebti SM and Cheng JQ: A small molecule inhibits Akt
through direct binding to Akt and preventing Akt membrane
translocation. J Biol Chem. 285:8383–8394. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li B, Ren H, Yue P, Chen M, Khuri FR and
Sun SY: The novel Akt inhibitor API-1 induces c-FLIP degradation
and synergizes with TRAIL to augment apoptosis independent of Akt
inhibition. Cancer Prev Res (Phila). 5:612–620. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohori M, Kinoshita T, Okubo M, Sato K,
Yamazaki A, Arakawa H, Nishimura S, Inamura N, Nakajima H, Neya M,
et al: Identification of a selective ERK inhibitor and structural
determination of the inhibitor-ERK2 complex. Biochem Biophys Res
Commun. 336:357–363. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Batista LF, Roos WP, Christmann M, Menck
CF and Kaina B: Differential sensitivity of malignant glioma cells
to methylating and chloroethylating anticancer drugs: p53
determines the switch by regulating xpc, ddb2, and DNA
double-strand breaks. Cancer Res. 67:11886–11895. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Boult J, Roberts K, Brookes MJ, Hughes S,
Bury JP, Cross SS, Anderson GJ, Spychal R, Iqbal T and Tselepis C:
Overexpression of cellular iron import proteins is associated with
malignant progression of esophageal adenocarcinoma. Clin Cancer
Res. 14:379–387. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Anadol E, Kanca H, Yar AS, Helvacioğlu F,
Menevşe S, Calgüner E and Erdoğan D: Prostaglandin F receptor
expression in intrauterine tissues of pregnant rats. J Vet Sci.
15:125–131. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pfaffl MW, Horgan GW and Dempfle L:
Relative expression software tool (REST) for group-wise comparison
and statistical analysis of relative expression results in
real-time PCR. Nucleic Acids Res. 30:e362002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brown KK and Toker A: The phosphoinositide
3-kinase pathway and therapy resistance in cancer. F1000Prime Rep.
7:132015. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Y, Kaiser CE, Frett B and Li HY:
Targeting mutant KRAS for anticancer therapeutics: A review of
novel small molecule modulators. J Med Chem. 56:5219–5230. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Britten CD: PI3K and MEK inhibitor
combinations: Examining the evidence in selected tumor types.
Cancer Chemother Pharmacol. 71:1395–1409. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cox AD, Fesik SW, Kimmelman AC, Luo J and
Der CJ: Drugging the undruggable RAS: Mission possible? Nat Rev
Drug Discov. 13:828–851. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baines AT, Xu D and Der CJ: Inhibition of
Ras for cancer treatment: The search continues. Future Med Chem.
3:1787–1808. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Takashima A and Faller DV: Targeting the
RAS oncogene. Expert Opin Ther Targets. 17:507–531. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gysin S, Salt M, Young A and McCormick F:
Therapeutic strategies for targeting ras proteins. Genes Cancer.
2:359–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Watanabe T, Kobunai T, Yamamoto Y, Matsuda
K, Ishihara S, Nozawa K, Iinuma H, Ikeuchi H and Eshima K:
Differential gene expression signatures between colorectal cancers
with and without KRAS mutations: Crosstalk between the KRAS pathway
and other signalling pathways. Eur J Cancer. 47:1946–1954. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Steelman LS, Chappell WH, Abrams SL, Kempf
RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F,
Mazzarino MC, et al: Roles of the Raf/MEK/ERK and
PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity
to therapy-implications for cancer and aging. Aging (Albany NY).
3:192–222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ye Q and She QB: Integration of AKT and
ERK signaling pathways in cancer: Biological and therapeutic
implications. J Pharmacol Clin Toxicol. 1:10092013.
|
|
42
|
Morrow CJ, Gray A and Dive C: Comparison
of phosphatidylinositol-3-kinase signalling within a panel of human
colorectal cancer cell lines with mutant or wild-type PIK3CA. FEBS
Lett. 579:5123–5128. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mut M, Lule S, Demir O, Kurnaz IA and
Vural I: Both mitogen-activated protein kinase (MAPK)/extra
ellular-signal-regulated kinases (ERK) 1/2 and
phosphatidylinositide-3-OH kinase (PI3K)/Akt pathways regulate
activation of E-twenty-six (ETS)-like transcription factor 1
(Elk-1) in U138 glioblastoma cells. Int J Biochem Cell Biol.
44:302–310. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Honda M, Kanno T, Fujita Y, Gotoh A,
Nakano T and Nishizaki T: Mesothelioma cell proliferation through
autocrine activation of PDGF-ββ receptor. Cell Physiol Biochem.
29:667–674. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ragusa M, Statello L, Maugeri M, Majorana
A, Barbagallo D, Salito L, Sammito M, Santonocito M, Angelica R,
Cavallaro A, et al: Specific alterations of the microRNA
transcriptome and global network structure in colorectal cancer
after treatment with MAPK/ERK inhibitors. J Mol Med (Berl).
90:1421–1438. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dirican A, Atmaca H, Bozkurt E, Erten C,
Karaca B and Uslu R: Novel combination of docetaxel and
thymoquinone induces synergistic cytotoxicity and apoptosis in
DU-145 human prostate cancer cells by modulating PI3K-AKT pathway.
Clin Transl Oncol. 17:145–151. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Doghman M and Lalli E: Efficacy of the
novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235 in a preclinical
model of adrenocortical carcinoma. Mol Cell Endocrinol.
364:101–104. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim JK and Diehl JA: Nuclear cyclin D1: An
oncogenic driver in human cancer. J Cell Physiol. 220:292–296.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Toulany M, Kehlbach R, Florczak U, Sak A,
Wang S, Chen J, Lobrich M and Rodemann HP: Targeting of AKT1
enhances radiation toxicity of human tumor cells by inhibiting
DNA-PKcs-dependent DNA double-strand break repair. Mol Cancer Ther.
7:1772–1781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Asakura T and Ohkawa K: Chemotherapeutic
agents that induce mitochondrial apoptosis. Curr Cancer Drug
Targets. 4:577–590. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hong S, Hwang I, Lee YS, Park S, Lee WK,
Fernandes-Alnemri T, Alnemri ES, Kim YS and Yu JW: Restoration of
ASC expression sensitizes colorectal cancer cells to genotoxic
stress-induced caspase-independent cell death. Cancer Lett.
331:183–191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nakagawa Y, Iinuma M, Naoe T, Nozawa Y and
Akao Y: Characterized mechanism of alpha-mangostin-induced cell
death: Caspase-independent apoptosis with release of endonuclease-G
from mitochondria and increased miR-143 expression in human
colorectal cancer DLD-1 cells. Bioorg Med Chem. 15:5620–5628. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tan BJ and Chiu GN: Role of oxidative
stress, endoplasmic reticulum stress and ERK activation in
triptolide-induced apoptosis. Int J Oncol. 42:1605–1612.
2013.PubMed/NCBI
|
|
55
|
Takuwa N and Takuwa Y: Regulation of cell
cycle molecules by the Ras effector system. Mol Cell Endocrinol.
177:25–33. 2001. View Article : Google Scholar : PubMed/NCBI
|