Introduction
Cervical cancer (CC) is the fourth most common
cancer in women, with 528,000 estimated new cases and 266,000
mortalities worldwide in 2012 (1). In
particular, 86% of mortalities caused by CC occur in developing
countries (2), due to the absence of
national CC screening programs (3,4).
Persistent infection by oncogenic, high-risk (HR)
strains of human papillomavirus (HPV) is strongly associated with
CC development (5). The oncogenic
properties of HR-HPV (including HPV16 and HPV18) have been mainly
attributed to the production of two viral oncoproteins, E7 and E6
(5). The primary activity of E7 and
E6 is to block the tumor suppressor functions of retinoblastoma
(Rb) (6) and p53 (7,8),
respectively, by targeting them for ubiquitin-mediated proteasome
degradation (9). The combination of
these effects leads to a lack of G1 cell cycle arrest (p53 and
Rb-mediated) and to an enhanced phase S entry (9).
CC treatment includes surgery, radiotherapy and
cisplatin-based chemotherapy, alone or in combination (10). However, 35% of CC patients develop
persistent or recurrent disease following treatment, and experience
a poor prognosis (10). Thus, the
requirement for innovative and effective therapies in refractory CC
remains a high priority.
The incorporation of human immunodeficiency virus
(HIV) protease inhibitors (PIs) into highly active antiretroviral
therapy (HAART) has deeply changed the natural history of HIV
infection (11). Furthermore, due to
the HIV-PIs' efficacy in treating HIV-related Kaposi's sarcoma
independently of their anti-HIV activity, the potential antitumor
properties of these drugs have been successfully investigated
against other solid and hematological malignancies (12). In this context, HIV-PIs may be
considered a new class of drugs with multiple antitumor effects,
including inhibition of tumor cell invasion and angiogenesis,
induction of apoptosis, and inhibition of cell proliferation and
proteasome activity (12–21). In particular, the selective inhibition
of the proteasomal function could represent an effective strategy
for the treatment of HR-HPV infections via E6 and E7 proteins
turnover regulation. However, there are very few references about
the antitumor effects of HIV-PIs in the context of CC, with only
two studies focused on pre-neoplastic (22) rather than on transfected (23) cellular models.
The aim of the present study was to evaluate the
antitumor effects of HIV-PIs, particularly indinavir, ritonavir and
saquinavir, on CC cell lines, by analyzing cell proliferation, cell
cycle, invasion, clonogenicity and radiosensitivity.
Materials and methods
CC cell lines
Two primary CC cell lines (CC1 and CC2) were
established in the Division of Obstetrics and Gynecology (‘Angelo
Nocivelli’ Institute for Molecular Medicine, University of Brescia,
Brescia, Italy) from primary tumors collected under approval of the
Institutional Review Board of the socio-sanitary territorial agency
(ASST) Civil Hospital (Brescia, Italy; study reference number,
NP1284). CC patients were enrolled at the Division of Obstetrics
and Gynecology (University of Brescia) between January and November
2008, and written informed consent was obtained prior to tumor
specimen collection. Tumor biopsies were mechanically minced and
incubated in RPMI medium (Euroclone SpA, Milan, Italy) with 0.14%
collagenase type I (125 U/mg; Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany) and 0.01% DNAse (2,000 kU/mg; Sigma-Aldrich;
Merck Millipore) for 1 h. Cell suspensions were cultured in
keratinocyte serum-free medium (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 35–50 µg/ml bovine pituitary
extract (Thermo Fisher Scientific, Inc.) and 5 ng/ml human
epidermal growth factor (Thermo Fisher Scientific, Inc.). Cell
cultures were grown at 37°C in an atmosphere of 5% CO2.
Subsequent to the 50th passage, the cell lines were maintained in
RPMI medium with 10% fetal bovine serum (FBS; Euroclone SpA). The
epithelial purity of the cell lines was evaluated by
immunocytochemical staining with antibodies against epithelial
membrane antigen (EMA; clone gp 1.4; Leica Biosystems; Danaher
Corporation, Washington, DC, USA) diluted at 1:150 (incubated at
37°C for 30 min) and pan cytokeratin (clone MNF116; Dako, Glostrup,
Denmark) diluted at 1:100 (incubated at 37°C for 30 min).
The CaSki, HeLa, HT3 and C33a CC cell lines were
obtained from the American Type Culture Collection (Manassas, VA,
USA) and cultured in RPMI medium supplemented with 10% FBS. Cell
cultures were maintained at 37°C with 5% CO2.
HPV genotyping and HPV DNA status
DNA was isolated from CC cell lines with the DNeasy
Blood & Tissue kit (Qiagen GmbH, Hilden, Germany), and HPV
genotyping was performed using the Linear Array HPV Genotyping Test
(Roche Diagnostics, Basel, Switzerland) following the
manufacturer's protocol. The physical status of HPV DNA (integrated
and/or episomal) was estimated by polymerase chain reaction (PCR)
amplification of the E2 gene using type-specific primers (Table I). Go Taq® DNA Polymerase
(Promega Corporation, Madison, WI, USA) was used for amplification,
and 30 consecutive cycles (94°C for 30 sec, 57°C for 30 sec and
72°C for 30 sec) were performed.
 | Table I.Type-specific primers for the HPV E2,
E6 and E7 genes of HPV16 and HPV18. |
Table I.
Type-specific primers for the HPV E2,
E6 and E7 genes of HPV16 and HPV18.
| Gene name | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| HPV16 E2 |
ATGGAGACTGTTTGCCAACG |
TCATATAGACATAAATCCAGTAG |
| HPV18 E2 |
ATGCAGACACCGAAGGAAAC |
TTACATTGTCATGTATCCCAC |
| HPV16 E6 |
TGATATAATATTAGAATGTGTGTACTGCAAGCAA |
GCATAAATCCCGAAAAGCAAAGTCA |
| HPV18 E6 |
GGTGTATAGAGACAGTATACCGCATG |
TGTCTCCATACACAGAGTCTGAATAATGT |
| HPV16 E7 |
GCTCAGAGGAGGAGGATGAAATAGA |
GAGTCACACTTGCAACAAAAGGTT |
| HPV18 E7 |
GTGAAGCCAGAATTGAGCTAGTAGT |
AGAAACAGCTGCTGGAATGCT |
RNA extraction and reverse
transcription PCR for E6/E7 transcript expression
Total RNA was isolated from HPV DNA-positive tumor
cell lines using the PureLinkTM RNA Mini kit (Thermo Fisher
Scientific, Inc.). RNA purity and quantity were evaluated
spectrophotometrically. Total RNA was treated with Turbo DNase
(Roche Diagnostics), and complementary DNA was synthesized using
random hexamers according to the SuperScript™ II Reverse
Transcriptase protocol (Thermo Fisher Scientific, Inc.). To verify
E6/E7 transcript expression, PCR was conducted with AmpliTaq
Gold® DNA Polymerase (Thermo Fisher Scientific, Inc.) on
a 2720 Thermal Cycler (Applied Biosystems; Thermo Fisher
Scientific, Inc.), using pairs of primers designed based on the
conserved sequence regions of the HPV16 and HPV18 genotypes
(Table I). HT3 cells served as
HPV-negative controls.
Treatment of CC cell lines with
HIV-PIs
Media containing HIV-PIs [National Institutes of
Health (NIH) Acquired Immune Deficiency Syndrome (AIDS) Research
and Reference Reagent Program, Germantown, MD, USA] or drug
diluents (dimethyl sulfoxide for saquinavir and ritonavir, and
H2O for indinavir) were added to exponentially growing
cell cultures every day during the whole experimental period. For
proliferation and proteasome assays, all CC cell lines were
incubated with 10 and 100 µM indinavir, and with ritonavir and
saquinavir ranging from 5 to 40 µM. For cell cycle, cell invasion,
clonogenicity and radiosensitivity assays, HeLa cells were
incubated in the presence of 10 or 19 µM saquinavir, which
corresponded to 50 and 100% of the inhibitory concentration
(IC)50 value, respectively.
HIV-PIs concentrations were set according to data
reported by Hampson et al (23) about the effects of HIV-PIs on CC cell
viability. In addition, experiments using the drug peak levels (10
µM) detectable in plasma of HIV-PIs treated, HIV-infected
individuals (24), or non-infected
patients experiencing complete remission or regression of
early-stage Kaposi's sarcoma with low or no toxicity (25), were included.
Proliferation assays
Cells were seeded in 96-well plates at a density of
500 cells/well for CaSki and CC1 cells, 250 cells/well for HeLa
cells, 2,000 cells/well for CC2 and C33a cells, and 1,750
cells/well for HT3 cells. Cellular growth during the treatment was
estimated by crystal violet staining (26) every day until the CC cell lines
reached ~80% confluence.
Proteasome assays
The three catalytic activities of the proteasome
(chymotrypsin-like, trypsin-like and caspase-like) were
investigated in all cell lines (27).
Cells were seeded in 96-well plates, as described above for the
proliferation assays. Following treatment, cells were tested for
proteasomal activity using Proteasome-Glo™ Cell-Based Assays
(Promega Corporation), according to the manufacturer's protocol.
Luminescence was measured using Infinite M200 (Tecan Group Ltd.,
Männedorf, Switzerland). A potent proteasome inhibitor treatment, 5
µM MG132, was used as a positive control. Additionally, the
proteasomal activity of HeLa cells was evaluated by immunoblotting
on whole-cell protein extracts.
Immunoblotting
After 2 h of treatment with 40, 60 and 80 µM
saquinavir, whole HeLa cell protein extracts were prepared in 150
mM NaCl, 1% Nonidet-40, 50 mM Tris-HCl (pH 7.5) and Halt Protease
Inhibitor Cocktail (Thermo Fisher Scientific, Inc.). Cell extracts
(20 µg) were resolved by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (NuPAGE® Novex® 4–12%
Bis-Tris gels; Thermo Fisher Scientific, Inc.) and blotted onto
polyvinylidene difluoride membranes (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). α-Tubulin was used as a protein loading
control. Following blocking in Tris-buffered saline containing 5%
non-fat milk, the blots were incubated with primary antibodies
against α-tubulin (dilution, 1:20,000; T5168; Sigma-Aldrich; Merck
Millipore) or ubiquitin (dilution, 1:200; P4D1; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) at 4°C for 12 hours, followed
by incubation with horseradish peroxidase-conjugated secondary
rabbit anti-mouse IgG (dilution, 1:10,000; catalog no., A9044;
Sigma-Aldrich; Merck Millipore) at room temperature for 1 h.
Signals were detected on a BioSpectrum Imaging System (UVP, Inc.,
Upland, CA, USA) with the LiteAblot® EXTEND (Euroclone
SpA). Images were processed with VisionWorks® LS Image
Acquisition and Analysis software version 7.0.1 (UVP, Inc.).
Cell cycle analysis by flow
cytometry
HeLa cells were seeded in 6-well plates (8,000
cells/well). Upon treatment, cells were counted and fixed in 70%
cold ethanol prior to staining with 5 µg/ml propidium iodide in
phosphate-buffered saline and 12.5 µl/ml RNAse A overnight at 4°C.
Flow cytometric cell cycle analysis was performed on a minimum of
20,000 cells using a BD FACSCalibur™ instrument (BD Biosciences,
Franklin Lakes, NJ, USA) equipped with a 488-nm laser; fluorescence
emission was detected using a filter for 620±35 nm. The percentages
of distribution of cells in the different phases of the cell cycle
were analyzed according to the method by Bertuzzi et al
(28).
Cell invasion assays
After 96 h of treatment, 60,000 HeLa cells were
seeded in the upper compartment of a Corning BioCoat Matrigel
Invasion Chamber 24-well plate (BD Biosciences) containing RPMI.
The lower compartment contained RPMI with 10% FBS as
chemoattractant. After 30 h, the migrated cells were evaluated by
microscopy.
Clonogenicity assay
Following 96 h of treatment, HeLa cells were seeded
into 6-well plates at a density of 250 cells/well, and were treated
with saquinavir or DMSO for an additional 6 days. The colonies were
stained with crystal violet solution and analyzed with an Entry
Level Image system (Immagini & Computers, Bareggio, Italy). A
background correction was performed, and the smallest control cell
colony (≥50 cells), was considered as the minimum value to set the
cut-off point.
Radiosensitivity
After 96 h of treatment, HeLa cells were seeded in
6-well plates at a density of 500 cells/well, exposed to a dose of
0, 2, 4, 6 and 8-Gy radiation, and treated for additional 6 days.
Colonies were analyzed as described above.
Statistical analysis
All experiments were repeated three times
independently, and all samples were tested in triplicate in each
experiment. Student's t-test was used for paired samples to
evaluate the differences between the means obtained from treated
and non-treated cells. SPSS version 16.0 (SPSS, Inc., Chicago, IL,
USA) was used for statistical analysis. P<0.05 was considered to
indicate a statistically significant difference.
Results
CC cell lines characterization
Two primary CC cell lines, CC1 and CC2, were
obtained from patients with squamous CC and adenosquamous invasive
CC (stage IB), respectively. The immunocytochemical staining with
antibodies against EMA and pan cytokeratin demonstrated that the
primary cultures contained >99% of epithelial cells.
HPV DNA was detected and genotyped using Linear
Array HPV Genotyping Test, which detected 37 high- and low-risk HPV
genotypes, revealing the presence of HPV-16 in CC1 and HPV-18 in
CC2, and confirming the presence of HPV-16 in CaSki and HPV-18 in
HeLa cells, and the absence of HPV in HT3 and C33a cells.
Furthermore, this analysis excluded the presence of multiple HPV
infections in all CC cell lines.
According to the assumption that HPV integration
disrupts the E2 gene (29),
type-specific E2 PCR was performed on HPV-positive cell lines,
revealing no E2 amplification in any of the samples and, thus,
suggesting the complete viral integration in the host genome.
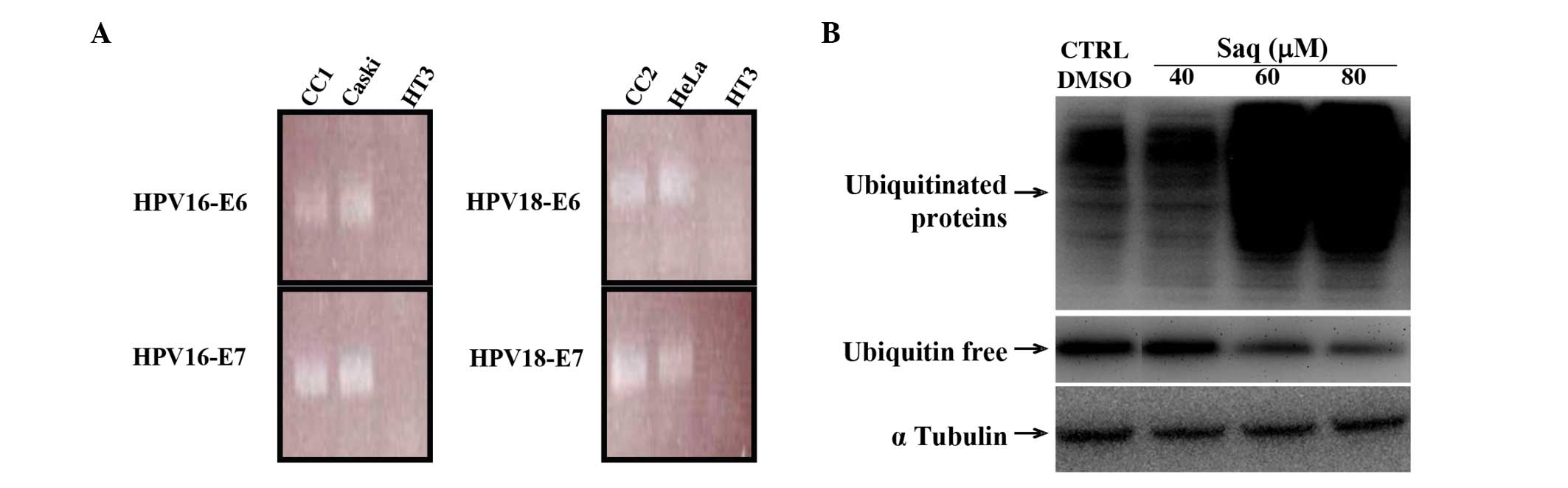
HPV16/18 E6 and E7 messenger RNA was uniformly detectable,
indicating that all HPV-positive cancer cell lines considered in
the present study were transcriptionally active and constitutively
expressed the oncogene transcripts (Fig.
1A).
HIV-PIs effects on cell
proliferation
In order to test the effects of indinavir, ritonavir
and saquinavir on cell proliferation, all CC cell lines were
cultured in the presence or absence of drugs at different
concentrations and for different exposure times. For all drugs, the
inhibition of cell growth was directly proportional to the drug
concentration and exposure time. This effect was observed in all CC
cell lines, as indicated in Table
II. Saquinavir was significantly more effective than ritonavir
in inhibiting cell proliferation. This effect was observed in all
CC cell lines and at all drug concentrations. On the contrary,
indinavir exerted a negligible and non-significant effect on cell
growth, even if used at high concentration (100 µM).
| Table II.Mean percentages of growth inhibition
of cervical cancer cell lines (CaSki, CC1, HeLa, CC2, C33a and HT3)
treated for 96 h with human immunodeficiency virus-protease
inhibitors. |
Table II.
Mean percentages of growth inhibition
of cervical cancer cell lines (CaSki, CC1, HeLa, CC2, C33a and HT3)
treated for 96 h with human immunodeficiency virus-protease
inhibitors.
|
| Indinavir (µM) | Ritonavir (µM) | Saquinavir
(µM) |
|---|
|
|
|
|
|
|---|
| Cells | 10 | 100 | 5 | 10 | 20 | 40 | 5 | 10 | 20 | 40 |
|---|
| CaSki | 3 | 5 | 3 | 7 | 30a | 67a | 10a | 15a | 47a | 100a |
| CC1 | 2 | 5 | 4 | 6 | 28a | 56a | 11a | 20a | 43a | 90a |
| HeLa | 6 | 8 | 4 | 15a | 40a | 77a | 10a | 17a | 56a | 100a |
| CC2 | 0 | 1 | 5 | 14a | 22a | 25a | 13a | 18a | 40a | 95a |
| C33a | 0 | 1 | 21a | 24a | 34a | 73a | 22a | 28a | 58a | 91a |
| HT3 | 0 | 5 | 8 | 14a | 40a | 60a | 21a | 26a | 58a | 100a |
HIV-PIs effects on proteasomal
activities
In order to evaluate if the observed effects of
HIV-PIs on cell proliferation could be correlated with a modulation
of the proteasome activity, all CC cell lines were treated with
saquinavir, indinavir and ritonavir under the same experimental
conditions used in the above proliferation assays (Table II). Chymotrypsin-like, trypsin-like
and caspase-like activities of the proteasome were analyzed by a
cell-based assay. Using these experimental conditions, no effect
was observed. On the contrary, when the concentrations of
saquinavir and ritonavir were increased to 60 and 80 µM,
respectively, and used to treat the cells for 2 h, a significant
modulation of proteasomal activities was observed (Table III). Treatment with 80 µM saquinavir
appeared to be the most effective one in inhibiting all three
proteasome activities for all CC cell lines. Saquinavir at 60 µM
and ritonavir at 80 µM inhibited the trypsin-like proteasome
activity in four cell lines (CC1, CC2, HeLa and C33a), as well as
the chymotrypsin-like proteasome activity in three cell lines (CC2,
CC1 and HeLa), and the caspase-like proteasome activity only in one
cell line (HeLa). Indinavir exerted a negligible and not
significant effect, even if used at 100 µM. Of note, among the CC
cell lines, CaSki and HT3 displayed a proteasome modulation only
with 80-µM saquinavir treatment. As a positive control, treatment
with 5 µM MG132 confirmed a strong inhibition of all proteasome
activities, ranging from 80 to 90% inhibition.
| Table III.Mean percentages of proteasome
inhibition of cervical cancer cell lines (CaSki, CC1, HeLa, CC2,
C33a and HT3) treated for 2 h with human immunodeficiency
virus-protease inhibitors. |
Table III.
Mean percentages of proteasome
inhibition of cervical cancer cell lines (CaSki, CC1, HeLa, CC2,
C33a and HT3) treated for 2 h with human immunodeficiency
virus-protease inhibitors.
|
|
| Ritonavir | Saquinavir |
|---|
|
|
|
|
|
|---|
|
| Indinavir ≤100
µM | ≤60 µM | 80 µM | ≤40 µM | 60 µM | 80 µM |
|---|
|
|
|
|
|
|
|
|
|---|
| Cells | All | All | Casp | ChyTry | Try | All | Casp | ChyTry | Try | Casp | ChyTry | Try |
|---|
| CaSki | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
9a | 26a | 18a |
| CC1 | 0 | 0 | 0 | 17a | 6a | 0 | 0 | 6–10a | 10a | 37a | 47a | 43a |
| HeLa | 0 | 0 | 13a | 20a | 15a | 0 |
41a | 53a | 51a | 49a | 56a | 54a |
| CC2 | 0 | 0 | 0 | 15a | 6a | 0 | 0 | 8a | 7a | 20a | 40a | 34a |
| C33a | 0 | 0 | 0 | 0 | 14a | 0 | 0 | 0 | 10a | 20a | 33a | 35a |
| HT3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 11a | 16a | 15a |
In order to confirm the effects of saquinavir on the
proteasome activities, the levels of ubiquitinated proteins were
evaluated in whole HeLa cell protein extracts by immunoblotting.
Consistent with the results of the proteasome cell-based assay,
treatment with indinavir or ritonavir had no or limited effects,
respectively, on the proteasome (data not shown), whereas treatment
with 60 or 80 µM saquinavir led to increased levels of
ubiquitinated proteins, indicating a significant inhibition of the
proteasomal machinery, as represented in Fig. 1B. The accumulation of ubiquitinated
proteins occurred with a concomitant decrease of ubiquitin. On the
contrary, treatment with low concentrations of saquinavir did not
reveal any significant change in the levels of ubiquitinated
proteins.
Effects of saquinavir on HeLa
cells
Based on the above data, saquinavir was selected as
the most effective drug, and HeLa cells were selected as the most
susceptible cell line, to analyze the effects of HIV-PIs on tumor
cell functions, including cell invasion, cell cycle, clonogenicity
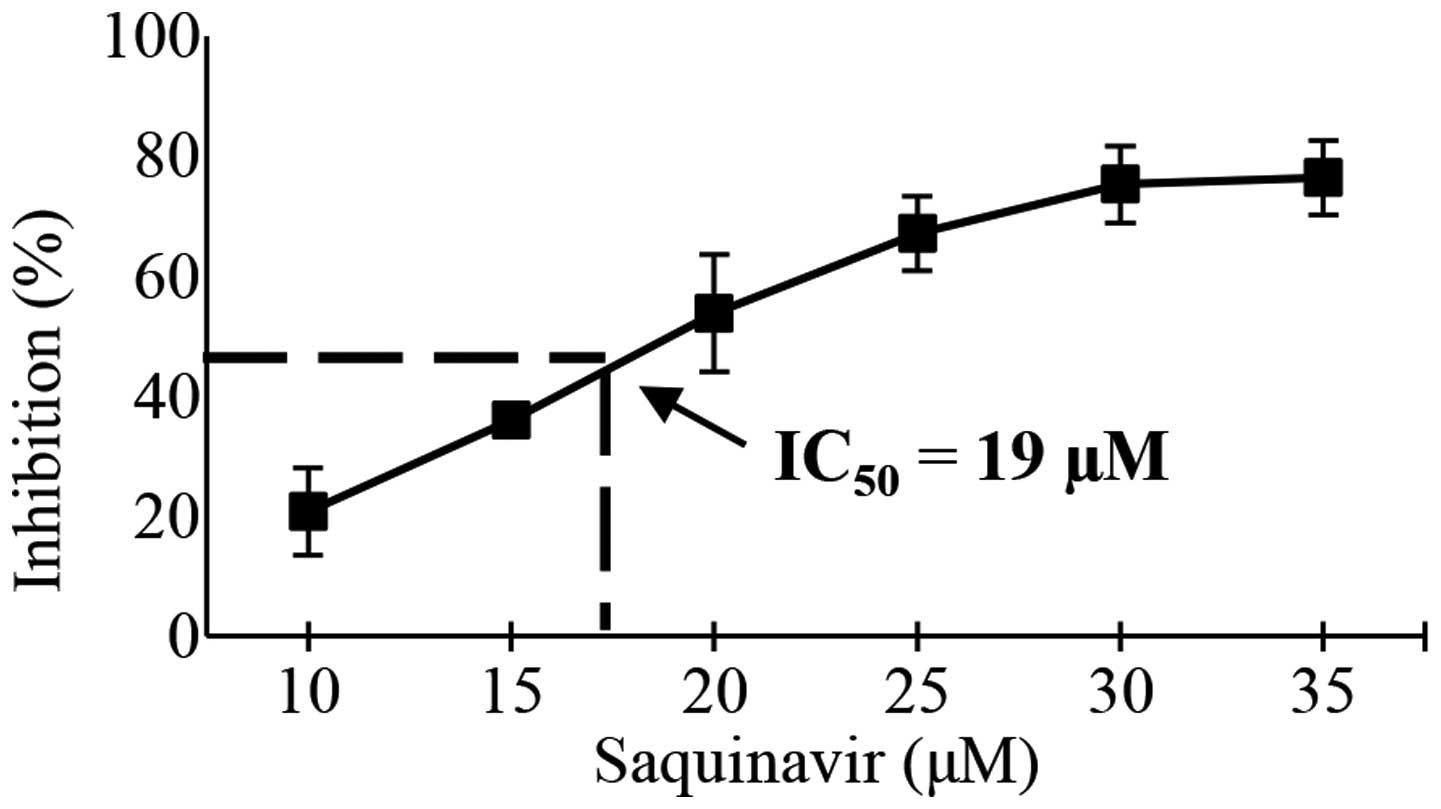
and radiosensitivity. First, the IC50 of saquinavir was
evaluated at 96 h, which was the maximal time observed for HeLa
cell growth in vitro. The extrapolated IC50 value
was 19 µM (Fig. 2). On this basis,
the following experiments were performed at concentrations of
saquinavir equal to 10 and 19 µM, corresponding to 50 and 100% of
its IC50 value, respectively.
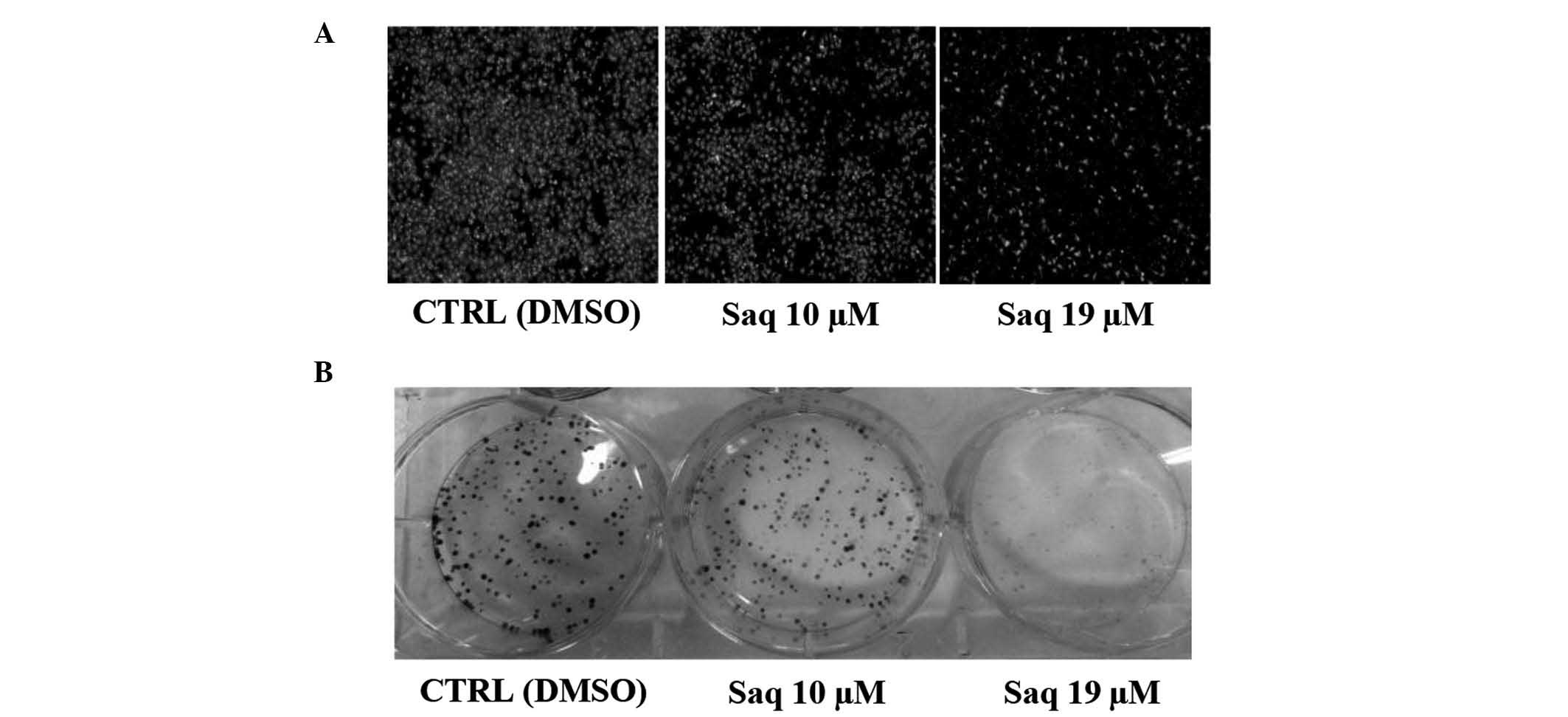
Cell invasion
To examine the effect of saquinavir on cell
invasion, a Corning BioCoat Matrigel Invasion Chamber assay was
conducted to determine the ability of HeLa cells to invade through
biological matrices in vitro. The treatment with saquinavir
significantly inhibited FBS-promoted HeLa cell invasion by 23%
(+/−10%) for 10-µM saquinavir, and by 61% (+/−18%) for 19 µM
saquinavir (P=0.0326 and P=0.0012, respectively) (Fig. 3A).
Clonogenicity
Treatment of HeLa cells with saquinavir reduced
their clonogenicity compared with control cells, and the effect was
dose-dependent. The treatment with saquinavir inhibited cell
clonogenicity by 39% (+/−12%) at 10 µM, and by 90% (+/−3%) at 19 µM
(P=0.0052 and P=0.0001, respectively) (Fig. 3B).
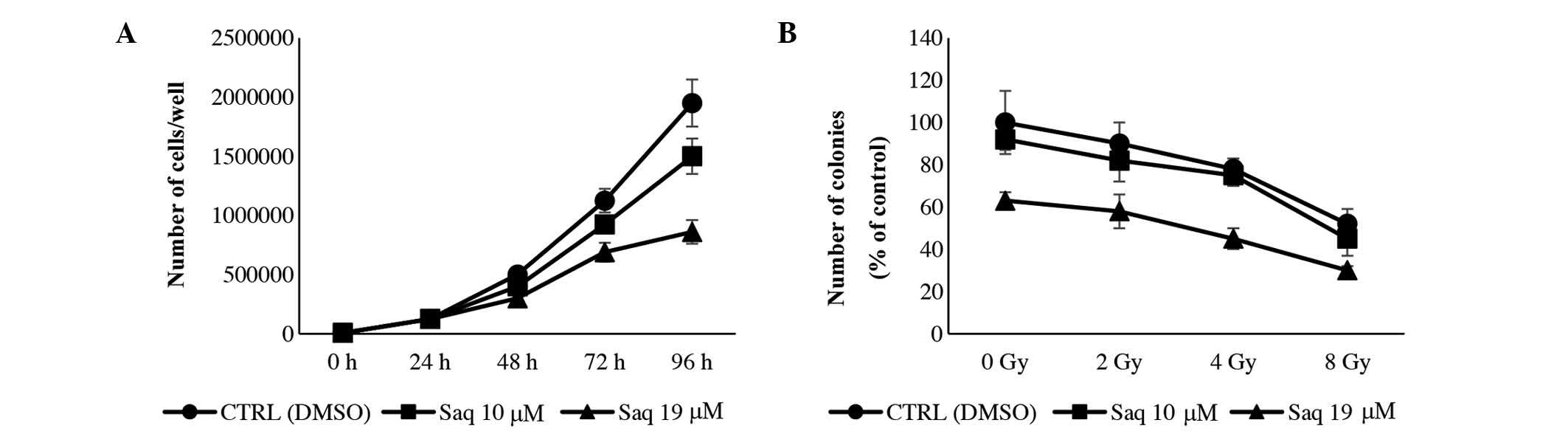
Cell cycle
To evaluate whether growth inhibition was associated
with an alteration in cell cycle phase distribution, HeLa cells
were exposed to 10 or 19 µM saquinavir for 24, 48, 72 and 96 h.
Saquinavir induced a growth inhibition effect in a dose-dependent
manner. After 96 h of treatment, saquinavir at a concentration of
19 µM caused a growth inhibitory effect of 56% compared with the
control cells (P=0.0012), while at 10 µM, the effect was less
prominent (P=0.0481), being the slope of the growth curve from 48
to 96 h similar to that of the controls (Fig. 4A). The analysis of the cell
distribution in the cell cycle phases revealed that saquinavir
caused a slight delay in crossing the G1 phase of the cell cycle.
At 72 h, the highest concentration of saquinavir tested (19 µM)
induced a temporary accumulation in S phase, which was then
repaired (Table IV).
| Table IV.Percentage of HeLa cells in various
phases of the cell cycle following treatment for 24, 48, 72 and 96
h with 10 and 19 µM Saq or dimethyl sulfoxide (CTRL). |
Table IV.
Percentage of HeLa cells in various
phases of the cell cycle following treatment for 24, 48, 72 and 96
h with 10 and 19 µM Saq or dimethyl sulfoxide (CTRL).
|
|
Treatment
duration (h) |
|---|
|
|
|
|---|
|
| 24 | 48 | 72 | 96 |
|---|
|
|
|
|
|
|
|---|
|
|
| Saq (µM) |
| Saq (µM) |
| Saq (µM) |
| Saq (µM) |
|---|
|
|
|
|
|
|
|
|
|
|
|---|
| Phase (% of
cells) | CTRL | 10 | 19 | CTRL | 10 | 19 | CTRL | 10 | 19 | CTRL | 10 | 19 |
|---|
| G1 | 48 | 53 | 59 | 52 | 57 | 59 | 51 | 55 | 42 | 50 | 54 | 49 |
| S | 39 | 32 | 30 | 36 | 32 | 32 | 35 | 33 | 47 | 35 | 31 | 40 |
| G2-M | 13 | 15 | 11 | 12 | 11 | 9 | 14 | 12 | 11 | 15 | 15 | 11 |
Radiosensitivity
The potential effects of saquinavir were analyzed on
HeLa cells exposed to increasing doses of X-rays in a clonogenic
survival assay. The treatment with saquinavir at 10 or 19 µM did
not contribute to an increase in the sensitivity of HeLa cells to
X-rays (Fig. 4B).
Discussion
Despite the use of screening programs and the
improvements in therapeutic approaches, CC remains the fourth most
lethal cancer among women worldwide (1,2). In an
effort to improve the efficacy of antitumor therapies, numerous
current medical strategies are aimed at designing novel inhibitors
of the relevant molecular pathways (30). In this context, the current study
focused on investigating the antitumor effects of HIV-PIs, a class
of drugs that reduce the incidence and/or promote the regression of
AIDS-associated cancers, independently of their anti-HIV or
immune-reconstituting activities (12–21). These
drugs, including indinavir, saquinavir and ritonavir, have been
demonstrated to target molecules with a key role in tumor
progression, such as matrix metalloproteinases (MMPs) or the
cellular proteasome, with antitumor effects (12–21).
However, to date, the effects of HIV-PIs in CC have yet to be
clarified. Accordingly, the present study evaluated the antitumor
effects of indinavir, saquinavir and ritonavir in primary CC cell
lines established in the Division of Obstetrics and Gynecology
(‘Angelo Nocivelli’ Institute for Molecular Medicine) as well as in
commercially available CC cell lines.
The present results indicated that saquinavir was
more effective than ritonavir and indinavir in reducing cell
proliferation and inhibiting the proteasome activities. These
effects were observed in all the CC cell lines tested, but with a
different degree of efficacy. HIV-PIs efficiency depends on the
access of these drugs to intracellular sites, which is mediated by
ATP-binding cassette (ABC) transporter family members such as
multidrug resistant-1 and multidrug resistance-associated proteins
(31). Therefore, it was speculated
that a differential ABC transporter family expression in the
present CC cell lines may be responsible for a different HIV-PIs
intracellular accumulation and, consequently, differential
efficacy.
The saquinavir concentrations required to modulate
the proteasome activities were higher than those that were
effective to inhibit cell proliferation. In fact, treatment with 40
µM saquinavir for 96 h inhibited cell proliferation by 90–100%
(P=0.0001), but it did not perturb any proteasome activity. Only
the treatment with 60–80 µM saquinavir was able to reduce the
proteasome functions by 10–50% (P<0.05). These results are in
agreement with the data of other studies conducted with similar
methods, which revealed a significant proteasomal effect of these
drugs only at high concentrations (15,16,32–36).
In contrast, previous studies that identified lower drug levels as
proteasome inhibitors, were conducted on cell extracts or purified
proteasomes (18,19,37).
However, proteasome activity profiles obtained by cell extracts are
known to be not necessarily representative of the in vivo
activity patterns, thus stressing the requirement for live
cell-based assays (38). Consistent
with this view, the current study investigated the effects of
HIV-PIs on the proteasome of intact cells. Saquinavir was selected
as the most effective drug, and HeLa cells as the most susceptible
cell line, to proceed with the analyses of HIV-PIs effects on tumor
cell functions. Saquinavir was strongly effective in inhibiting the
invasion, clonogenicity and proliferation of HeLa cells. However,
the cell cycle analysis results revealed that the growth inhibition
was not associated with a strong alteration in cell cycle phase
distribution. This suggested that, in HeLa cells, probably
saquinavir did not influence a specific cell cycle check point, but
slowed down the progress of cells through all the phases of the
cell cycle. Of note, the saquinavir-mediated actions on invasion,
clonogenicity and cell cycle were observed at concentrations much
lower (10 and 19 µM) than those required to perturb the proteasomal
activities. Our observation of these proteasome-independent effects
of saquinavir suggested its potential role in other oncogenic
pathways. It is possible to speculate that saquinavir may therefore
influence different CC cell functions modulating ≥1 pathways such
as Akt, signal transducer and activator of transcription 3 and p21,
as previously reported for other tumor models (35,36).
Concerning CC, two oncoproteins produced during
HR-HPV infection, E6 and E7, subvert the cell growth-regulatory
pathways and modify the cellular environment by labeling p53 and Rb
for ubiquitin-dependent proteasomal degradation (6–9).
Therefore, stabilization of p53 and Rb by preventing their
degradation, could be a useful strategy to revert CC cell behavior.
In the present study, saquinavir exhibited antitumor activities in
a proteasome-independent way. Thus, the effects of saquinavir
observed at low concentrations in the current study were likely not
associated with the stabilization of p53 or Rb by proteasome
inhibition.
Previous reports (14,15)
suggested that the ubiquitin-dependent proteasomal pathway plays a
crucial role in enhancing radiosensitivity, by inhibiting nuclear
factor (NF)-κB activation. Activated NF-κB induces the expression
of genes involved in protecting cells against apoptosis response to
genotoxic stresses such as ionizing radiation (14). The inhibition of the
ubiquitin-proteasome pathway may suppress NF-κB activation through
the stabilization of the inhibitory protein inhibitor of kappa
light chain gene enhancer in B cells α, which would otherwise be
degraded upon exposure to genotoxic stresses (14). Based on this hypothesis, and according
to the fact that radiotherapy is frequently used in both early- and
late-stage CC treatment, the potential effects of saquinavir on the
sensitivity to X-rays were analyzed in the present study. It was
observed that, when HeLa cells were cultured with saquinavir at 10
and 19 µM, the cells did not become more sensitive to X-rays. This
result was consistent with the fact that the treatment of HeLa
cells at these concentrations of saquinavir did not decrease their
proteasomal activities.
In conclusion, to the best of our knowledge, the
present is the first study evaluating the potential effects of
three HIV-PIs in primary and established CC cell lines,
comprehensively investigating the proteasome functions. The present
study demonstrated that saquinavir is active and may consistently
reduce proliferation, cell invasion and clonogenicity in a
proteasome-independent way in CC cell lines. Considering the
saquinavir dose-dependent effect observed in the present cell
lines, additional studies evaluating its concentration in CC
tissues in vivo in HIV+ patients receiving HAART
therapy are warranted prior to potentially translating the present
findings in a novel treatment strategy for
HPV+HIV− CC patients with disease resistant
to standard treatment modalities.
Acknowledgements
HIV-PIs were a kind gift of the NIH AIDS Research
and Reference Reagent Program (Germantown, MD, USA). The present
study was supported by grants from the Italian Ministry of Health,
Oncology Research 2006 (Rome, Italy; grant no. RF-ISS-2006-406810)
awarded to S.P.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arbyn M, Castellsagué X, de Sanjosé S,
Bruni L, Saraiya M, Bray F and Ferlay J: Worldwide burden of
cervical cancer in 2008. Ann Oncol. 22:2675–2686. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lăără E, Day NE and Hakama M: Trends in
mortality from cervical cancer in the Nordic countries: Association
with organised screening programmes. Lancet. 1:1247–1249. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hakama M and Louhivuori K: A screening
programme for cervical cancer that worked. Cancer Surv. 7:403–416.
1988.PubMed/NCBI
|
|
5
|
Bodily J and Laimins LA: Persistence of
human papillomavirus infection: Keys to malignant progression.
Trends Microbiol. 19:33–39. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Münger K, Werness BA, Dyson N, Phelps WC,
Harlow E and Howley PM: Complex formation of human papillomavirus
E7 proteins with the retinoblastoma tumor suppressor gene product.
EMBO J. 8:4099–4105. 1989.PubMed/NCBI
|
|
7
|
Scheffner M, Werness BA, Huibregtse JM,
Levine AJ and Howley PM: The E6 oncoprotein encoded by human
papillomavirus types 16 and 18 promotes the degradation of p53.
Cell. 63:1129–1136. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Crook T, Tidy JA and Vousden KH:
Degradation of p53 can be targeted by HPV E6 sequences distinct
from those required for p53 binding and trans-activation. Cell.
67:547–556. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boyer SN, Wazer DE and Band V: E7 protein
of human papilloma virus-16 induces degradation of retinoblastoma
protein through the ubiquitin-proteasome pathway. Cancer Res.
56:4620–4624. 1996.PubMed/NCBI
|
|
10
|
DiSaia PJ and Creasman WT: Cervical
CancerClinical Gynecologic Oncology. 5th. Mosby; Maryland Heights:
pp. 1–106. 1997
|
|
11
|
Deeks SG, Smith M, Holodniy M and Kahn JO:
HIV-1 protease inhibitors. A review for clinicians. JAMA.
277:145–153. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sgadari C, Barillari G, Toschi E, Carlei
D, Bacigalupo I, Baccarini S, Palladino C, Leone P, Bugarini R,
Malavasi L, et al: HIV protease inhibitors are potent
anti-angiogenic molecules and promote regression of Kaposi sarcoma.
Nat Med. 8:225–232. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Monini P, Sgadari C, Toschi E, Barillari G
and Ensoli B: Antitumour effects of antiretroviral therapy. Nat Rev
Cancer. 4:861–875. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Russo SM, Tepper JE, Baldwin AS Jr, Liu R,
Adams J, Elliott P and Cusack JC Jr: Enhancement of
radiosensitivity by proteasome inhibition: Implications for a role
of NF-kappaB. Int J Radiat Oncol Biol Phys. 50:183–193. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pajonk F, Himmelsbach J, Riess K, Sommer A
and McBride WH: The human immunodeficiency virus (HIV)-1 protease
inhibitor saquinavir inhibits proteasome function and causes
apoptosis and radiosensitization in non-HIV-associated human cancer
cells. Cancer Res. 62:5230–5235. 2002.PubMed/NCBI
|
|
16
|
Gaedicke S, Firat-Geier E, Constantiniu O,
Lucchiari-Hartz M, Freudenberg M, Galanos C and Niedermann G:
Antitumor effect of the human immunodeficiency virus protease
inhibitor ritonavir: Induction of tumor-cell apoptosis associated
with perturbation of proteasomal proteolysis. Cancer Res.
62:6901–6908. 2002.PubMed/NCBI
|
|
17
|
Pati S, Pelser CB, Dufraine J, Bryant JL,
Reitz MS Jr and Weichold FF: Antitumorigenic effects of HIV
protease inhibitor ritonavir: Inhibition of Kaposi sarcoma. Blood.
99:3771–3779. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
André P, Groettrup M, Klenerman P, de
Giuli R, Booth BL Jr, Cerundolo V, Bonneville M, Jotereau F,
Zinkernagel RM and Lotteau V: An inhibitor of HIV-1 protease
modulates proteasome activity, antigen presentation, and T cell
responses. Proc Natl Acad Sci USA. 95:13120–13124. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schmidtke G, Holzhütter HG, Bogyo M,
Kairies N, Groll M, de Giuli R, Emch S and Groettrup M: How an
inhibitor of the HIV–I protease modulates proteasome activity. J
Biol Chem. 274:35734–35740. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Piccinini M, Rinaudo MT, Chiapello N,
Ricotti E, Baldovino S, Mostert M and Tovo PA: The human 26S
proteasome is a target of antiretroviral agents. AIDS. 16:693–700.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Goldberg AL and Rock K: Not just research
tools-proteasome inhibitors offer therapeutic promise. Nat Med.
8:338–340. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Barillari G, Iovane A, Bacigalupo I,
Palladino C, Bellino S, Leone P, Monini P and Ensoli B: Ritonavir
or saquinavir impairs the invasion of cervical intraepithelial
neoplasia cells via a reduction of MMP expression and activity.
AIDS. 26:909–919. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hampson L, Kitchener HC and Hampson IN:
Specific HIV protease inhibitors inhibit the ability of HPV16 E6 to
degrade p53 and selectively kill E6-dependent cervical carcinoma
cells in vitro. Antivir Ther. 11:813–825. 2006.PubMed/NCBI
|
|
24
|
Justesen US: Protease inhibitor plasma
concentrations in HIV antiretroviral therapy. Dan Med Bull.
55:165–185. 2008.PubMed/NCBI
|
|
25
|
Monini P, Sgadari C, Grosso MG, Bellino S,
Di Biagio A, Toschi E, Bacigalupo I, Sabbatucci M, Cencioni G,
Salvi E, et al: Clinical course of classic Kaposi's sarcoma in
HIV-negative patients treated with the HIV protease inhibitor
indinavir. AIDS. 23:534–538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Flick DA and Gifford GE: Comparison of in
vitro cell cytotoxic assays for tumor necrosis factor. J Immunol
Methods. 68:167–175. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tanaka K, Mizushima T and Saeki Y: The
proteasome: Molecular machinery and pathophysiological roles. Biol
Chem. 393:217–234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bertuzzi A, Gandolfi A, Germani A, Spanò
M, Starace G and Vitelli R: Analysis of DNA synthesis rate of
cultured cells from flow cytometric data. Cytometry. 5:619–628.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kalantari M, Karlsen F, Kristensen G, Holm
R, Hagmar B and Johansson B: Disruption of the E1 and E2 reading
frames of HPV 16 in cervical carcinoma is associated with poor
prognosis. Int J Gynecol Pathol. 17:146–153. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu J and Westin SN: Rational selection of
biomarker driven therapies for gynecologic cancers: The more we
know, the more we know we don't know. Gynecol Oncol. 141:65–71.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee CG, Gottesman MM, Cardarelli CO,
Ramachandra M, Jeang KT, Ambudkar SV, Pastan I and Dey S: HIV-1
protease inhibitors are substrates for the MDR1 multidrug
transporter. Biochemistry. 37:3594–3601. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kraus M, Bader J, Overkleeft H and
Driessen C: Nelfinavir augments proteasome inhibition by bortezomib
in myeloma cells and overcomes bortezomib and carfilzomib
resistance. Blood Cancer J. 3:e1032013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kraus M, Malenke E, Gogel J, Müller H,
Rückrich T, Overkleeft H, Ovaa H, Koscielniak E, Hartmann JT and
Driessen C: Ritonavir induces endoplasmic reticulum stress and
sensitizes sarcoma cells toward bortezomib-induced apoptosis. Mol
Cancer Ther. 7:1940–1948. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Taura M, Kariya R, Kudo E, Goto H, Iwawaki
T, Amano M, Suico MA, Kai H, Mitsuya H and Okada S: Comparative
analysis of ER stress response into HIV protease inhibitors:
Lopinavir but not darunavir induces potent ER stress response via
ROS/JNK pathway. Free Radic Biol Med. 65:778–788. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gupta AK, Cerniglia GJ, Mick R, McKenna WG
and Muschel RJ: HIV protease inhibitors block Akt signaling and
radiosensitize tumor cells both in vitro and in vivo. Cancer Res.
65:8256–8265. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ikezoe T, Saito T, Bandobashi K, Yang Y,
Koeffler HP and Taguchi H: HIV-1 protease inhibitor induces growth
arrest and apoptosis of human multiple myeloma cells via
inactivation of signal transducer and activator of transcription 3
and extracellular signal-regulated kinase 1/2. Mol Cancer Ther.
3:473–479. 2004.PubMed/NCBI
|
|
37
|
Laurent N, de Boüard S, Guillamo JS,
Christov C, Zini R, Jouault H, Andre P, Lotteau V and Peschanski M:
Effects of the proteasome inhibitor ritonavir on glioma growth in
vitro and in vivo. Mol Cancer Ther. 3:129–136. 2004.PubMed/NCBI
|
|
38
|
Berkers CR, Verdoes M, Lichtman E,
Fiebiger E, Kessler BM, Anderson KC, Ploegh HL, Ovaa H and Galardy
PJ: Activity probe for in vivo profiling of the specificity of
proteasome inhibitor bortezomib. Nat Methods. 2:357–362. 2005.
View Article : Google Scholar : PubMed/NCBI
|