Introduction
Multiple myeloma (MM) accounts for 10% of all
hematological malignancies and is a B-cell malignancy characterized
by a clonal proliferation of abnormal plasma cells in the bone
marrow (1,2). MM occurs most frequently in patients
between the ages of 50 and 80 years (3). MM is characterized by the presence of
monoclonal immunoglobulin (Ig) in the serum, of which IgG and IgA
are the most frequently observed subtypes (4). IgM MM is an extremely rare
lymphoproliferative disease associated with an aggressive clinical
course, which accounts for <0.5% of all MM cases (4,5).
Distinguishing IgM MM from Waldenström's macroglobulinemia (WM) is
extremely important, but is often challenging due to similar
characteristics (6). Differentiation
may be established based on clinical presentation or bone marrow
morphology (7). Diagnosis of IgM MM
is usually established based on the International Myeloma Working
Group criteria, which state that IgM MM includes: i) The presence
of M-protein in the serum and/or urine; ii) >10% bone marrow
clonal plasma cells or plasmacytoma; and iii) associated organ or
tissue impairment (end organ damage, including bone lesions)
(8). The median reported survival for
IgM MM is 36 months (9). In this
study, the case of a 41-year-old man who presented with
immunoglobulin (Ig)M multiple myeloma (MM) with an unusually
non-aggressive clinical course is presented. A watch-and-wait
strategy was adopted, however clinical progression occurred after
eight years. At present the patient remains alive nine years after
the initial diagnosis. Written informed consent was obtained from
the patient for publication of this case report and any
accompanying images.
Case report
In October 2005, a 41-year-old man with an
unremarkable medical history presented to the Coffs Harbour Health
Campus, Coffs Harbour, Australia, with the complaint of tingling
and burning paresthesia in both feet and exertional dyspnea. No
hepatosplenomegaly or lymphadenopathy was identified and physical
examination was otherwise unremarkable. The laboratory examination
results at initial presentation were as follows: White blood cell
count, 6.2×109/l (reference range,
4.0–12.0×109/l); hemoglobin, 150 g/l (reference range,
130–180 g/l); platelet count, 281×109/l (reference
range, 150–400×109/l); erythrocyte sedimentation rate,
115 mm/h (reference range, 1–10 mm/h); creatinine, 0.10 mmol/l
(reference range, 0.05–0.12 mmol/l), blood urea nitrogen, 6.7
mmol/l (reference range, 2.5–6.5 mmol/l); calcium, 2.61 mmol/l
(reference range, 2.22–2.65 mmol/l); lactate dehydrogenase, 171
IU/l (reference range, 60–200 IU/l). All other routine laboratory
parameters tested were within the normal ranges. Serum
electrophoresis revealed a homogeneous peak in the gamma fraction,
which was identified as IgM kappa by immunofixation. Nephelometry
identified an elevated concentration of IgM (49.00 g/l; reference
range, 0.44–2.76 g/l) and reduced IgA (0.25 g/l; reference range,
0.80–4.12 g/l) and IgG (3.89 g/l; reference range, 6.45–13.90 g/l)
concentrations. β2-microglobulin levels were 1.42 mg/l
(reference range, 0–20 mg/l). Magnetic resonance imaging of the
spine revealed no evidence of lytic bone lesions. Consecutive bone
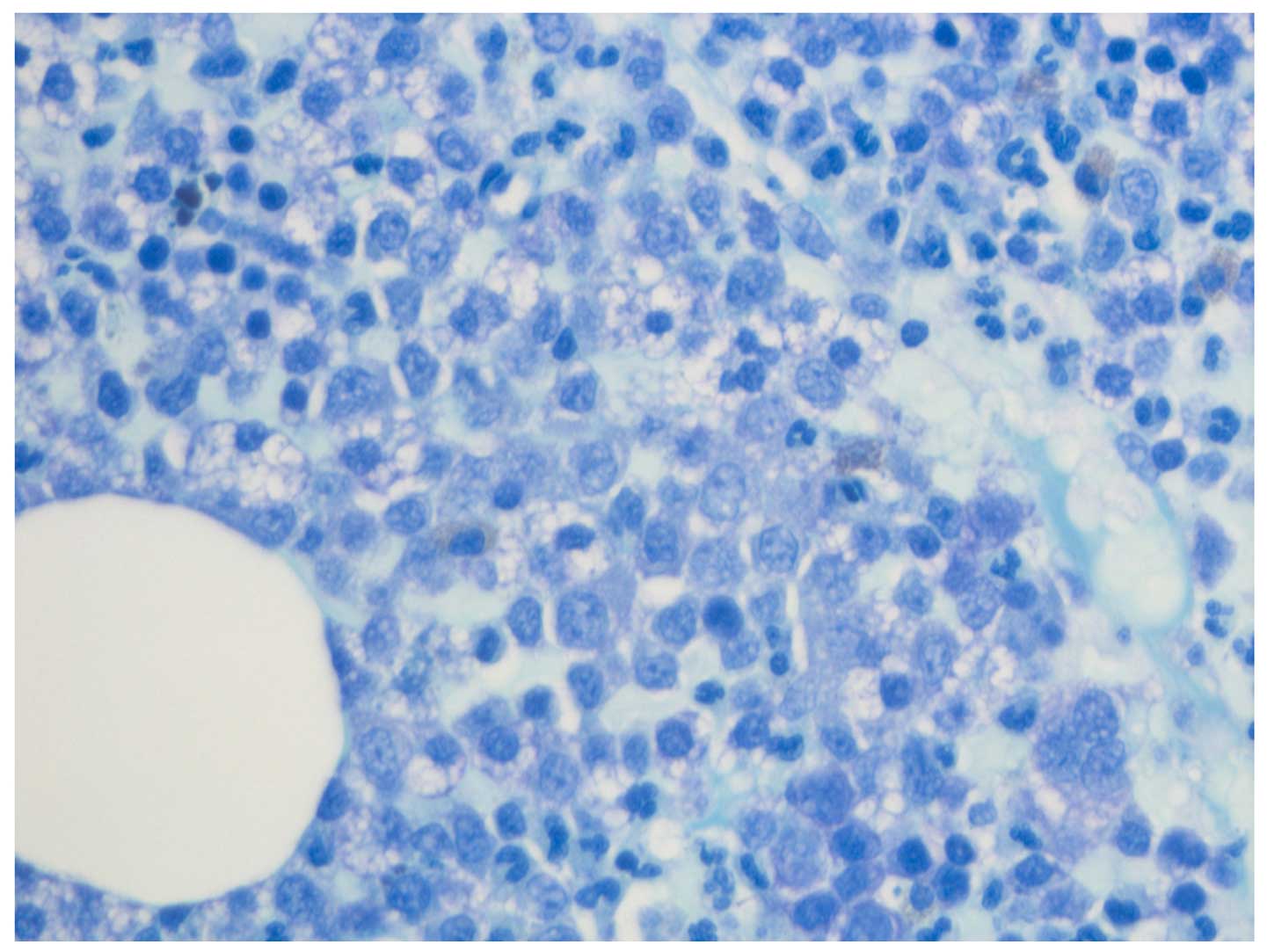
marrow (BM) aspirate morphology revealed moderate diffuse
infiltration with 12% plasma cells. A small number of plasma cells
exhibited atypical multinucleated forms (≤3 nuclei) or cytoplasmic
vacuolation. BM biopsy demonstrated diffuse proliferation of plasma
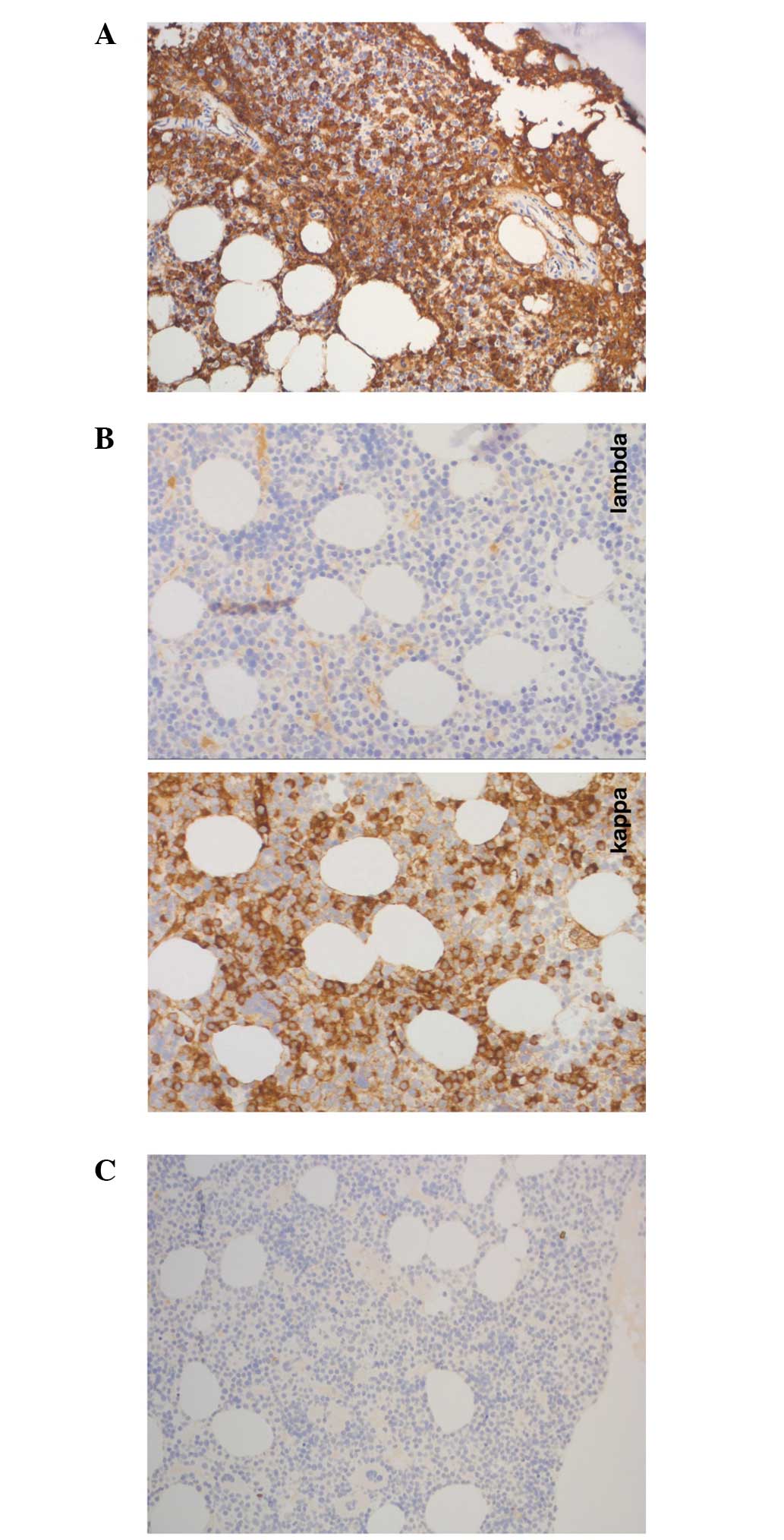
cells expressing cluster of differentiation (CD)38 and IgM
exhibiting kappa restriction. Lymphoplasmocytic lymphoma/WM was
excluded by morphology and immunohistochemistry. No cytogenetic
abnormalities were identified. Diagnosis of symptomatic IgM kappa
MM was established based on the presence of an M-protein in the
serum, plasma cell infiltration in the BM and associated tissue
impairment (polyneuropathy), according to the International Myeloma
Working Group consensus statement and guidelines (8). Due to the mild clinical presentation and
mild end organ damage observed, a watch-and-wait strategy was
selected. During the following 8 years, the patient exhibited an
extremely benign course of the disease. However, as a result of
continuously increasing IgM levels and progressive polyneuropathy,
BM aspirate/biopsy was repeated in November 2012 and March 2014.
Giemsa staining, which was performed at room temperature overnight,
was used for morphological analysis. Immunohistochemical analyses
were performed using standard techniques and antibodies to CD3,
CD4, CD5, CD8, CD10, CD19, CD20, CD56, CD38, CD45 and CD138.
Morphology demonstrated diffuse infiltration with 10% abnormal
plasma cells, which were positive for IgM kappa (Figs. 1 and 2).
The immunophenotype observed was consistent with the presence of
plasma cells with abnormal cells exhibiting positivity for CD38,
CD138, CD10 and negativity for CD19 and CD56, as demonstrated by
flow cytometric analysis. In addition, interphase fluorescence
in situ hybridization analysis was performed using samples
enriched for CD138-positive plasma cells. The results revealed a
translocation involving myeloma overexpressed (11q13) and
immunoglobulin heavy locus (IGH) (14q32), and therefore positivity
for t(11;14) (q13;q32), an additional signal for IGH (14q32), loss
of one copy of MAF bZIP transcription factor (16q23), deleted in
lymphocytic leukemia 1 (13q14) and fibroblast growth factor
receptor 4 (4p16). In May 2014, elevated calcium levels (2.75
mmol/l; reference range, 2.1–2.55 mmol/l), deteriorating
polyneuropathy and the identification of IGH locus rearrangement
resulted in the initiation of chemotherapy treatment. The patient
was administered 4 cycles of induction chemotherapy: velcade (1.3
g/m2 subcutaneously; days 1, 4, 8 and 11),
cyclophosphamide (500 mg/m2 intravenously; days 1 and 8)
and dexamethasone (40 mg; days 1, 2, 4, 5, 8, 9, 11 and 12) with
cycle 2 starting at day 22, cycle 3 at day 43 and cycle 4 at day
64. No evidence of lytic bone lesions was identified on whole body
bone computed tomography. In July 2014, the patient had completed
his last cycle of induction chemotherapy with VCD. At the time of
writing this manuscript (December 2014), the patient remains in
good health and the symptoms of polyneuropathy have improved
slightly following initiation of chemotherapy.
Discussion
Distinguishing IgM MM from WM is critical; however,
it may be difficult. IgM MM and WM are two distinct hematological
entities with the common symptom of an IgM monoclonal gammopathy
(6). Differentiation may be
established based on clinical presentation or BM morphology. The
clinical symptoms of anemia, hypercalcemia, renal impairment, lytic
bone lesions, and plasma cell infiltration of BM clearly indicate
the rare diagnosis of IgM MM (7).
However, the above-mentioned criteria are not highly sensitive.
Lymphadenopathy and hepatosplenomegaly, two symptoms of WM, are
typically present in only 20–40% of all WM cases (6). Furthermore, bone lesions are not always
present in IgM MM (10). Recently,
the presence of t(11;14) in IgG MM has been demonstrated to be
highly specific (11). Translocation
t(11;14) leads to dysregulation of cyclin D1 and has been
identified in 7/8 patients with IgM myeloma in a study by
Avet-Loiseau et al (11),
while no such translocation was identified in all 17 cases of WM.
In addition, recently a mutation in exon 5 of the MYD88 gene
(MYD88 L265P), which is absent in IgM MM patients, was demonstrated
to be specific for WM with a specificity of >90% (12). Due to the extremely rare incidence of
IgM MM, only a few case series have been reported in the literature
to date (4,5,9). Notably,
IgM MM appears to be more aggressive than IgG or IgA MM, as well as
WM, with an overall median survival time of 36 months (9). In addition, IgM MM has a poor clinical
outcome in the context of high-dose therapy (13). To date, the longest reported survival
time of a patient with IgM myeloma was 60 months (5). At present, treatment guidelines
recommend the same therapeutic approach for IgM MM and IgG or IgA
myelomas (14). Notably, IgM
monoclonal gammopathy of unknown significance has a benign course
with no progression to IgM MM (15).
In the present case, the clinical presentation was
atypical; the patient initially presented with polyneuropathy,
without hypercalcemia, anemia, renal impairment or lytic bone
lesions. However, diagnosis of symptomatic MM was established
according to the International Myeloma Working Group consensus
statement and guidelines (8). Due to
the mild symptoms observed, no therapy was initiated and the
patient was closely followed up. However, 8 years after initial
diagnosis clinical, morphological and genetic progression occurred,
with the development of hypercalcemia, progressively deteriorating
polyneuropathy, clonal expansion of plasma cells up to 50 of
hematopoietic cells and demonstration of the typical t(11;14)
translocation (IGH locus rearrangement). In conclusion, to the best
of our knowledge, the survival time of the symptomatic IgM MM
patient in this case is the longest to be reported in the
literature to date, which contradicts previous evidence that
suggests IgM MM exhibits an aggressive clinical course (9). IgM MM may have a more heterogeneous
disease course than was previously understood. However, larger
observational studies are required in order to identify the
potential risk and protecting factors that result in an aggressive
or comparatively non-aggressive IgM MM course, respectively.
References
|
1
|
Kyle RA and Rajkumar SV: Multiple myeloma.
Blood. 111:2962–2972. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kyle RA: Diagnostic criteria of multiple
myeloma. Hematol Oncol Clin North Am. 6:347–358. 1992.PubMed/NCBI
|
|
3
|
Boccadoro M and Pileri A: Diagnosis,
prognosis, and standard treatment of multiple myeloma. Hematol
Oncol Clin North Am. 11:111–131. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
DeGramont A, Grosbois B, Michaux JL, Peny
AM, Pollet JP, Smadja N, Krulik M, Debray J, Bernard JF and
Monconduit M: IgM myeloma: 6 cases and a review of the literature.
Rev Med Interne. 11:13–18. 1990.(In French). PubMed/NCBI
|
|
5
|
Dierlamm T, Laack E, Dierlamm J, Fiedler W
and Hossfeld DK: IgM myeloma: A report of four cases. Ann Hematol.
81:136–139. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dimopoulos MA, Panayiotidis P, Moulopoulos
LA, Sfikakis P and Dalakas M: Waldenström's macroglobulinemia:
Clinical features, complications, and management. J Clin Oncol.
18:214–226. 2000.PubMed/NCBI
|
|
7
|
Schuster SR, Rajkumar SV, Dispenzieri A,
Morice W, Aspitia AM, Ansell S, Kyle R and Mikhael J: IgM multiple
myeloma: Disease definition, prognosis, and differentiation from
Waldenstrom's macroglobulinemia. Am J Hematol. 85:853–855. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dimopoulos M, Terpos E, Comenzo RL, Tosi
P, Beksac M, Sezer O, Siegel D, Lokhorst H, Kumar S, Rajkumar SV,
et al: IMWG: International myeloma working group consensus
statement and guidelines regarding the current role of imaging
techniques in the diagnosis and monitoring of multiple myeloma.
Leukemia. 23:1545–1556. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Annibali O, Petrucci MT, Del Bianco P,
Gallucci C, Levi A, Foà R and Avvisati G: IgM multiple myeloma:
Report of four cases and review of the literature. Leuk Lymphoma.
47:1565–1569. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Haghighi B, Yanagihara R and Cornbleet PJ:
IgM myeloma: Case report with immunophenotypic profile. Am J
Hematol. 59:302–308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Avet-Loiseau H, Garand R, Lodé L,
Harousseau JL and Bataille R: Intergroupe Francophone du Myélome:
Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE, and
nonsecretory multiple myeloma variants. Blood. 101:1570–1571. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu L, Hunter ZR, Yang G, Zhou Y, Cao Y,
Liu X, Morra E, Trojani A, Greco A, Arcaini L, et al: MYD88 L265P
in Waldenström macroglobulinemia, immunoglobulin M monoclonal
gammopathy, and other B-cell lymphoproliferative disorders using
conventional and quantitative allele-specific polymerase chain
reaction. Blood. 121:2051–2058. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Morris C, Drake M, Apperley J, Iacobelli
S, van Biezen A, Bjorkstrand B, Goldschmidt H, Harousseau JL,
Morgan G, de Witte T, et al: Myeloma Subcommittee of Chronic
Leukaemia Working Party of EBMT: Efficacy and outcome of autologous
transplantation in rare myelomas. Haematologica. 95:2126–2133.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moreau P, San Miguel J, Ludwig H, Schouten
H, Mohty M, Dimopoulos M and Dreyling M: ESMO Guidelines Working
Group: Multiple myeloma: ESMO Clinical Practice Guidelines for
diagnosis, treatment and follow-up. Ann Oncol. 24(Suppl 6):
vi133–vi137. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kyle RA, Therneau TM, Rajkumar SV,
Remstein ED, Offord JR, Larson DR, Plevak MF and Melton LJ III:
Long-term follow-up of IgM monoclonal gammopathy of undetermined
significance. Blood. 102:3759–3764. 2003. View Article : Google Scholar : PubMed/NCBI
|