Introduction
Ovarian carcinoma is the seventh most common cancer
in women, worldwide, and the leading cause of mortality from
gynecological malignancies (1).
Despite the development of surgical techniques and the emergence of
novel chemotherapy agents, the 5-year overall survival rate remains
poor at ~30% (2). High-grade serous
ovarian carcinoma (HGSOC) is a dominant subgroup of ovarian
carcinomas with rapid evolution, late diagnosis and an extremely
poor prognosis; these factors justify the requirement for novel
therapeutic strategies (3). Numerous
genetic alteration events are involved in the molecular mechanisms
that underlie cancer development, progression and metastasis.
Understanding and elucidating these genetic aberrations in HGSOCs
could provide innovative therapeutic options with novel targeted
agents.
Tumor protein 53 (P53) alterations are frequently
detected in HGSOCs and are associated with high tumor cell
proliferation (3). With the exception
of its potential as a prognostic marker, P53 alterations could
serve as a theranostic biomarker in HGSOC. For example, one study
reported the destabilization and degradation of mutant P53 by the
histone deacetylase (HDAC) 6 inhibitor, vorinostat (4), leading to antitumor activity. In
addition, tumor angiogenesis could be inhibited through the
downregulation of vascular endothelial growth factor (VEGF),
induced by HDAC-mediated hypoxia inducible factor inhibition
(5). Therefore, synergistic antitumor
activity has been reported with combined inhibition of VEGF and
HDAC. HDAC may be induced by pazopanib and vorinostat, which
demonstrates better antitumor activity with a significantly longer
progression-free survival (PFS) and overall survival (OS) in
patients bearing mutant P53 solid tumors, including ovarian
cancers, compared with in P53 wild-type tumors (6). Stratifying HGSOC patients on P53
alteration status has, therefore, been proposed as a molecular
rationale for the addition of vorinostat to anti-VEGF maintenance
therapy, and may maximize the clinical benefits that limit the
significant toxicities associated with the use of cytotoxic
chemotherapy (7).
RAS/mitogen-activated protein kinase (MAPK) and
phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/protein
kinase B (AKT) and are two major intracellular signaling
transduction pathways that can be activated due to: A loss of
phosphatase and tensin homolog (PTEN) function; genetic mutations
in phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
α (PIK3CA), Kirsten rat sarcoma viral oncogene homolog
(KRAS), neuroblastoma RAS viral oncogene homolog
(NRAS) or B-Raf proto-oncogene, serine/threonine kinase
(BRAF) (8); or the activation
of mutations in MET proto-oncogene, receptor tyrosine kinase
(MET) receptors (9) that are
involved in the carcinogenesis of HGSOC. These genetic
abnormalities are associated with a decreased responsiveness to
conventional chemotherapy and a poor prognosis (10–12).
The PI3K/AKT pathway is activated in ~70% of ovarian
carcinomas, and activation of this pathway is associated with
increased invasive and migratory capacities of tumor cells and
resistance to cytotoxic chemotherapy (13).
PTEN is a major negative regulator of the PI3K
signaling pathway and PTEN loss has been reported as a common
driver event and prognostic classifier in HGSOC (14). One previous study showed that
immunohistochemistry (IHC) is preferable for identifying a loss of
PTEN function (15). However, the
association between loss of PTEN and patient outcome remains
controversial (14,16).
Although RAS mutations have been considered
the major molecular feature of low-grade ovarian cancer,
NRAS, a member of the RAS family, was found to be an
oncogenic driver in HGSOC, which indicates a degree of overlap
across the molecular profiles of low- and high-grade ovarian
carcinomas (17).
The pathological activation of MET through
MET gene mutation is well characterized as a driver in
oncogenesis (9) and has been reported
to promote tumor proliferation, invasive growth and angiogenesis,
which are widely observed in HGSOC (8,18). In
addition, MET activation can promote angiogenesis by activating
common signaling pathways such as PI3K/AKT and RAS/MAPK, and the
hypoxic environment induced by anti-angiogenic agents has been
shown to potentially promote the MET-dependent spreading of cancer
cells (19). This finding suggests
that the selective targeting of one pathway may induce the
compensatory upregulation of another. This may explain that in
clinical trials evaluating bevacizumab in ovarian carcinoma, no
significant increase of overall survival was observed (20,21), and
that the dual inhibition of MET/vascular endothelial growth factor
receptor 2 in MET-mutated papillary renal carcinoma
(22) demonstrated clinical benefit,
as observed in ovarian cancer models (23).
In the present study, in addition to the analysis of
oncogenic alterations of P53 and PTEN expression, KRAS,
NRAS, BRAF, PIK3CA and MET mutations
were screened as putative sources of RAS/MAPK and PI3K/AKT
signaling pathways activators. The association of these alterations
with the clinical outcome of patients with HGSOC was then
evaluated.
Patients and methods
Patients
Patients treated between January 2007 and December
2012 in Cancer Institute of Lorraine (Vandoeuvre-les-Nancy, France)
with a proven diagnosis of HGSOC were selected. In all cases, the
final diagnosis was established according to the International
Federation of Gynecology and Obstetrics (FIGO) (24) and World Health Organization (25,26)
criteria. All women received cytoreductive surgery and a
platinum-based chemotherapy and were followed-up in the same
institution. Tumor samples were obtained prior to chemotherapy. The
present study was approved by the Scientific Committee of the
Cancer Institute of Lorraine and all the patients provided formal
consent for the study.
Tumor samples
Formalin-fixed paraffin-embedded (FFPE) specimens
were used for IHC. Hematoxylin-eosin (H&E) staining was
performed on 5-µm sections and the tissue was validated by a senior
pathologist (Dr C. Barlier, Cancer Institute of Lorraine). Only
specimens with >20% tumor cells were used in the study.
Frozen tumor fragments were used for mutation
analysis. Cryosections (5-µm) were immediately fixed in alcohol,
formalin and acetic acid, prior to being H&E-stained and
validated by a senior pathologist to ensure the tumor cell content.
Unfixed frozen macrodissected regions with >20% tumor cells were
then used for DNA extraction using the QIAmp DNA mini kit (Qiagen
GmbH, Hilden, Germany). Extracted DNA was then purified and DNA
concentration was measured by NanoVue (GE Healthcare Life Sciences,
Chalfont, UK) and finally stored at −80°C until processed for
polymerase chain reaction (PCR) and next generation sequencing
(NGS).
IHC
Tissue sections (5-µm) were cut from each block and
deposited on an IHC-specific slide with a drop of distilled water,
prior to being dried on a hot plate and placed overnight in a stove
at 56°C. The sections were deparaffinized and rehydrated using EZ
Prep solution (Ventana Medical Systems, Inc., Tucson, AZ, USA) and
then restored with Cell Conditioner 1 (Ventana Medical Systems,
Inc.) for protein confirmation. Primary mouse anti-human PTEN
antibody (dilution, 1:125; catalog no., 9188; clone D4.3; Dako,
Glostrup, Denmark) was incubated for 1 h at room temperature and
primary mouse anti-human P53 antibody (dilution, 1:50; catalog no.,
M7001; clone DO-7; Dako) was incubated for 32 min at 42°C. The
procedure was performed in a BenchMark Ultra® with
UltraView Universal DAB Detection kit (Ventana Medical Systems,
Inc.) and the sections were lightly counterstained with hematoxylin
and bluing reagent (Ventana Medical Systems, Inc.). The IHC results
were recorded as follows: Cytoplasmic staining of PTEN was
considered as positive; nuclear staining of P53 was considered as
positive. The interpretation of staining was blinded from the
clinical outcome data.
Mutation analysis by high resolution
melting-PCR (HRM-PCR) and NGS
PCR assays were performed to screen somatic
mutations in KRAS and NRAS at exons 2, 3 and 4,
BRAF at exon 15, and PIK3CA at exons 10 and 21. PCR
fragments were stained with Resolight® DNA fluorescent
intercalant probe (Roche Diagnostics, Meylan, France) and then
amplified using LightCycler® 480 High Resolution Melting
Master kit (Roche Diagnostics) in a LightCycler® 480
thermocycler (Roche Diagnostics) for 35 cycles under the following
conditions: 95°C for 10 sec; 65°C for 15 sec; and 72°C for 30
sec.
Appropriate positive and negative controls were
included for each of the exons evaluated. The cell lines used as
positive controls were: Lovo, Calu6 and ML2 for KRAS, exons
2, 3 and 4); Molt 4, MZ2 and Nalm6 for NRAS, exons 2, 3 and
4; HT29 for BRAF, exon 15; and MCF7 and HCT116 for
PIK3CA, exons 10 and 21. The cell lines used as negative
controls were: WIDR for KRAS and NRAS, exons 2, 3 and
4; Lovo for BRAF, exon 15; and MDA-MB231 for PIK3CA,
exons 10 and 21.
NGS was performed using ultra-deep biparallel
pyrosequencing (GS Junior; Roche Diagnostics) to confirm and
identify the somatic alterations detected by HRM-PCR and to further
investigate PIK3CA, exon 5, and MET, exons 14, 16,
17, 18 and 19. Firstly, for the library preparation, sequences of
interest were amplified by TAG-PCR and MID-PCR using FastStart High
Fidelity PCR System, dNTPack kit (Roche Diagnostics). The procedure
was performed using Nexus Mastercycler® (Eppendorf AG,
Hamburg, Germany). The PCR products were then controlled by 2%
agarose gel electrophoresis and quantitated using the Quant-iT™
PicoGreen dsDNA Assay kit (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). DNA concentration was finally adjusted to 106
DNA molecules/µl. Secondly, the prepared library was amplified by
emulsion-PCR using the GS Junior Titanium emPCR kit (Roche
Diagnostics) and enriched with the emPCR Bead Recovery Reagents kit
(Roche Diagnostics) prior to sequencing with the GS Junior Titanium
Sequencing kit (Roche Diagnostics). Data were treated with Amplicon
Variant Analyzer software, version 3.0 (454 Life Sciences; Roche
Diagnostics, Branford, CT, USA). Sequences were aligned with the
NM_002524.4, NM_006218.2 and NM_000245.2 nucleotides for references
for NRAS, PIK3CA and MET sequences, respectively, and
variant calling was processed. At ×1,000 depth, NGS sensitivity was
1%. A second data analysis using Burrows-Wheeler Aligner 0.7.12
(maximal exact match algorithm; default parameters; distributed
under GPLv3) for mapping and sorting and indexing using SAMtools
(SAMtools; GitHub, Inc., San Francisco, CA, USA) was performed.
VarScan2 (mpileup2snp algorithm; filters-min-coverage 100-minreads
20-min-var-freq 0.01-P-value 0.05) was used for variant calling, as
reported previously (27).
Statistics
Quantitative variables were described with the mean,
standard deviation and range; qualitative variables by frequency
and percentage.
The association between P53 expression, PTEN loss of
expression, KRAS, NRAS, PIK3CA, MET
mutations and patient age, FIGO stage, chemosensitivity to platinum
drugs (defined as lack of tumor progression at 6 months) was
analyzed using Chi-squared test or Fisher's exact test. The
progression-free survival (PFS) was defined as the time interval
between the termination of frontline therapy and the first
recurrence either by radiological findings or a rising of CA-125
level by two serial tests ≥1 month apart. Patients alive and free
of recurrence were censored at the last follow-up. The overall
survival (OS) was defined as the time interval between termination
of frontline therapy and fatality (all causes). All causes of
fatality were counted as failures. The PFS and OS curves, according
to the expression status of P53 and PTEN and the mutational status,
were established using logistic regression.
The PFS and OS were described with the Kaplan Meier
method. Results are described with cumulative incidence and 95%
confidence interval.
For each outcome, prognostic factors were tested
with the Cox proportional hazard model in univariate analysis.
Parameters with a P-value of <0.1 were introduced in a
multivariate Cox proportional hazard model with stepwise selection
(with a significance level for entering effects at 0.1 and for
removing effects at 0.05).
All statistical analyses were performed using SAS
version 9.3 (SAS, Cary, NC, USA). The significance level was set at
0.05.
Results
Patient characteristics
In total, 45 patients were included in the present
study. The characteristics of the patient population are summarized
in Table I. Seven BRCA
germline mutations (5 with a BRCA1 mutation, 2 with a
BRCA2 mutation) were identified in 17 patients that
underwent oncogenetic tests due to a familial and/or personal
history of ovarian or breast carcinoma. Successful primary
treatment was achieved in all patients through a combination of
surgery and 5–8 cycles of platinum-based chemotherapy.
 | Table I.Clinicopathological characteristics
of the patient population. |
Table I.
Clinicopathological characteristics
of the patient population.
| Cohort | No. of patients
(%) |
|---|
| Total | 45 |
| Age, years |
|
| Mean
(SD) | 59.1 (12.1) |
|
Range | 25–87 |
| Familial
history |
|
|
Yes | 19 (42) |
| No | 26 (58) |
| Personal
history |
|
|
Yes | 7
(16) |
| No | 38 (84) |
| Oncogenetic
counsultation |
|
|
Yes | 17 (38) |
| No | 28 (62) |
| BRCA mutation
(n=17) |
|
| No
mutation | 10 (59) |
| BRCA
1+ | 5
(29) |
| BRCA 2+ | 2
(12) |
| Tumor stage |
|
|
I–II | 5
(11) |
|
III–IV | 40 (89) |
| Surgery
modality |
|
| Primary
cytoreductive | 36 (80) |
|
Neoadujuvant chemotherapy | 9
(20) |
| Surgical
outcome |
|
|
Completed | 21 (47) |
| Not
competed | 24 (53) |
| Adjuvant
chemotherapy regimen |
|
| Single
agent platinum | 8
(18) |
|
Platinum/taxane doublet | 31 (69) |
|
Bevacizumab included | 6
(13) |
| Response to
platinum-based chemotherapy |
|
|
Sensitivea | 31 (69) |
|
Resistantb | 14 (31) |
IHC
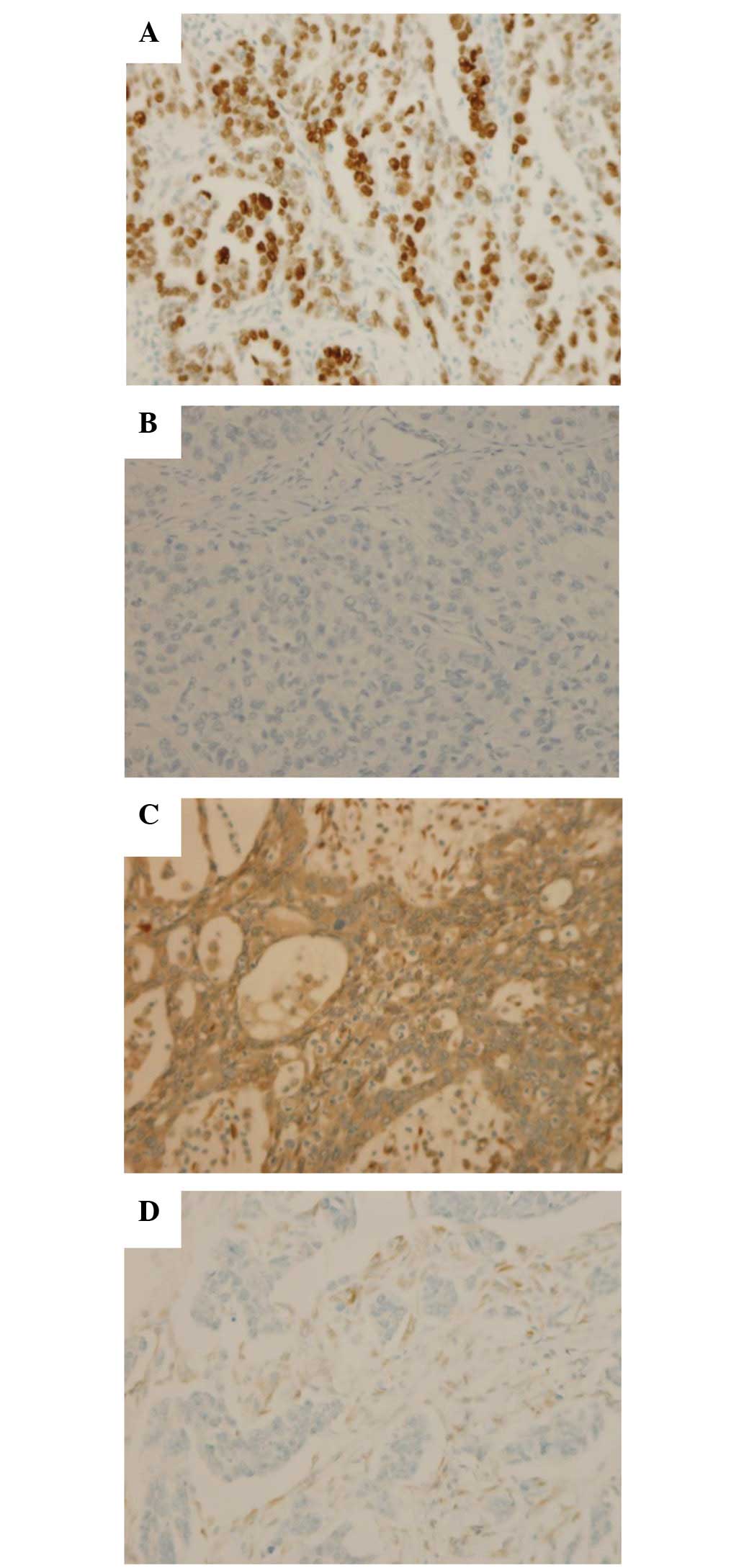
P53 overexpression was observed in 30/45 tumors
(67%) and PTEN loss of expression was observed in 15/39 tumors
(38%) (Fig. 1). In addition, 6 cases
with negative stromal cell staining for PTEN were considered to be
a result of unsuccessful IHC reactions and were excluded from the
statistical analysis.
The overexpression of P53 was found to be associated
with sensitivity to platinum-based chemotherapy (P=0.039) (Table II). No other clinicopathological
feature was associated with P53 status. No clinicopathological
feature was associated with PTEN status.
| Table II.Association between P53 and
PTENa protein expression
and the clinicopathological features. |
Table II.
Association between P53 and
PTENa protein expression
and the clinicopathological features.
|
|
P53+ |
P53− | P-value |
PTEN+ |
PTEN− | P-value |
|---|
| Expression, n
(%) | 30 (67) | 15 (33) |
| 24 (62) | 15 (38) |
|
| Age, n (%) |
|
| 0.546 |
|
| 0.525 |
|
<59 | 17 (57) | 7
(47) |
| 14 (58) | 7
(47) |
|
|
>59 | 13 (43) | 8
(53) |
| 10 (42) | 8
(53) |
|
| FIGO stage, n
(%) |
|
| 1.000 |
|
| 1.000 |
|
I–II | 3
(10) | 2
(13) |
| 3
(13) | 1 (7) |
|
|
III–IV | 27 (90) | 13 (87) |
| 21 (87) | 14 (93) |
|
| Response to
platinum-based chemotherapy, n (%) |
|
| 0.039 |
|
| 1.000 |
|
Sensitive | 24 (80) | 7
(47) |
| 15 (63) | 10 (67) |
|
|
Resistant | 6
(20) | 8
(53) |
| 9
(37) | 5
(33) |
|
Mutation analysis
HRM-PCR assay was performed on the tumor DNA
extracts from 50 tumors. Out of these 50 samples, 2 cases were
found to bear the NRAS, exon 3, mutation, a no samples
possessed KRAS, BRAF or PIK3CA mutations
(Table III).
| Table III.Mutation analysis. |
Table III.
Mutation analysis.
|
|
| HRM-PCR | NGS |
|
|---|
|
|
|
|
|
|
|---|
| Gene | Chromosome | Exon | No. of cases | Exon | c. | p. | No. of cases | Mutated allele
frequency, % | Clinical
significanced |
|---|
|
NRASa | 1 | 3 | 2 | 3 |
181C>A | Gln61Lys | 2 | 24.7/24.1 | Pathogenic |
|
PIK3CAb | 3 |
|
| 5 | 1037T>A | Leu346Gln | 1 | 60.3 | Newly identified
mutation |
|
|
|
|
| 10 | 1571G>A | Arg524Lys | 2 | 17.1/5.0 | Missense mutation,
uncertain |
|
METc | 7 |
|
| 14 | 2962C>T | Arg988Cys | 1 | 65.3 | Missense mutation
uncertain |
|
|
|
|
|
| 3029C>T | Thr1010Ile | 3 | 45.1/82.8/58.7 | Missense mutation,
uncertain |
|
|
|
|
| 18 | 3619G>A | Ala1207Thr | 1 | 32.9 | Newly identified
mutation |
Using NGS, all the mutations found by PCR were
confirmed. In addition, 3 mutations of PIK3CA (2 in exon 10
and 1 in exon 5) and 5 mutations of MET (4 in exon 14 and 1
in exon 18) were detected (Table
III).
The group of patients harboring one of these
mutations showed no survival differences compared with the patients
without any of these mutations.
Clinical outcome
The median follow-up time was 38 months (range, 6–93
months). Out of the 45 patients, 35 patients showed disease
progression and 25 patients succumbed during the follow-up. The
2-year PFS rate was 28% (range, 16–42%) and 5-year OS rate was 37%
(range, 21–53%).
In the univariate analysis, residual disease was
significantly identified as predictive factor for a shorter PFS
[hazard ratio (HR), 2.134; 95% confidence interval (CI),
1.080–4.214] and P53 overexpression was found to predict a longer
PFS (HR, 0.440; 95% CI, 0.216–0.894) (Table IVA). There was a trend for a longer
PFS among patients belonging to the subgroup with a personal
history of breast cancer (HR, 0.319; 95% CI, 0.097–1.045) or
BRCA germline mutations (HR, 0.321; 95% CI, 0.098–1.054). In
the multivariate analysis, P53 overexpression remained
significantly associated with a longer PFS (HR, 0.351; 95% CI,
0.167–0.739) (Table IVA).
| Table IV.Prognostic factors of (A)
progression-free survival and (B) overall survival. |
Table IV.
Prognostic factors of (A)
progression-free survival and (B) overall survival.
| A, Progression-free
survival |
|---|
|
|---|
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Variable | HR and 95% CI | P-value | HR and 95% CI | P-value |
|---|
| Age, years |
| 0.1888 |
|
|
|
<59 | 1 |
|
|
|
|
>59 | 1.564
(0.803–3.045) |
|
|
|
| Family
historya |
| 0.9632 |
|
|
| No | 1 |
|
|
|
|
Yes | 0.984
(0.499–1.942) |
|
|
|
| Personal
historyb |
| 0.0592 |
| 0.0286 |
| No | 1 |
| 1 |
|
|
Yes | 0.319
(0.097–1.045) |
| 0.261
(0.078–0.869) |
|
| BRCA
mutationsc |
| 0.0610 |
|
|
| No | 1 |
|
|
|
|
Yes | 0.321
(0.098–1.054) |
|
|
|
| Stage |
| 0.2443 |
|
|
|
I+II | 1 |
|
|
|
|
III+IV | 2.030
(0.616–6.687) |
|
|
|
| Surgery
modality |
| 0.1014 |
|
|
| Upfront
surgery | 1 |
|
|
|
|
Interval surgery | 1.962
(0.876–4.395) |
|
|
|
| Residual
disease |
| 0.0029 |
| 0.0346 |
|
Complete | 1 |
| 1 |
|
|
Non-complete | 2.134
(1.080–4.214) |
| 2.127
(1.056–4.286) |
|
|
Bevacizumab-containing chemotherapy |
| 0.8372 |
|
|
| No | 1 |
|
|
|
|
Yes | 1.105
(0.426–2.870) |
|
|
|
| P53
overexpression |
| 0.0233 |
| 0.0058 |
| No | 1 |
| 1 |
|
|
Yes | 0.440
(0.216–0.894) |
| 0.351
(0.167–0.739) |
|
| PTEN loss |
| 0.7452 |
|
|
| No | 1 |
|
|
|
|
Yes | 0.886
(0.426–1.841) |
|
|
|
| Mutation
statusd |
| 0.5259 |
|
|
| No | 1 |
|
|
|
|
Yes | 0.763
(0.331–1.758) |
|
|
|
|
| B, Overall
survival |
|
| Age, years |
| 0.2974 |
|
|
|
<59 | 1 |
|
|
|
|
>59 | 1.524
(0.690–3.369) |
|
|
|
| Family
historya |
| 0.7463 |
|
|
| No | 1 |
|
|
|
|
Yes | 0.876
(0.393–1.954) |
|
|
|
| Personal
historya |
| 0.1043 |
| 0.0261 |
| No | 1 |
| 1 |
|
|
Yes | 0.299
(0.069–1.284) |
| 0.168
(0.035–0.809) |
|
| BRCA
mutationsc |
| 0.1002 |
|
|
| No | 1 |
|
|
|
|
Yes | 0.186
(0.025–1.382) |
|
|
|
| Stage |
| 0.1804 |
|
|
|
I+II | 1 |
|
|
|
|
III+IV | 3.933
(0.530–29.158) |
|
|
|
| Surgery
modality |
| 0.5155 |
|
|
| Upfront
surgery | 1 |
|
|
|
|
Interval surgery | 1.440
(0.480–4.318) |
|
|
|
| Residual
disease |
| 0.0961 |
|
|
|
Complete | 1 |
|
|
|
|
Non-complete | 1.983
(0.885–4.442) |
|
|
|
|
Bevacizumab-containing chemotherapy |
| 0.6308 |
|
|
| No | 1 |
|
|
|
|
Yes | 0.739
(0.216–2.533) |
|
|
|
| P53
overexpression |
| 0.0619 |
| 0.0078 |
| No | 1 |
| 1 |
|
|
Yes | 0.450
(0.195–1.041) |
| 0.269
(0.102–0.708) |
|
| PTEN loss |
| 0.9501 |
|
|
| No | 1 |
|
|
|
|
Yes | 1.027
(0.442–2.388) |
|
|
|
| Mutation
statusd |
| 0.9655 |
|
|
| No | 1 |
|
|
|
|
Yes | 1.022
(0.381–2.741) |
|
|
|
In the univariate analysis, no significant
predictive factor of OS was identified (Table IVB), although a trend for longer OS
time was observed (HR, 0.450; 95% CI, 0.195–1.041) in patients with
tumors demonstrating P53 overexpression. In the multivariate Cox
proportional hazard model regression analysis, P53 overexpression
(HR, 0.269; 95% CI, 0.102–0.708) and a personal history of breast
cancer (HR, 0.168; 95% CI, 0.035–0.809) were identified as
predictive factors of a longer OS (Table
IVB).
Discussion
The median PFS and OS times of the cohort examined
in the present study are consistent with previous studies (28).
Considering the IHC of P53, the antibody used in the
present study is able to recognize wild-type and non-null mutant
P53 (29). When assessed by IHC,
non-null mutant P53 often exhibits an intense nuclear staining in a
high percentage of tumor cells, whereas wild-type P53 exhibits a
moderate nuclear staining in a small percentage of tumor cells.
Null-mutant P53 is truncated and unstable, and does not produce
nuclear staining (30). In the
present study, all P53-positive tumors exhibited diffuse and
intense P53 nuclear staining, which may present the non-null mutant
P53 group; by contrast, the P53-negative tumors showed a complete
absence of nuclear staining, which may present a null-mutant P53
group (31). This may reflect that
P53 alterations occur in almost all HGSOCs, presented as null or
non-null mutant P53 (10).
P53 overexpression was observed in 30/45 (67%)
tumors, while the complete absence of P53 expression was observed
in 15/45 (33%) tumors, which is similar to a previous study
(32).
The association between the overexpression of P53
and the patient outcome remains controversial, which could be due
to the various tumor populations explored, differing methodologies
of detection and interpretation of P53 alterations (33). The present study found that P53
overexpression was associated with a longer PFS and longer OS
compared with the group with a complete absence of P53 expression,
which is consistent with a broad-scale study (32). Although this finding can be explained
by the fact that null-mutant P53 is often truncated and has no
wild-type P53 function, while non-null P53 retains part of the
wild-type P53 function (31), the
full the mechanism underlying this difference in outcome is
uncertain and requires further investigation.
Association between loss of PTEN and HGSOC patient
outcome remains controversial (14,16). In
the present study, PTEN loss was observed in 38% of tumors, which
is similar to a previous study (16).
A recent study that evaluated PTEN expression in HGSOC reported an
association between loss of PTEN expression and a longer PFS
(16). However, other studies suggest
that the association of PTEN loss and better prognosis may be
partially explained by defected homologous recombination DNA repair
function that could be caused by PTEN deficiency (34). This homologous recombination defect
could sensitize tumor cells to anticancer drugs, such as platinum
or poly ADP ribose polymerase inhibitors (35), similarly to the observations of
BRCA-deficient tumors. In the present study, the lack of
association of PTEN loss with PFS or OS may be due to the small
sample size.
The rate of MET mutations (11%) observed in
the current study is consistent with a previous study (36), and also confirms the juxtamembrane
exon 14 mutations, c.2962C>T p.Arg988Cys and c.3029C>T
p.Thr1010Ile. These mutations have been reported to induce the
constitutive activation of MET in lung cancer cells (37) but were also described as single
nucleotide polymorphisms (rs34589476 and rs56391007, respectively)
in the ClinVar database (National Institutes of Health, Bethesda,
MA, USA). The c.3029C>T p.Thr1010Ile variation has also been
associated with ethnic polymorphism in people with Italian
heritage, which was the case in one of the patients in the present
study, with a mutated allele frequency of 45.0%. In addition, in
other studies, these mutations did not enhance any transforming
capacity (38), and did not exhibit
oncodriver activity but could sensitize tumor cells to MET
inhibitors (39). This suggests that
these mutational activations of MET could be either an oncogene
addiction as an oncodriver in certain cancers or an oncogene
expedience as a second event that aggravates the malignant
properties of already transformed cells in others (40). In addition, the present study
identified a novel mutation in the tyrosine kinase domain
(c.3619G>A p.Ala1207Thr on exon 18), for which the association
with disease progression and response to therapy remains to be
elucidated. The impact of these MET nucleotide variations in
the biological or clinical behavior of HGSC requires additional
elucidation, as MET inhibition is of great interest for use as a
novel therapeutic target, and numerous MET inhibitors are currently
under investigation in other types of cancer (40).
Large-scale study showed that the mutation of
PIK3CA is usually observed in endometrioid carcinomas
(10). In the present study, 2
PIK3CA mutations were detected in 3 patients (7%) with 1
mutation newly identified (c.1037T>A p.Leu346Gln) and requiring
further investigation into its impact on protein function and tumor
behavior. This mutation frequency is consistent with another study
that performed a small sample size analysis on 2 types of ovarian
cancers (41). In the present study,
PTEN loss was detected in 1/3 of tumors with PIK3CA
mutations. This frequency was previously reported in a study
whereby PIK3CA mutation associated with PTEN loss was
sufficient to initiate ovarian tumors in animal models (42). In addition, tumors with PTEN loss were
reported to regress when exposed to drugs targeting the PI3K
pathway (43). Since >1/3 of the
HGSOC samples presented with a loss of PTEN expression, PTEN loss
could be a predictive marker of PI3K pathway activation and the
response of PI3K pathway inhibitors such as NVP-BYL719 (44). Numerous clinical trials (NCT00877773,
NCT01219699, NCT01449058, NCT 01501604, NCT01708161, NCT01928459,
NCT02439489, NCT02449538 and NCT02449564) of PI3K pathway
inhibitors are ongoing in selected patients with tumors harboring
PIK3CA mutations or PTEN loss, including HGSOC patients.
Although the RAS/MAPK pathway was considered to be
mostly altered in type I ovarian carcinoma (3), the present study has shown that HGSOC
could share a similar molecular profile. The NRAS mutation
(c.181C>A p.Gln61Lys) identified in 2 patients in the present
study had already been reported as an oncogene driver in HGSOC
(17). Furthermore, no KRAS or
BRAF mutations were found in the present study. This finding
confirmed the less frequent dysregulation of the RAS/RAF pathway
and the potential interest of NRAS as a biological marker for
selecting patients that may benefit from targeted therapy (44). Since the emerging targeted therapy
showed antitumor activity in NRAS-mutated cancer (45), the role of NRAS mutations in
HGSOC warrants further investigation.
HGSOCs are a group of heterogeneous malignancies
that are associated with P53 alteration, PTEN loss and alterations
of the PI3K/AKT and RAS/MAPK signaling pathways. Routine therapies
have almost reached the efficacy limit and novel therapeutic
strategies are urgently required. Mutations in P53 and the loss of
PTEN expression are frequent events in HGSOCs. The present study
showed that these events are associated with patient outcome; thus,
mutations in P53 and the loss of PTEN expression may serve as
predictive markers for stratifying patients for appropriate
therapy. The observed alterations in the PI3K/AKT and RAS/MAPK
pathways elucidate the potential for the clinical application of
targeted and personalized therapy, as has been used for other type
of cancer.
Acknowledgements
Dr ShuHui Chen is supported by the China Scholarship
Council (grant no. 201208420600; Wuhan, China).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kurman RJ and Ie M Shih: Pathogenesis of
ovarian cancer: Lessons from morphology and molecular biology and
their clinical implications. Int J Gynecol Pathol. 27:151–160.
2008.PubMed/NCBI
|
|
4
|
Li D, Marchenko ND and Moll UM: SAHA shows
preferential cytotoxicity in mutant p53 cancer cells by
destabilizing mutant p53 through inhibition of the HDAC6-Hsp90
chaperone axis. Cell Death Differ. 18:1904–1913. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ellis L, Hammers H and Pili R: Targeting
tumor angiogenesis with histone deacetylase inhibitors. Cancer
Lett. 280:145–153. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fu S, Hou MM, Naing A, Janku F, Hess K,
Zinner R, Subbiah V, Hong D, Wheler J, Piha-Paul S, et al: Phase I
study of pazopanib and vorinostat: A therapeutic approach for
inhibiting mutant p53-mediated angiogenesis and facilitating mutant
p53 degradation. Ann Oncol. 26:1012–1028. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matulonis U, Berlin S, Lee H, Whalen C,
Obermayer E, Penson R, Liu J, Campos S, Krasner C and Horowitz N:
Phase I study of combination of vorinostat, carboplatin, and
gemcitabine in women with recurrent, platinum-sensitive epithelial
ovarian, fallopian tube, or peritoneal cancer. Cancer Chemother
Pharmacol. 76:417–423. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bast RC, Hennessy B and Mills GB: The
biology of ovarian cancer: New opportunities for translation. Nat
Rev Cancer. 9:415–428. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Skead G and Govender D: Gene of the month:
MET. J Clin Pathol. 68:405–9. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cancer Genome Atlas Research Network, .
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang J, Zhang L, Greshock J, Colligon TA,
Wang Y, Ward R, Katsaros D, Lassus H, Butzow R, Godwin AK, et al:
Frequent genetic abnormalities of the PI3K/AKT pathway in primary
ovarian cancer predict patient outcome. Genes Chromosomes Cancer.
50:606–618. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Choi KC, Auersperg N and Leung PC:
Mitogen-activated protein kinases in normal and (pre)neoplastic
ovarian surface epithelium. Reprod Biol Endocrinol. 1:712003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bai H, Li H, Li W, Gui T, Yang J, Cao D
and Shen K: The PI3K/AKT/mTOR pathway is a potential predictor of
distinct invasive and migratory capacities in human ovarian cancer
cell lines. Oncotarget. 6:25520–25532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Martins FC, Santiago ID, Trinh A, Xian J,
Guo A, Sayal K, Jimenez-Linan M, Deen S, Driver K, Mack M, et al:
Combined image and genomic analysis of high-grade serous ovarian
cancer reveals PTEN loss as a common driver event and prognostic
classifier. Genome Biol. 15:5262014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Djordjevic B, Hennessy BT, Li J, Barkoh
BA, Luthra R, Mills GB and Broaddus RR: Clinical assessment of PTEN
loss in endometrial carcinoma: Immunohistochemistry outperforms
gene sequencing. Mod Pathol. 25:699–708. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bakkar RM, Xie SS, Urbauer DL, Djordjevic
B, Vu K and Broaddus RR: Intact PTEN expression by
immunohistochemistry is associated with decreased survival in
advanced stage ovarian/primary peritoneal high-grade serous
carcinoma. Int J Gynecol Pathol. 34:497–506. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Emmanuel C, Chiew YE, George J,
Etemadmoghadam D, Anglesio MS, Sharma R, Russell P, Kennedy C,
Fereday S, Hung J, et al: Genomic classification of serous ovarian
cancer with adjacent borderline differentiates RAS pathway and
TP53-mutant tumors and identifies NRAS as an oncogenic driver. Clin
Cancer Res. 20:6618–6630. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gentile A, Trusolino L and Comoglio PM:
The Met tyrosine kinase receptor in development and cancer. Cancer
Metastasis Rev. 27:85–94. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gherardi E, Birchmeier W, Birchmeier C and
Woude G Vande: Targeting MET in cancer: Rationale and progress. Nat
Rev Cancer. 12:89–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oza AM, Cook AD, Pfisterer J, Embleton A,
Ledermann JA, Pujade-Lauraine E, Kristensen G, Carey MS, Beale P,
Cervantes A, et al: Standard chemotherapy with or without
bevacizumab for women with newly diagnosed ovarian cancer (ICON7):
Overall survival results of a phase 3 randomised trial. Lancet
Oncol. 16:928–936. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aghajanian C, Goff B, Nycum LR, Wang YV,
Husain A and Blank SV: Final overall survival and safety analysis
of OCEANS, a phase 3 trial of chemotherapy with or without
bevacizumab in patients with platinum-sensitive recurrent ovarian
cancer. Gynecol Oncol. 139:10–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Choueiri TK, Vaishampayan U, Rosenberg JE,
Logan TF, Harzstark AL, Bukowski RM, Rini BI, Srinivas S, Stein MN,
Adams LM, et al: Phase II and biomarker study of the dual
MET/VEGFR2 inhibitor foretinib in patients with papillary renal
cell carcinoma. J Clin Oncol. 31:181–186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zillhardt M, Park SM, Romero IL, Sawada K,
Montag A, Krausz T, Yamada SD, Peter ME and Lengyel E: Foretinib
(GSK1363089), an orally available multikinase inhibitor of c-Met
and VEGFR-2, blocks proliferation, induces anoikis, and impairs
ovarian cancer metastasis. Clin Cancer Res. 17:4042–4051. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeppernick F and Meinhold-Heerlein I: The
new FIGO staging system for ovarian, fallopian tube, and primary
peritoneal cancer. Arch Gynecol Obstet. 290:839–842. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kurman RJ, Carcangiu ML, Herrington CS and
Young RH: WHO Classification of Tumours of Female Reproductive
Organs. Fourth. World Health Organization; Geneva: 2014
|
|
26
|
Meinhold-Heerlein I, Fotopoulou C, Harter
P, Kurzeder C, Mustea A, Wimberger P, Hauptmann S and Sehouli J:
The new WHO classification of ovarian, fallopian tube, and primary
peritoneal cancer and its clinical implications. Arch Gynecol
Obstet. 293:695–700. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Harlé A, Filhine-Tresarrieu P, Husson M,
Boidot R, Rouyer M, Dubois C, Leroux A and Merlin JL: Rare RAS
mutations in metastatic colorectal cancer detected during routine
RAS genotyping using next generation sequencing. Target Oncol.
11:363–370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hennessy BT, Coleman RL and Markman M:
Ovarian cancer. Lancet. 374:1371–1382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bonsing BA, Corver WE, Gorsira MC, van
Vliet M, Oud PS, Cornelisse CJ and Fleuren GJ: Specificity of seven
monoclonal antibodies against p53 evaluated with Western blotting,
immunohistochemistry, confocal laser scanning microscopy, and flow
cytometry. Cytometry. 28:11–24. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Psyrri A, Kountourakis P, Yu Z,
Papadimitriou C, Markakis S, Camp RL, Economopoulos T and
Dimopoulos MA: Analysis of p53 protein expression levels on ovarian
cancer tissue microarray using automated quantitative analysis
elucidates prognostic patient subsets. Ann Oncol. 18:709–715. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hashimoto T, Tokuchi Y, Hayashi M,
Kobayashi Y, Nishida K, Hayashi S, Ishikawa Y, Tsuchiya S, Nakagawa
K, Hayashi J and Tsuchiya E: p53 null mutations undetected by
immunohistochemical staining predict a poor outcome with
early-stage non-small cell lung carcinomas. Cancer Res.
59:5572–5577. 1999.PubMed/NCBI
|
|
32
|
Köbel M, Reuss A, du Bois A, Kommoss S,
Kommoss F, Gao D, Kalloger SE, Huntsman DG and Gilks CB: The
biological and clinical value of p53 expression in pelvic
high-grade serous carcinomas. J Pathol. 222:191–198. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kmet LM, Cook LS and Magliocco AM: A
review of p53 expression and mutation in human benign, low
malignant potential, and invasive epithelial ovarian tumors.
Cancer. 97:389–404. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
McEllin B, Camacho CV, Mukherjee B, Hahm
B, Tomimatsu N, Bachoo RM and Burma S: PTEN loss compromises
homologous recombination repair in astrocytes: Implications for
glioblastoma therapy with temozolomide or poly(ADP-ribose)
polymerase inhibitors. Cancer Res. 70:5457–5464. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mendes-Pereira AM, Martin SA, Brough R,
McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ and Ashworth A:
Synthetic lethal targeting of PTEN mutant cells with PARP
inhibitors. EMBO Mol Med. 1:315–322. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tang C, Jardim DLF, Falchook GS, Hess K,
Fu S, Wheler JJ, Zinner RG, Naing A, Tsimberidou AM, De Melo
Galgiato D, et al: MET nucleotide variations and amplification in
advanced ovarian cancer: Characteristics and outcomes with c-Met
inhibitors. Oncoscience. 1:5–13. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ma PC, Kijima T, Maulik G, Fox EA, Sattler
M, Griffin JD, Johnson BE and Salgia R: c-MET mutational analysis
in small cell lung cancer: Novel juxtamembrane domain mutations
regulating cytoskeletal functions. Cancer Res. 63:6272–6281.
2003.PubMed/NCBI
|
|
38
|
Tyner JW, Fletcher LB, Wang EQ, Yang WF,
Rutenberg-Schoenberg ML, Beadling C, Mori M, Heinrich MC, Deininger
MW, Druker BJ and Loriaux MM: MET receptor sequence variants R970C
and T992I lack transforming capacity. Cancer Res. 70:6233–6277.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Arriola E, Cañadas I, Arumí-Uría M, Dómine
M, Lopez-Vilariño JA, Arpí O, Salido M, Menéndez S, Grande E,
Hirsch FR, et al: MET phosphorylation predicts poor outcome in
small cell lung carcinoma and its inhibition blocks HGF-induced
effects in MET mutant cell lines. Br J Cancer. 105:814–823. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Comoglio PM, Giordano S and Trusolino L:
Drug development of MET inhibitors: Targeting oncogene addiction
and expedience. Nat Rev Drug Discov. 7:504–516. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Garrido-Castro AC, Argilés G, Moreno D,
Rodriguez-Freixinos V, Vilaro M, Macarulla T, Zambrano C Cruz,
Azaro A, Adamo B, Alsina M, et al: Molecular profiling in
gynecologic cancer and matched targeted therapy: A step toward
improving personalized medicine. J Clin Oncol 32 (Suppl 5s).
55782014.
|
|
42
|
Kinross KM, Montgomery KG, Kleinschmidt M,
Waring P, Ivetac I, Tikoo A, Saad M, Hare L, Roh V, Mantamadiotis
T, et al: An activating Pik3ca mutation coupled with Pten loss is
sufficient to initiate ovarian tumorigenesis in mice. J Clin
Invest. 122:553–557. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Janku F, Wheler JJ, Westin SN, Moulder SL,
Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna
I, et al: PI3K/AKT/mTOR inhibitors in patients with breast and
gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol.
30:777–782. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fritsch C, Huang A, Chatenay-Rivauday C,
Schnell C, Reddy A, Liu M, Kauffmann A, Guthy D, Erdmann D, De
Pover A, et al: Characterization of the novel and specific PI3Kα
inhibitor NVP-BYL719 and development of the patient stratification
strategy for clinical trials. Mol Cancer Ther. 13:1117–1129. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ascierto PA, Schadendorf D, Berking C,
Agarwala SS, van Herpen CM, Queirolo P, Blank CU, Hauschild A, Beck
JT, St-Pierre A, et al: MEK162 for patients with advanced melanoma
harbouring NRAS or Val600 BRAF mutations: A non-randomised,
open-label phase 2 study. Lancet Oncol. 14:249–256. 2013.
View Article : Google Scholar : PubMed/NCBI
|