Introduction
Pituicytoma (previously referred to as
infundibuloma) is a rare tumor of the sellar and suprasellar
regions, originating from specialized glial cells in the
neurohypophysis and infundibulum (1).
The tumor is slow growing and benign, and histologically
corresponds to World Health Organization (WHO) grade I (2,3). Only 78
cases of pituicytoma have been reported since it was first
described in 1955 (4). Due to its
rarity, the clinical manifestations, radiological characteristics,
histopathological features and prognoses have yet to be fully
elucidated. Pituicytoma is typically challenging to distinguish
from other sellar and suprasellar lesions, including granular cell
tumor, pituitary adenoma, pilocytic astrocytoma and lymphocytic
hypophysitis (2,3). Surgical treatment may be challenging,
owing to the hyper vascularity of the tumor. The present case study
reports three cases of histopathologically diagnosed pituicytoma.
Clinical presentations, surgical strategies and treatment outcomes
are presented and the relevant published literature is
discussed.
Case reports
Case 1
A 41-year-old male presented to the First Hospital
of Jilin University (Changchun, China) in February 2015 with a
20-day history of headaches and dizziness. The physical examination
revealed bitemporal hemianopsia. Brain magnetic resonance imaging
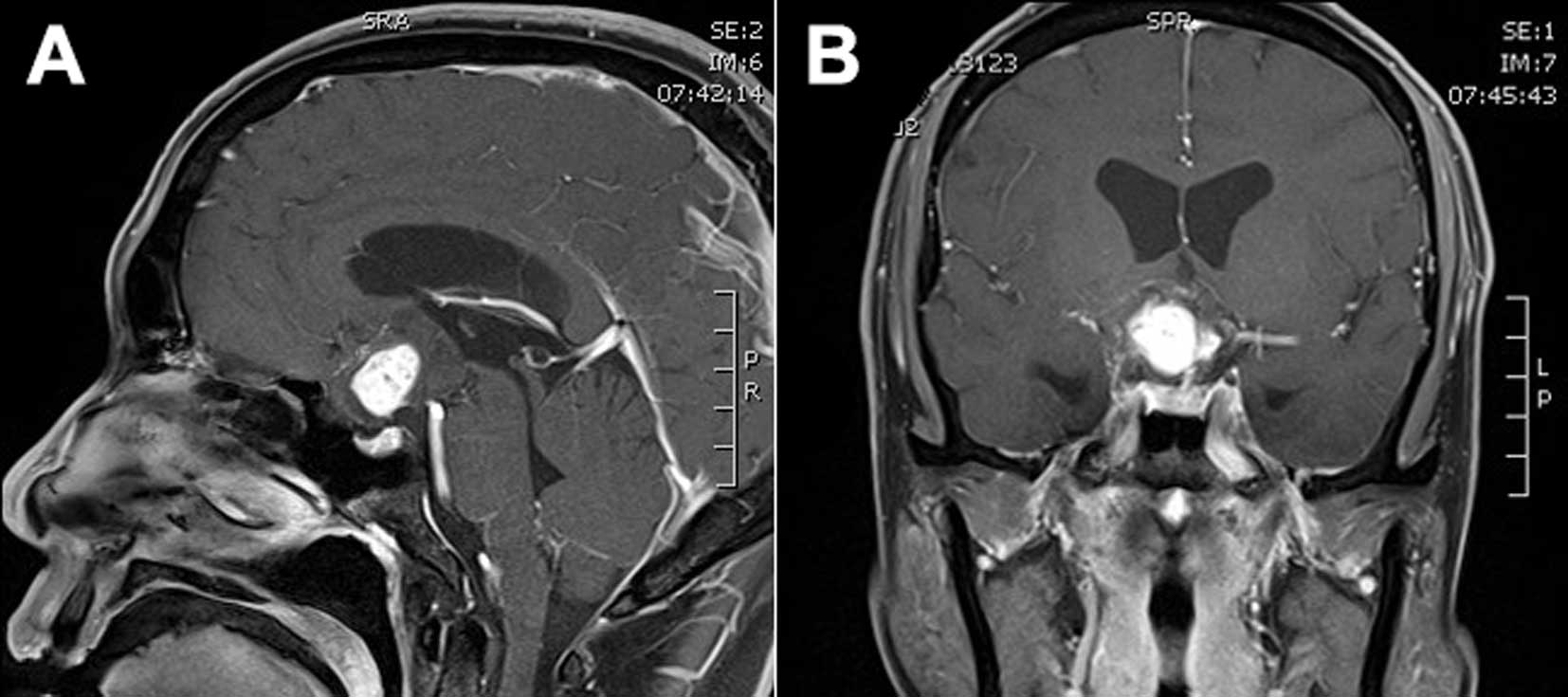
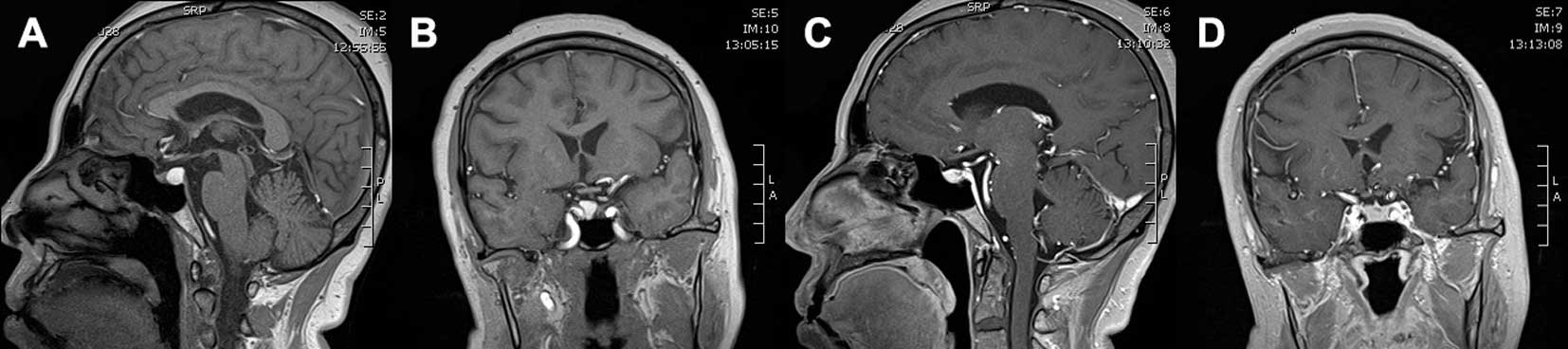
(MRI) revealed a contrast-enhanced 3.7×2.2×2.5-cm mass compressing
the pituitary stalk and optic chiasma (Fig. 1A and B). The mass appeared isointense
on T1-weighted images (T1WI), and there was signal heterogeneity on
T2-weighted images (T2WI); the central homogeneous enhancement was
remarkable following gadolinium-diethylenetriamine pentaacetic acid
(Gd-DTPA) administration. Subsequent investigation revealed normal
endocrine hormone levels (including TSH, FT3, FT4, PRL, FSH, GH,
LH, serum hydrocortisone, and urinary free cortisol). A
preoperative diagnosis of craniopharyngioma was determined.
Examinations of the anterior segments of the eyes
and fundus were normal. No dermatological abnormalities that are
typically associated with neurofibromatosis were detected. A
further ophthalmologic examination revealed binocular ametropia,
whereas the binocular visual field was normal. Right eye proptosis
and right sixth nerve palsy were clearly discernible. No further
neurological abnormalities were identified.
A craniotomy was performed via a right
frontal-temporal approach. Intraoperatively, a hard gray-white mass
was observed, which exhibited solid and cystic components, hyper
vascularity and was adhered to the optic nerve. The tumor was
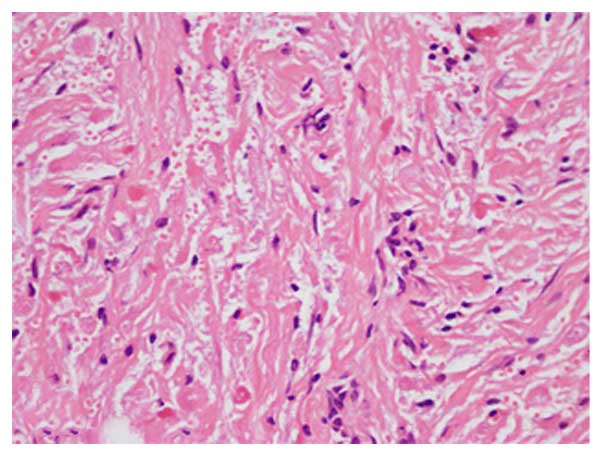
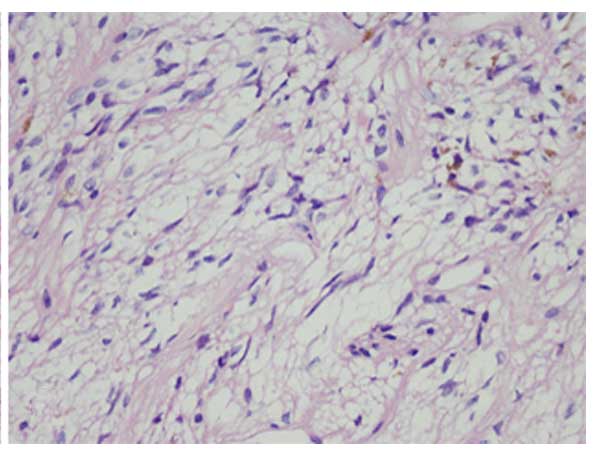
resected gradually and a diagnosis of pituicytoma was subsequently
confirmed following histopathological examination (Fig. 2). Immunohistochemical staining
(5) revealed that the tumor was
positive for glial fibrillary acidic protein (GFAP),
oligodendrocyte transcription factor 2 (Oligo-2), vimentin and
S-100 protein, but negative for tumor protein 53 (p53), isocitrate
dehydrogenase 1 R132H (IDH1R132H), myelin basic protein (MBP),
epidermal growth factor receptor, synaptophysin (Syn), chromogranin
A (CgA), neuronal nuclear antigen and epithelial membrane antigen
(EMA). The positive expression rate for antigen Ki-67 was ~2%, and
for O-6-methylguanine-DNA methyltransferase (MGMT) it was
<5%.
Postoperatively, the patient experienced nausea and
vomiting, and his skin was abnormally dry. Investigation revealed a
decreased serum sodium ion level (120–130 mmol/l; normal range,
136–146 mmol/l), a decreased free triiodothyronine level (1.73
pmol/l; normal range, 3.1–6.8 pmol/l), an elevated free thyroxine
level (8.99 pmol/l; normal range, 3.1–6.8 pmol/l), a decreased
adrenocorticotropic hormone level (0 h level, 0.22 pmol/l; 16 h
level, 0.28 pmol/l; normal range, 1.6–13.9 pmol/l) and decreased
serum testosterone level (1.39 nmol/l; normal range, 6.07–27.1
nmol/l). Hypopituitarism was diagnosed and complete remission was
achieved following treatment with hydrocortisone (Shanghai Sine
Pharmaceutical Laboratories, Co., Ltd., Shanghai, China; 20 mg at 8
am and 2 pm for 2 weeks) and euthyrox (Merck KGaA, Darmstadt,
Germany; 25 µg once a day for one month following hydrocortisone
treatment for a week). At the 17-month postsurgical follow-up,
binocular vision was markedly improved. No recurrence of the tumor
was noted, and all aforementioned hormone levels were observed to
be within the normal ranges.
Case 2
A 44-year-old female presented with a 4-year history
of irregular menstruation. The physical examination revealed no
evident abnormalities. A brain MRI revealed a contrast-enhancing
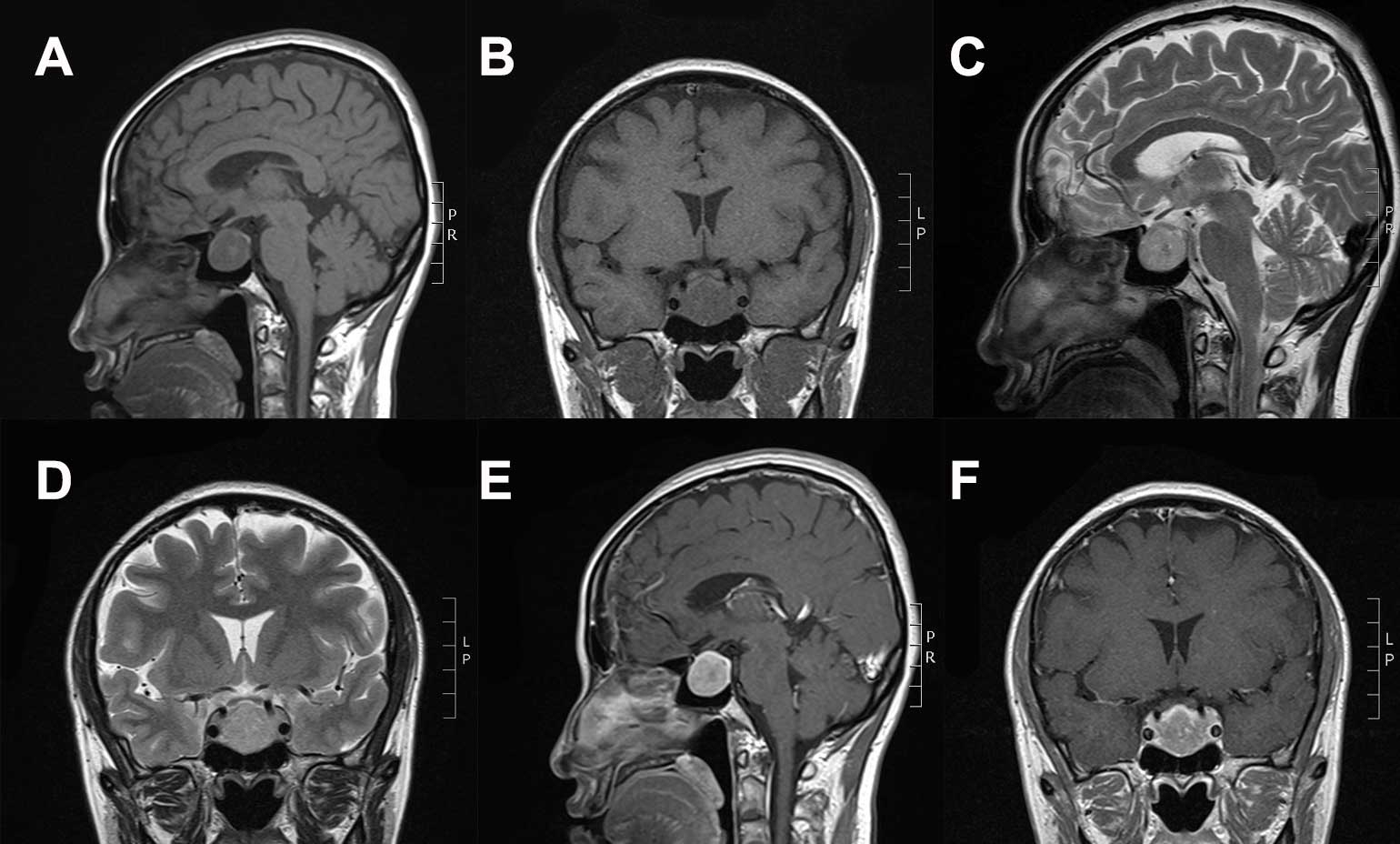
2.2×2.2×2.1-cm mass in the sellar and suprasellar regions (Fig. 3A-F). The mass appeared hypo intense on
T1WI and exhibited heterogeneous hyper intensity on T2WI; the tumor
demonstrated clear homogeneous enhancement following Gd-DTPA
administration. The tumor was located adjacent to the bilateral
internal carotid arteries; the pituitary stalk was displaced to the
left, and the optic chiasma displaced antrorsely. Endocrine hormone
levels were observed to be within normal ranges: TSH, 0.465 µIU/ml
(normal range, 0.27–4.2 µIU/ml); FT3, 3.2 pmol/l (normal range,
3.1–6.8 pmol/l); FT4, 12.4 pmol/l (normal range, 12–22 pmol/l);
PRL, 61.11 mIU/l (normal range, 70.81–566.5 mIU/l); FSH, 21.46
mIU/l (normal range, 4.54–22.51 mIU/l); GH, 0.327 ng/ml (normal
range, 0.01–3.607 ng/ml); LH, 9.74 mIU/ml (normal range, 2.12–10.89
mIU/ml); serum hydrocortisone (8 am, 292.07 nmol/l; 0 am, 513.54
nmol/l; normal range, 240–619 nmol/l; 4 pm, 523.7 nmol/l; normal
range, <276.0); urinary free cortisol, 905 nmol/24 h (normal
range, 108–961 nmol/24 h). The preoperative diagnosis was pituitary
adenoma.
The tumor was completely resected via a
transsphenoidal surgical approach. Intraoperatively, the gray-white
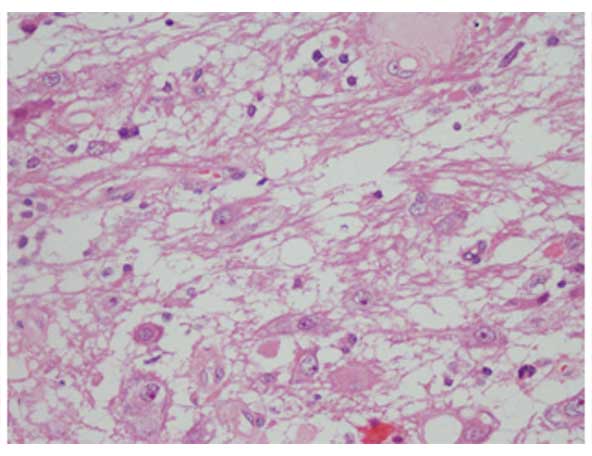
mass was soft and hypovascular. Histopathological examination
confirmed pituicytoma (Fig. 4).
Immunohistochemical staining revealed that the tumor was positive
for Oligo-2, Syn, neural cell adhesion molecule (CD56), CgA and
neuro-specific enolase but negative for GFAP, p53, MBP and S-100
protein. The positive expression rate for MGMT was 30–40%.
Postoperatively, the patient experienced transient
diabetes insipidus that resolved following 6 IU hypophysin
treatment (Tianjin Biochemical Pharmaceutical Co., Ltd., Tianjin,
China). Repeat investigations revealed normal hormone levels (TSH,
FT3, FT4, PRL, FSH, GH, LH, serum hydrocortisone and urinary free
cortisol). Regular menstruation was restored during the 43-month
follow-up period and no recurrence of the tumor was observed.
Case 3
A 61-year old female presented with a 6-month
history of frequent headaches, nausea and vomiting. The physical
examination revealed no discernable abnormalities. A brain MRI
identified a well-defined 0.9×0.8×0.9-cm mass in the sellar region,
compressing the pituitary gland to the left (Fig. 5). The mass exhibited hyperintensity on
T1WI and isointensity on T2WI, with a cystic component in the
central section of the pituitary; the tumor was markedly enhanced
following Gd-DTPA administration. Subsequent investigation
revealedan elevated serum thyroid stimulating hormone (TSH) level
(5.73 µIU/ml; normal range, 0.27–4.2 µIU/ml). A preoperative
diagnosis of pituitary adenoma was determined.
The tumor was completely removed using a
transsphenoidal approach. Intraoperatively, the gray-white mass was
soft and hypervascular. Histopathological examination confirmed
pituicytoma (Fig. 6).
Immunohistochemical staining indicated that the tumor was positive
for GFAP, vimentin and S-100 protein, but negative for reticular
fibers. The positive expression rate for antigen Ki-67 was
<1%.
Postoperative recovery was satisfactory and
subsequent examination revealed normal TSH levels (mean, 0.475
µIU/ml). During the follow-up period of 45 months, no recurrence of
the tumor was observed.
Discussion
Pituicytomas are rare, primary tumors originating
from the so-called pituicytes in neurohypophysis and pituitary
stalk (2). Pituicytes are glial cells
that support the large axons of vasopressin- and oxytocin-producing
hypothalamic neurons, and comprise major, dark, oncocytic,
ependymal and granular cell types; the majority of pituicytomas are
considered to derive from the first two types (6). The first case of this type of glioma was
identified in the posterior lobe of the pituitary gland and was
described by Scothorne in 1955 (4).
Brat et al (2) reported nine
cases of low-grade glioma of the neurohypophysis in 2000, for which
the term pituicytoma was proposed. Pituicytoma was previously
regarded as a condition with a wide clinical spectrum, including
pituitary astrocytoma, posterior lobe glioma, choristoma and
infundibuloma in the sellar and suprasellar regions. However, the
tumor was named as a separate entity in the 2007 WHO classification
of central nervous system tumors (3).
A total of 78 published case reports of pituicytoma
were retrieved in a search of the published literature. A summary
of these cases is presented below (Table
I). Pituicytomas occur predominantly in adults, with a mean age
of 46.9 years (range, 7–83 years) at the time of diagnosis. The
current study indicated that the peak age was between the fourth
and fifth decade, accounting for 50.6% of all patients. Out of 78
patients, only 3 were in the pediatric age group (age, 7–13 years)
(7–9).
A total of 44 male and 33 female subjects were included in the
present study; the male: Female ratio was 1.3:1. The clinical
details were not available in one case. The most common symptoms of
pituicytoma were vision and visual field disorders (44 cases,
56.4%), headaches (34 cases, 43.6%), hypopituitarism (17 cases,
21.8%), hyposexuality (16 cases, 20.5%), sexual dysfunction (7
cases, 9.0%), menstrual disorder (7 cases, 9.0%), dizziness (6
cases, 7.7%), diabetes insipidus (3 cases, 3.8%) (10–12),
epilepsy (3 cases, 3.8%) (13–15),
gynecomastia (3 cases, 3.8%) (10,16,17) and
spontaneous tumor hemorrhage (1 case, 1.3%) (18). There were also sporadic cases that
presented with nausea (6), vomiting
(9) or edema (9). In one case the pituicytoma was
incidentally revealed at autopsy (19).
 | Table I.The summary of previous cases with
pituicytomas. |
Table I.
The summary of previous cases with
pituicytomas.
| Characteristics | Case number (N) | Percentage |
|---|
| Gender | N=77a |
|
| Male | 44 | 57.1% |
|
Female | 33 | 42.9% |
| Main clinical
symptoms | N=78 |
|
| Vision
and visual field disorders | 44 | 56.4% |
|
Headache | 34 | 43.6% |
|
Hypopituitarism | 17 | 21.8% |
|
Hyposexuality | 16 | 20.5% |
| Sexual
dysfunction | 7 | 9.0% |
| Menstrual
disorder | 7 | 9.0% |
|
Dizziness | 6 | 7.7% |
| Diabetes
insipidus | 3 | 3.8% |
|
Epilepsy | 3 | 3.8% |
|
Gynecomastia | 3 | 3.8% |
|
Spontaneous tumor
hemorrhage | 1 | 1.3% |
| Locations | N=77 |
|
| The
sellar region | 15 | 19.4% |
| The
suprasellar region | 34 | 44.2% |
| The
sellar and suprasellar regions | 28 | 36.4% |
| Operations | N=67 |
|
|
Craniotomy | 24 | 35.9% |
|
Transsphenoidal operation | 41 | 61.1% |
|
Endoscopic endonasal
transsphenoidal operation | 2 | 3.0% |
The clinical symptoms are typically attributable to
the local effects of tumor, and therefore, depend on the tumor size
and location. For instance, optic chiasm compression may cause
bitemporal hemianopsia; hypophysis compression may lead to headache
and hypopituitarism; infundibular compression may result in
hypothalamic dopamine delivery disorders, inducing
hyperprolactinemia, amenorrhea, hyposexuality and sexual
dysfunction. The three cases detailed in this study presented with
combinations of bitemporal hemianopsia, headache, dizziness,
amenorrhea, nausea and vomiting, which is consistent with the
previous case reports. The duration of the symptoms prior to the
diagnosis ranged from a few months to several years. One patient
had acute symptomatic onset due to spontaneous tumor hemorrhage
into the third ventricle (17). There
was one case of accidental head trauma in which a diagnosis of
pituicytoma was made (20).
In various patients, there was a history of
endocrine disorders prior to the onset of pituicytoma. These
included parathyroid adenomas and follicular carcinoma of the
thyroid (21), strumectomy (16), amyotrophic lateral sclerosis (17), orchiectomy (22), orchidorrhaphy (18), diabetes (23) and Cushing's disease (24). Furthermore, a solitary case was
reported that exhibited genomic copy-number imbalances, including
losses on chromosome arms 1p, 14q and 22q, and gains on 5p
(11).
The imaging characteristics of pituicytoma are
nonspecific. The radiological profiles were available in 77 cases;
15 of these 77 cases revealed pituicytoma in the sellar region, 34
in the suprasellar region, and in 28 both the sellar and
suprasellar regions were involved. In 12 cases, a computed
tomography scan revealed a solid, isointense mass with homogenous
enhancement and no accompanying evidence of calcification,
necrosis, bony erosion or hyperostosis. On the MRI examination,
pituicytomas commonly presented as well-defined, solid, round or
oval masses in the sellar region, with or without suprasellar
extension. The tumors usually appeared hypointense-isointense on
T1WI, low-moderately hyperintense on T2WI, and with homogenous or
heterogeneouscontrast enhancement (6,13,25). Only four cases had solid cystic
pituicytomas (2,7,26). In the
present study, cases 1 and 3 exhibited solid cystic tumors. The
differential diagnoses had included pituitary adenoma, meningioma,
craniopharyngioma, hemangiopericytoma, pilocytic astrocytoma,
granulocyte tumor, ganglioglioma, germinoma, hamartoma and
metastatic tumors (6,27).
Surgical resection is the preferred treatment for
pituicytoma, with an extremely low recurrence rate (4.3%) following
complete resection. Among all the reported cases treated with gross
total resection, only one experienced tumor recurrence (28). In the literature, 67 surgeries,
involving 60 patients, were described. Craniotomy was performed in
24 patients, of which gross total resection was achieved in 7
patients; complications were noted in 11 patients. Transsphenoidal
surgery was performed on 41 patients, of which gross total
resection was achieved in 14 patients; complications were noted in
8 patients. An endoscopic endonasal transsphenoidal approach was
employed in 2 patients, and gross total resection was achieved in
the 2 cases (29). Intraoperatively,
the tumors were revealed to be pink and solid. The majority of
pituicytomas were well demarcated and of a benign nature. In a
minority of cases, the tumor had a tight dural attachment at the
diaphragma sellae (22). Pituicytomas
with a soft texture or a cystic component were infrequently
observed (17,30). Hypervascularity is a common
intraoperative challenge, as it can hinder the success of gross
total resection. However, hypovascular entities have also been
described in the literature (31). In
certain cases, carotid angiography was helpful in surgical planning
(17,23). To achieve complete tumor resection, a
comprehensive preoperative assessment is essential.
Current mainstream surgical approaches include the
aforementioned frontotemporal craniotomy and the transsphenoidal
approach. Feng et al (29)
reported complete resection of recurring pituicytomas when the
surgery was performed via an expanded endoscopic, endonasal,
transsphenoidal and transplanum approach. However, the efficacy and
safety of the expanded transsphenoidal procedure has yet to be
fully established as there is currently insufficient clinical
evidence (29). The most common
postoperative complications included diabetes insipidus (9 cases),
hypopituitarism (7 cases), visual impairment (6 cases) and
hypothyroidism (3 cases). These complications were considered to be
associated with the iatrogenic trauma to contiguous structures.
Adjuvant radiotherapy was administered in 7 patients, however the
respective follow-up data was not available. In the current case
series, no adjuvant radiotherapy, or chemotherapy, was performed
and there were no tumor recurrences observed during a maximal
follow-up period of 45 months.
The accurate diagnosis of pituicytoma continues to
depend on histopathological evidence. Microscopically, pituicytomas
are composed of round to spindle-shaped cells with a fascicular or
storiform growth pattern. The tumor cells have an abundant
eosinophilic cytoplasm and a rich capillary network is visible.
Tumor cell nuclei are round to oval, without evident atypia or
mitotic figures. However, Zhi et al (28) reported a pituicytoma with atypical
histological features, including sparse intercellular reticulin
surrounding the tumor cells, absent Rosenthal fibers and
eosinophilic granular bodies, which usually help to distinguish
between pituicytomas and pilocytic astrocytomas (32). In previous studies, pituicytomas
demonstrated positive immunofluorescence staining for S-100 and
vimentin protein, negative or low-moderately positive staining for
GFAP (2,10,21,31), and
negative staining for EMA, Syn, chromogranin, cytokeratin and
neurofilament protein (26). Using
electron microscopy, cytoplasmic intermediate filaments and tumor
vessel basal lamina were typically observed in pituicytomas, but
desmosomes and pericellular basal lamina were absent (10,31). In
the cases presented in this study, the immunohistochemical features
are consistent with those reported previously. In addition, the low
proliferation index is indicative that pituicytomas may be
consistently benign.
The recurrence interval following subtotal tumor
resection is usually long and no instances of malignant
transformation or cerebrospinal dissemination have been reported.
Since the radiological characteristics and clinical manifestations
are nonspecific, pituicytomas are liable to be initially
misdiagnosed. The definitive diagnosis still depends on
pathological examination. The role and efficacy of adjuvant
radiotherapy and chemotherapy is unclear at present and requires
further study. Considering the local recurrence following subtotal
resection, postoperative radiotherapy should be recommended
inpatients where gross total resection is not feasible. Even for
those patients undergoing complete tumor resection, a close MRI
follow-up is essential.
Pituicytomas are extremely rare entities. The
surgical resection should be designed for optimal functional
preservation, and an MRI may provide guidance in this respect.
Furthermore, understanding of the surrounding anatomical structures
is crucial in facilitating complete tumor resection and nerve
preservation.
Acknowledgements
Written, informed consent has been obtained from
each patient for the publication of this case report and the
accompanying images. The authors would like to thank the patients
and all the physicians and staff who assisted in this study. The
authors also thank Medjaden Bioscience Ltd. (Hong Kong, China) for
assisting in the preparation of this study.
References
|
1
|
Ulm AJ, Yachnis AT, Brat DJ and Rhoton AL
Jr: Pituicytoma: Report of two cases and clues regarding
histogenesis. Neurosurgery. 54:753–758. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brat DJ, Scheithauer BW, Staugaitis SM,
Holtzman RN, Morgello S and Burger PC: Pituicytoma: A distinctive
low-grade glioma of the neurohypophysis. Am J Surg Pathol.
24:362–368. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brat DJ, Scheithauer BW, Fuller GN and
Tihan T: Newly codified glial neoplasms of the 2007 WHO
Classification of Tumours of the Central Nervous System:
Angiocentric glioma, pilomyxoid astrocytoma and pituicytoma. Brain
Pathol. 17:319–324. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scothorne CM: A glioma of the posterior
lobe of the pituitary gland. J Pathol Bacteriol. 69:109–112. 1955.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cenacchi G, Giovenali P, Castrioto C and
Giangaspero F: Pituicytoma: Ultrastructural evidence of a possible
origin from folliculo-stellate cells of the adenohypophysis.
Ultrastruct Pathol. 25:309–312. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shah B, Lipper MH, Laws ER, Lopes MB and
Spellman MJ Jr: Posterior pituitary astrocytoma: A rare tumor of
the neurohypophysis: A case report. AJNR Am J Neuroradiol.
26:1858–1861. 2005.PubMed/NCBI
|
|
7
|
Yilmaz Ö, Turan A, Yiğit H, Duymuş M and
Koşar U: Case of pituicytoma in childhood. Childs Nerv Syst.
28:11–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tian Y, Yue S, Jia G and Zhang Y:
Childhood giant pituicytoma: A report and review of the literature.
Clin Neurol Neurosurg. 115:1943–1950. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chakraborti S, Mahadevan A, Govindan A,
Sridhar K, Mohan NV, Satish IR, Rudrappa S, Mangshetty S and
Shankar SK: Pituicytoma: Report of three cases with review of
literature. Pathol Res Pract. 209:52–58. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Figarella-Branger D, Dufour H, Fernandez
C, Bouvier-Labit C, Grisoli F and Pellissier JF: Pituicytomas, a
mis-diagnosed benign tumor of the neurohypophysis: Report of three
cases. Acta Neuropathol. 104:313–319. 2002.PubMed/NCBI
|
|
11
|
Phillips JJ, Misra A, Feuerstein BG,
Kunwar S and Tihan T: Pituicytoma: Characterization of a unique
neoplasm by histology, immunohistochemistry, ultrastructure, and
array-based comparative genomic hybridization. Arch Pathol Lab Med.
134:1063–1069. 2010.PubMed/NCBI
|
|
12
|
Mao Z, Xiao W, Wang H, Li Z, Huang Q, He D
and Zhu Y: Pituicytoma: Report of two cases. Oncol Lett. 2:37–41.
2011.PubMed/NCBI
|
|
13
|
Furtado SV, Ghosal N, Venkatesh PK, Gupta
K and Hegde AS: Diagnostic and clinical implications of
pituicytoma. J Clin Neurosci. 17:938–943. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hammoud DA, Munter FM, Brat DJ and Pomper
MG: Magnetic resonance imaging features of pituicytomas: Analysis
of 10 cases. J Comput Assist Tomogr. 34:757–761. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang Y, Quan J, Su H, Hu HX and Wang F:
40-year old female with a sellar mass. Brain Pathol. 22:871–874.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kowalski RJ, Prayson RA and Mayberg MR:
Pituicytoma. Ann Diagn Pathol. 8:290–294. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Benveniste RJ, Purohit D and Byun H:
Pituicytoma presenting with spontaneous hemorrhage. Pituitary.
9:53–58. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Newnham HH and Rivera-Woll LM: Images in
clinical medicine. Hypogonadism due to pituicytoma in an identical
twin. N Engl J Med. 359:28242008.
|
|
19
|
Takei H, Goodman JC, Tanaka S,
Bhattacharjee MB, Bahrami A and Powell SZ: Pituicytoma incidentally
found at autopsy. Pathol Int. 55:745–749. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Grote A, Kovacs A, Clusmann H, Becker AJ
and Niehusmann P: Incidental pituicytoma after accidental head
trauma-case report and review of literature. Clin Neuropathol.
29:127–133. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schultz AB, Brat DJ, Oyesiku NM and Hunter
SB: Intrasellar pituicytoma in a patient with other endocrine
neoplasms. Arch Pathol Lab Med. 125:527–530. 2001.PubMed/NCBI
|
|
22
|
Gibbs WN, Monuki ES, Linskey ME and Hasso
AN: Pituicytoma: Diagnostic features on selective carotid
angiography and MR imaging. AJNR Am J Neuroradiol. 27:1639–1642.
2006.PubMed/NCBI
|
|
23
|
Thiryayi WA, Gnanalingham KK, Reid H,
Heald A and Kearney T: Pituicytoma: A misdiagnosed benign tumour of
the posterior pituitary. Br J Neurosurg. 21:47–48. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schmalisch K, Schittenhelm J, Ebner FH,
Beuschlein F, Honegger J and Beschorner R: Pituicytoma in a patient
with Cushing's disease: Case report and review of the literature.
Pituitary. 15(Suppl 1): S10–S16. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Islamian A Pirayesh, Buslei R, Saeger W
and Fahlbusch R: Pituicytoma: Overview of treatment strategies and
outcome. Pituitary. 15:227–236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Secci F, Merciadri P, Rossi DC, D'Andrea A
and Zona G: Pituicytomas: Radiological findings, clinical behavior
and surgical management. Acta Neurochir (Wien). 154:649–657. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hurley TR, D'Angelo CM, Clasen RA,
Wilkinson SB and Passavoy RD: Magnetic resonance imaging and
pathological analysis of a pituicytoma: Case report. Neurosurgery.
35:314–317. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhi L, Yang L, Quan H and Bai-Ning L:
Pituicytoma presenting with atypical histological features.
Pathology. 41:505–509. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Feng M, Carmichael JD, Bonert V, Bannykh S
and Mamelak AN: Surgical management of pituicytomas: Case series
and comprehensive literature review. Pituitary. 17:399–413. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Uesaka T, Miyazono M, Nishio S and Iwaki
T: Astrocytoma of the pituitary gland (pituicytoma): Case report.
Neuroradiology. 44:123–125. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zygourakis CC, Rolston JD, Lee HS, Partow
C, Kunwar S and Aghi MK: Pituicytomas and spindle cell oncocytomas:
Modern case series from the University of California, San
Francisco. Pituitary. 18:150–158. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nakasu Y, Nakasu S, Saito A, Horiguchi S
and Kameya T: Pituicytoma. Two case reports. Neurol Med Chir
(Tokyo). 46:152–156. 2006. View Article : Google Scholar : PubMed/NCBI
|