Introduction
Cachexia, which is the loss of body mass that cannot
be reversed by nutrition, is frequently observed in patients with
cancer. Cancer cachexia is a leading cause of morbidity and
mortality worldwide (1). The
prominent clinical feature of cancer cachexia is the continuous
loss of skeletal muscle that cannot be fully reversed by
conventional nutritional support, leading to progressive functional
impairment (2,3). The skeletal muscle loss is associated
with a reduced quality of life, as well as poor survival (4). The pathophysiology of cancer cachexia is
characterized by a negative protein and energy balance (5); however, the underlying mechanism remains
largely unknown. Previous studies have demonstrated that numerous
signaling molecules and transcription factors are involved in the
regulation of skeletal muscle mass (6,7).
Mitochondrial function is crucial for the
maintenance of the skeletal muscle (8). It has been demonstrated that suppression
of mitochondrial function is sufficient to cause muscle wasting in
adult animals (8). Mitochondrial
function requires the coordination of mitochondrial fusion and
fission processes that are referred to as mitochondrial dynamics
(9,10). A system of pro-fusion and pro-fission
proteins regulates mitochondrial morphology and subcellular
localization (9,10). The fusion proteins mitofusin-1 (Mfn1)
and mitofusin-2 (Mfn2) promote mitochondrial elongation and
activity (11). As a major
determinant of the fusion process, Mfn2 is a large GTPase that is
integral to the mitochondrial outer membrane (12). It is essential for mitochondrial
fusion during embryonic development and neuronal differentiation
(13). In humans, mutations in the
Mfn2 locus have been associated with numerous diseases, including
Charcot-Marie-Tooth type 2A neuropathy, diabetes and Alzheimer's
disease (14). Mfn2 is robustly
expressed in muscle cells and its insufficiency has been associated
with the fragmentation of the mitochondrial network, which is
essential for the normal mitochondrial function (14). Inhibition of mitochondrial fission has
been shown to inhibit muscle loss during fasting (11,15). In
addition, downregulation of Mfn2 has been observed in the muscle of
obese and non-obese type 2 diabetic subjects (16,17), and
decreased mRNA expression levels of Mfn2 were observed in the
skeletal muscle of tumor-bearing mice with severe cachexia
(11). These results suggested that
Mfn2 is involved in muscle wasting associated with diseases such as
cancer cachexia.
The role of Mfn2 in skeletal muscle loss in cancer
cachexia remains largely unknown. Therefore, the present study
aimed to investigate the association between Mfn2 expression in
skeletal muscle and body weight loss in patients with cancer
cachexia. In addition, the potential role of Mfn2 in skeletal
muscle atrophy and mass loss was evaluated by in vitro
methods using the C2C12 mouse myoblast cell line and in vivo
animal experiments.
Materials and methods
Reagents
Monoclonal mouse Mfn2 antibody (cat. no. sc-100560;
1:100) was purchased from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA). Polyclonal rabbit cytochrome c oxidase subunit IV
(COX IV; cat. no. AC610; 1:1,000) and monoclonal mouse cytochrome
c (Cyto C; cat. no. AC909; 1:200) antibodies were obtained
from Beyotime Institute of Biotechnology (Haimen, China).
Monoclonal mouse β-actin antibody (cat. no. A1978; 1:200) and tumor
necrosis factor-α (TNF-α) were purchased from Sigma-Aldrich (Merck
Millipore, Darmstadt, Germany). Polyclonal goat anti-rabbit (cat.
no. 35552; 1:5,000) anti-mouse (cat. no. 35502; 1:5,000) IgG
secondary antibodies were purchased from Thermo Fisher Scientific,
Inc., (Waltham, MA, USA). All other chemical reagents were standard
commercial products of analytical-reagent grade.
Human skeletal muscle samples of
cancer cachexia
Rectus abdominis muscle tissues were obtained from
21 patients undergoing primary surgical resection of
gastrointestinal cancer at Zhongshan Hospital of Fudan University
School of Medicine (Shanghai, China) between March and September
2012. The inclusion criteria were as follows: i) Patients had
histologically documented cancers; ii) patients had not received
prior anticancer treatment; and iii) patients had exhibited weight
loss in the prior 6 months. Muscle tissue samples (5×5×5 mm) were
fresh-frozen in liquid nitrogen immediately following resection,
and were stored at −80°C. The present study was approved by the
Ethics Committee of Zhongshan Hospital of Fudan University. Written
informed consent approving tissue donation for research purposes
was obtained from all patients prior to tissue collection.
Lentivirus
The vector plasmid for Mfn2 overexpression and
control vector plasmid were prepared by GeneChem, Co., Ltd.
(Shanghai, China). The lentivirus was produced by transient
transfection of 293T cells with the vector plasmids, gag-pol
packaging plasmid (Shanghai Institutes for Biological Sciences,
Chinese Academy of Sciences, Shanghai, China) and envelope plasmid
(Shanghai Institutes for Biological Sciences, Chinese Academy of
Sciences) using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's
protocol.
Cell culture
The C2C12 mouse myoblast and HCT116 human colon
cancer cell lines (Cell Bank of Shanghai Institutes for Biological
Sciences, Chinese Academy of Sciences) were maintained in
Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific,
Inc.) supplemented with 10% fetal bovine serum (Thermo Fisher
Scientific, Inc.), 2 mM L-glutamine, 100 U/ml penicillin and 100
µg/ml streptomycin. For the differentiation of C2C12 cells, the
cells (2.5×105) were grown in DMEM supplemented with 2%
(v/v) horse serum (Thermo Fisher Scientific, Inc.), 2 mM
L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. Cells
were then incubated with TNF-α (0, 2.5 and 5 ng/ml) and cultured at
37°C in an incubator with 5% CO2 for 7 days.
Animals
Four-week-old male nude mice (BALB/cA-nu/nu) (n=22;
weight, 14.96±1.39 g) were obtained from the Shanghai Laboratory
Animal Center (Shanghai Institutes for Biological Sciences, Chinese
Academy of Sciences) and maintained under pathogen-free conditions.
The mice were housed in laboratory cages at 23°C and exposed to 12
h light/dark cycles with free access to food and water. All
protocols invovling animals were approved by the Institutional
Animal Care and Use Committee at the Institute for Nutritional
Sciences (Shanghai, China). The mice were separated into four
groups (5–6 mice/group), as follows: i) Tumor-bearing and blank
lentivirus-treated mice (group 1); ii) tumor-bearing and Mfn2
overexpression lentivirus-treated mice (group 2); iii) tumor-free
and blank lentivirus-treated mice (group 3); and iv) tumor-free and
Mfn2 overexpression lentivirus-treated mice (group 4). The
tumor-bearing mice were subcutaneously injected with
5×106 HCT116 cells (500 µl) at each flank, whereas the
tumor-free mice were injected with 500 µl phosphate-buffered saline
(PBS) as a control. Body weights, food intake, and tumor formation
were assessed every 3 days. The mice were categorized as having
severe cachexia when the body weights were >2 standard
deviations (SDs) away from the means of the age-matched control
mice. The mice with cachexia were treated with Mfn2 overexpressing
lentivirus (108 in 100 µl PBS), which was injected
intramuscularly into the gastrocnemius muscles every 3 days, four
times in total. All mice were maintained for 40 days prior to
euthanasia with CO2. Immediately following sacrifice,
the gastrocnemius muscles were dissected and weighed, and then
fresh-frozen in liquid nitrogen or fixed in paraformaldehyde. All
mice were maintained and used in accordance with the guidelines of
the Institutional Animal Care and Use Committee of the Institute of
Nutritional Sciences.
Western blot analysis
Western blot analysis was performed as described
previously (18). Briefly, frozen
muscle tissue samples or cells were homogenized in Mueller buffer
and the protein concentration was determined by the Bradford
method. Crude tissue homogenate (40 µg) was fractionated on 8–13%
SDS-polyacrylamide gels, and the proteins were transferred onto
polyvinylidene fluoride membranes overnight. The membranes were
stained with Ponceau solution to verify that each gel had been
equally loaded. Subsequently, the membranes were blocked overnight
with 5% milk in PBS containing 0.1% Tween-20 (PBS-T), followed by
incubation for 1 h at room temperature with anti-Mfn2 (86 kDa;
1:100), anti-COX IV (17 kDa; 1:1,000), anti-Cyto C (15 kDa; 1:200)
and anti-β-actin (43 kDa; 1:200) primary antibodies. The membranes
were then incubated for 1 h at 25°C with horseradish
peroxidase-conjugated anti-rabbit and anti-mouse IgG (diluted
1:5,000 in 5% milk in PBS-T), followed by detection of the bound
secondary antibodies with enhanced chemiluminescence (GE Healthcare
Life Sciences, Piscataway, NJ, USA). Images were digitally scanned,
and blots were quantified by densitometry using ImageJ 1.46r
software (National Institutes of Health, Bethesda, MA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Isolation of RNA from the rectus abdominis muscle
tissues was performed using the EZ-10 Spin Column Total RNA
Mini-Preps Super Kit (Bio Basic Inc., Amherst, NY, USA). RT-PCR was
performed on an Applied Biosystems Two-Step Real-Time PCR System
(Thermo Fisher Scientific, Inc.) using DNAse I (Thermo Fisher
Scientific, Inc.) using the SYBR Green Realtime PCR Master Mix
(Toyobo, Osaka, Japan). PCR was performed under the following
conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15
sec and 58°C for 60 sec. The primer sequences were as follows: Mfn2
forward, 5′-GCTCGGAGGCACATGAAAGT-3′ and reverse,
5′-ATCACGGTGCTCTTCCCATT-3′; and β-actin forward,
5′-GACAGTGTTGTGGGTGTAGGTACTAAC-3′ and reverse,
5′-CCGCTTTACACCAGCGTCAT-3′. β-actin was used as the internal
control. Relative mRNA expression levels of Mfn2 were determined
using the 2−ΔΔCq method, as described previously
(19).
Electron microscopy
The mitochondria in C2C12 cells were observed by
electron microscopy using conventional fixation-embedding
procedures (20).
Hematoxylin-eosin (HE) staining
The gastrocnemius muscles from the mice were
embedded in paraffin and cut into 6-µm tissue sections, which were
then stained with HE (Beyotime Institute of Biotechnology),
according to the manufacturer's protocol. Subsequently, the tissue
sections were observed under a light microscope.
Statistical analysis
Statistical analyses involved the unpaired
two-tailed Student's t-test, two-way analysis of variance and the
Student-Newman-Keuls method, which were performed using GraphPad
Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA).
Data are presented as the mean ± standard error of the mean of
three independent experiments. P<0.05 was considered to indicate
a statistically significant difference.
Results
Downregulation of Mfn2 in the rectus
abdominis is associated with body weight loss in patients with
gastrointestinal cancer
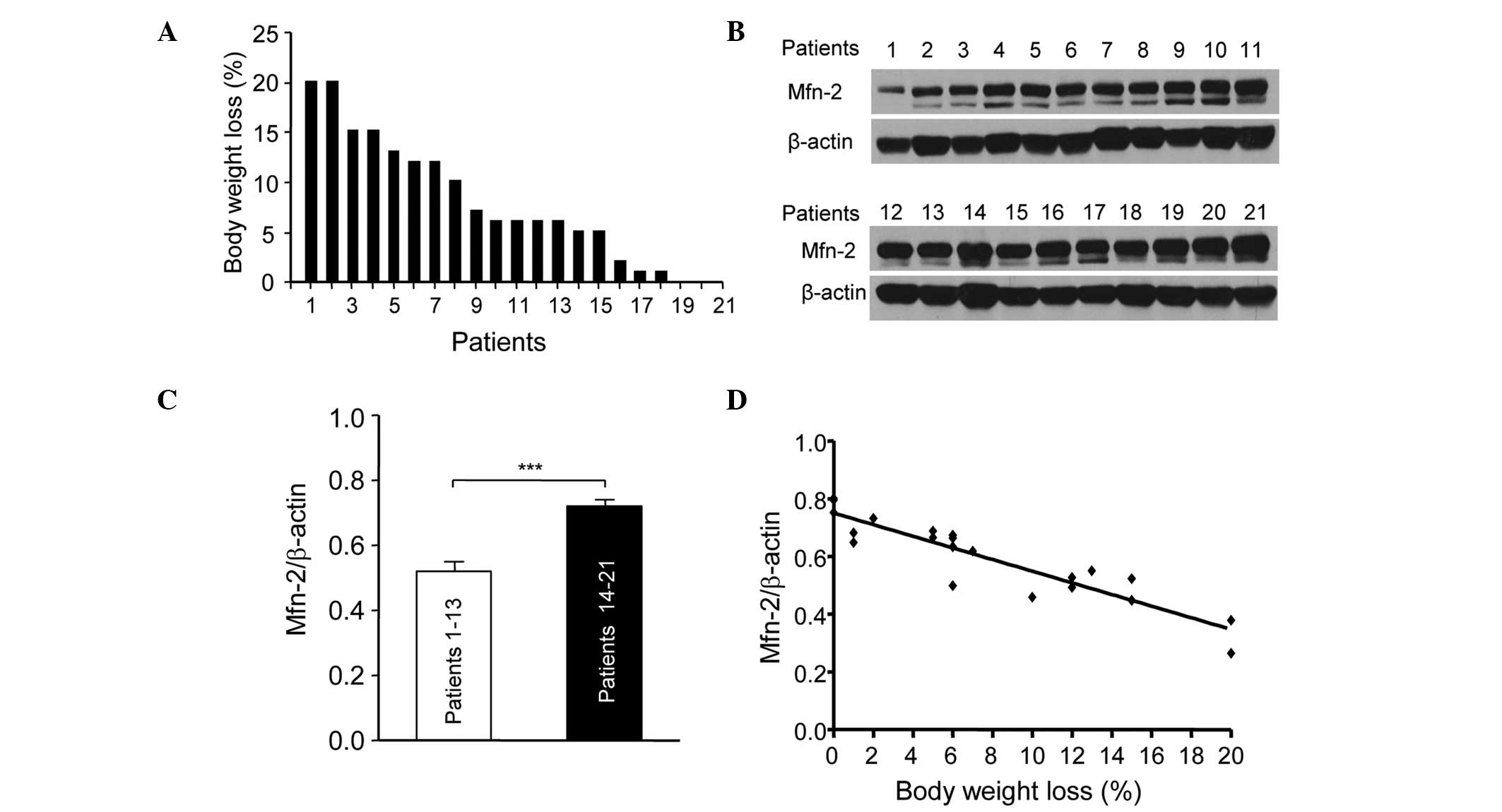
The present study assessed the association between
Mfn2 levels in the rectus abdominis and the body weight loss in 21
patients with gastric or colorectal cancer. Fig. 1 shows the percentage body weight loss
in the 21 patients at 6 months prior to surgery. The patients were
numbered according to their weight loss. Patient 1 lost ~20% body
weight, while patients 19, 20 and 21 showed little weight loss
(Fig. 1A). The expression levels of
Mfn2 in the rectus abdominis of the 21 patients were determined by
western blotting (Fig. 1B). The
patients were divided into two groups: Patients (nos. 1–13) who
showed >5% weight loss were in the cachexia period and those who
showed <5% weight loss were in the pre-cachexia period. The
relative protein expression levels of Mfn2 in the patients with
cachexia were significantly lower compared with the pre-cachexia
patients (P<0.001; Fig. 1C). In
addition, the downregulation of Mfn2 expression in the rectus
abdominis of patients with cachexia was associated with weight loss
(Fig. 1D). These results suggest that
Mfn2 is involved in cachexia and its downregulation is associated
with the progression of cachexia.
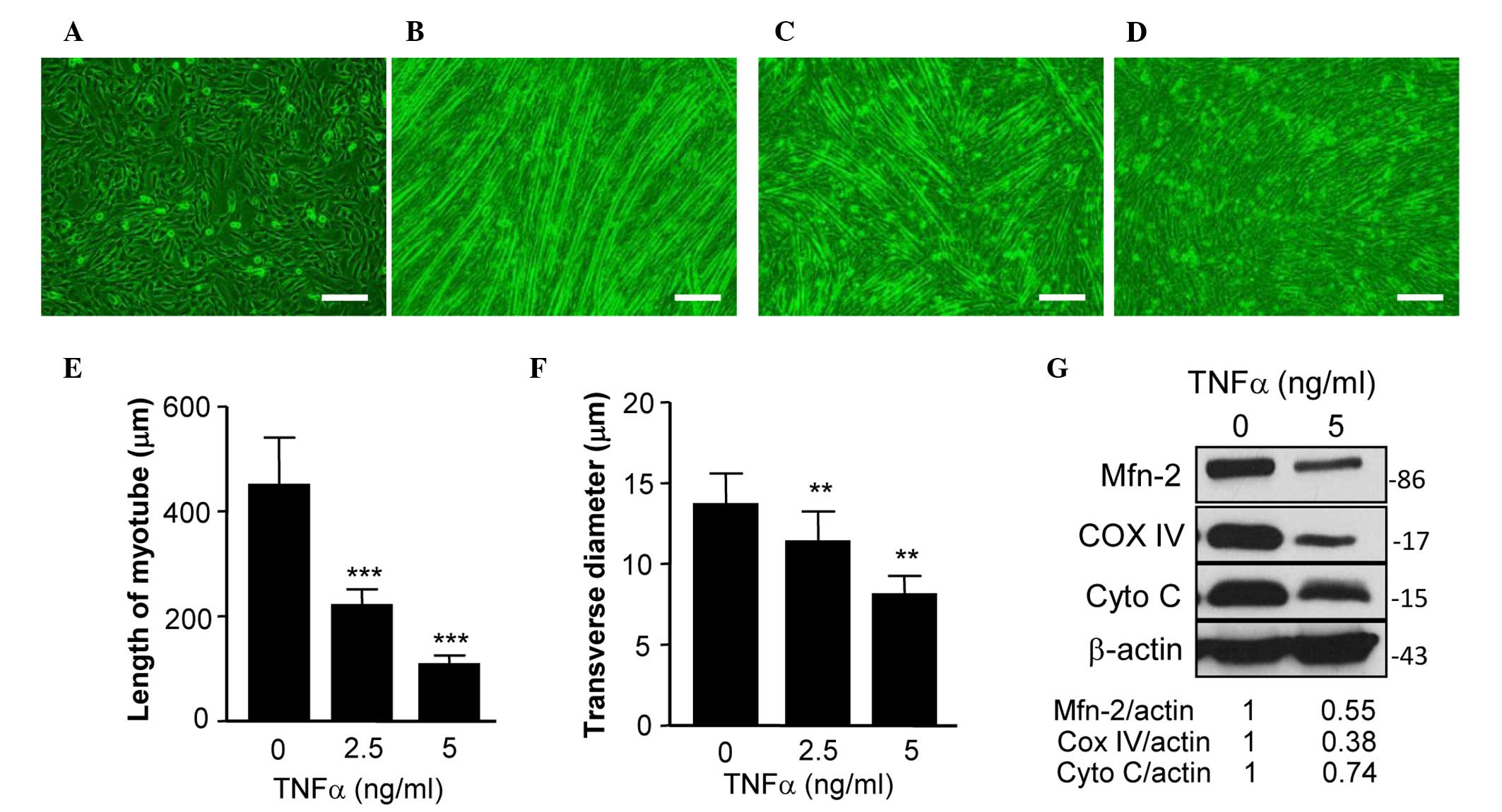
Expression of Mfn2 is downregulated in
TNF-α-induced myotube atrophy of C2C12 cells
C2C12 cells are an immortalized cell line of mouse
skeletal muscle cells that fuse and differentiate into myotube
under low-serum conditions (21).
TNF-α has a critical role in muscle loss during cancer cachexia
(22), and TNF-α-treated
differentiated C2C12 cells have previously been used as an in
vitro model of muscle atrophy (23). The present study used this model to
assess the expression of Mfn2 in differentiated C2C12 cells treated
with or without TNF-α. Fig. 2A shows
C2C12 cells prior to differentation and Fig. 2B shows the differentiated cells with
normal myotube formation. In a previous study, TNF-α was able to
reduce the expression of Mfn2 in cultured cells (16). In the present study, the addition of
TNF-α prevented the formation of normal myotube (Fig. 2C and D). Specifically, TNF-α treatment
reduced the length (Fig. 2E) and
transverse diameter (Fig. 2F) of
myotube, suggesting that TNF-α induces myotube atrophy.
Furthermore, the expression levels of Mfn2 in TNF-α-treated C2C12
cells were examined, and it was demonstrated that TNF-α inhibited
the expression of Mfn2 (Fig. 2G).
The mitochondrial proteins COX IV and Cyto C are two
key proteins of the mitochondrial respiratory chain, which has a
critical role in mitochondrial function (11). The expression levels of COX IV and
Cyto C have been used to assess the mitochondrial function of cells
in previous studies (11,24). Therefore, the present study determined
the expression levels of COX IV and Cyto C in C2C12 cells, and
demonstrated that the expression of these two proteins were
decreased (Fig. 2H), indicating that
the mitochondrial function of the cells was impaired.
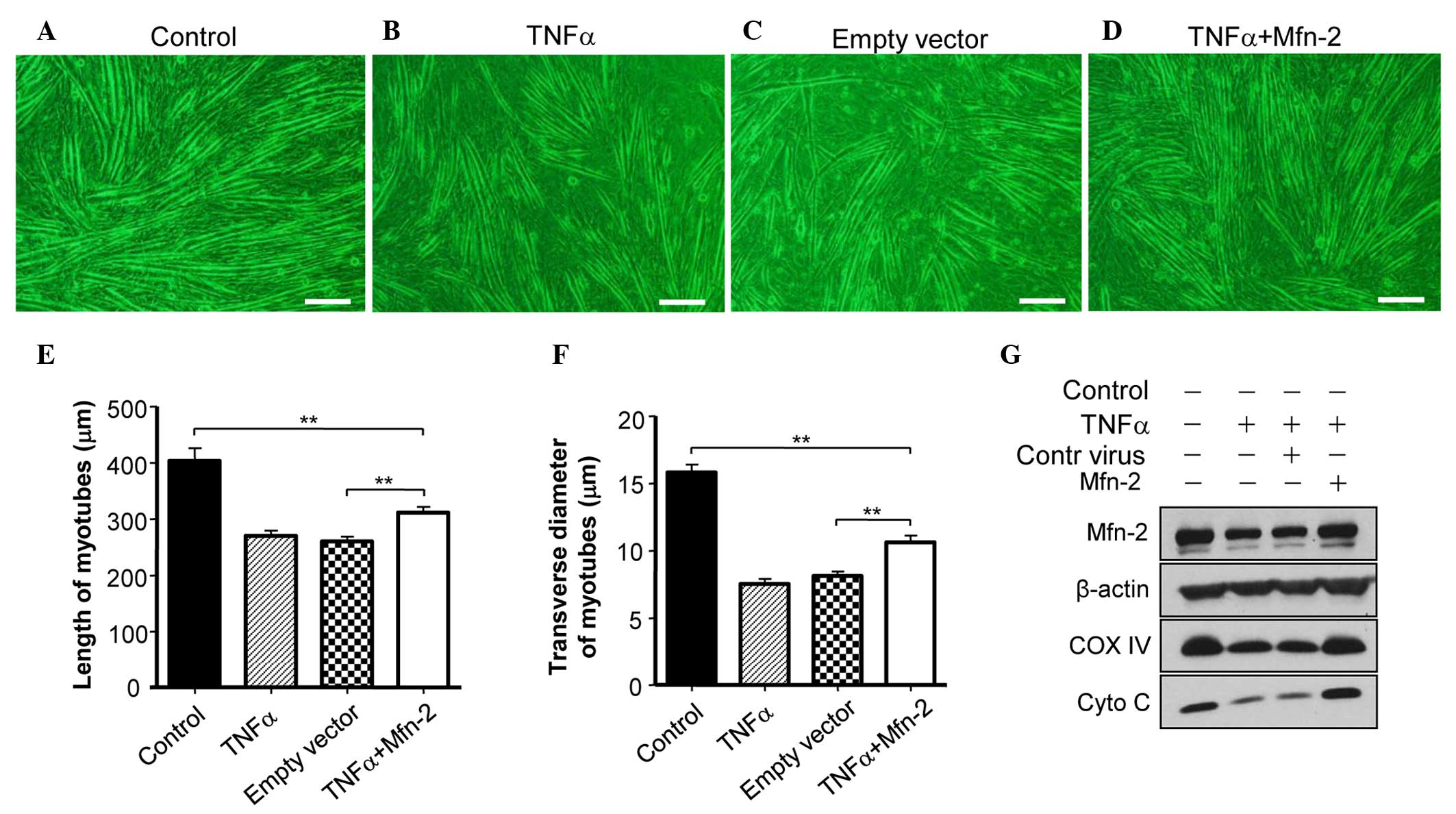
Overexpression of Mfn2 ameliorates
TNF-induced myotube atrophy of C2C12 cells
To assess whether Mfn2 is required for the
TNF-α-induced myotube atrophy of C2C12 cells, Mfn2 was
overexpressed in C2C12 cells. It was observed that overexpression
of Mfn2 prevented the TNF-α-mediated induction of myotube atrophy
(Fig. 3A and B). In addition,
overexpression of Mfn2 upregulated the expression of COX IV and
Cyto C in C2C12 cells (Fig. 3C),
suggesting the recovery of mitochondrial function. Furthermore, the
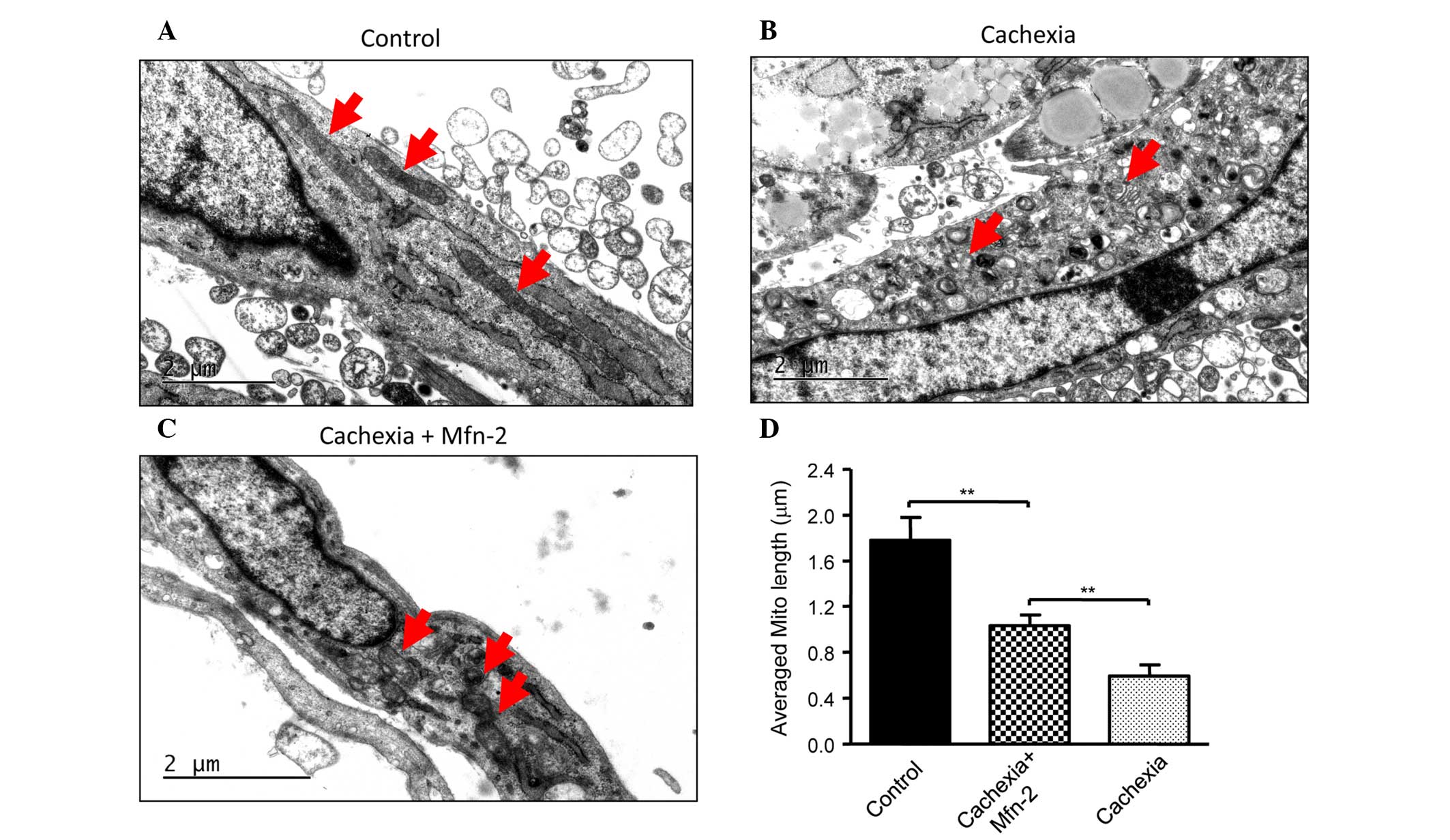
appearance of the mitochondria in C2C12 cells prior to and
following TNF-α treatment was examined. Prior to treatment, C2C12
cells had mitochondria of a normal size with cristae (Fig. 4A), whereas TNF-α treatment led to
abnormal and fragmented mitochondria in cells (Fig. 4B). Overexpression of Mfn2 in C2C12
cells partially attenuated the damage to mitochondria (Fig. 4C). In addition, the lengths of
mitochondria were measured (Fig. 4D),
and TNF-α was shown to decrease the average length of mitochondria
in C2C12 cells, which was reversed in part by overexpression of
Mfn2.
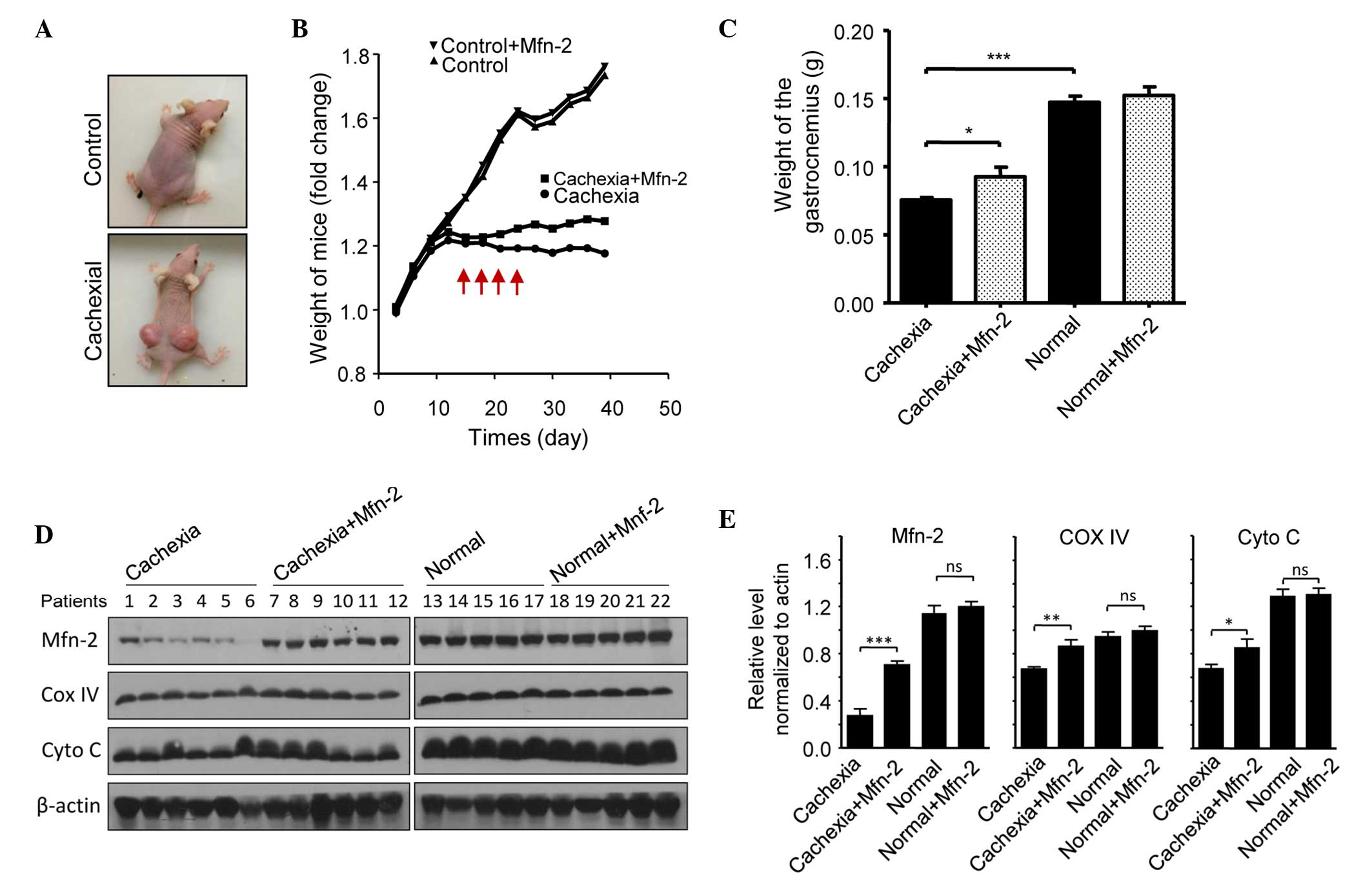
Overexpression of Mfn2 prevents
cachexia-induced gastrocnemius loss in mice
The potential role of Mfn2 in cachexia was assessed
using a nude mouse model. Mice were injected subcutaneously with
HCT116 cells to promote the growth of tumor xenografts. Notably,
xenograft growth was associated with weight loss in the mice
(Fig. 5A). Mice were categorized as
having severe cachexia if the body weights were >2 SDs away from
the means of age-matched control mice. Empty or Mfn2 lentivirus
were injected into the gastrocnemius of these mice every 3 days,
four times in total (Fig. 5A). After
40 days, the mice were sacrificed and the gastrocnemius muscles
were excised and weighed. The protein expression levels of Mfn2 in
the mouse gastrocnemius muscles are shown in Fig. 5B. It was observed that the weight of
the gastrocnemius muscle in mice with cachexia was significantly
decreased, as compared with the control mice (P<0.001; Fig. 5C). Notably, treatment of the cachexia
mice with Mfn2 lentivirus significantly increased the weight of the
gastrocnemius muscle, as compared with the untreated cachexia mice
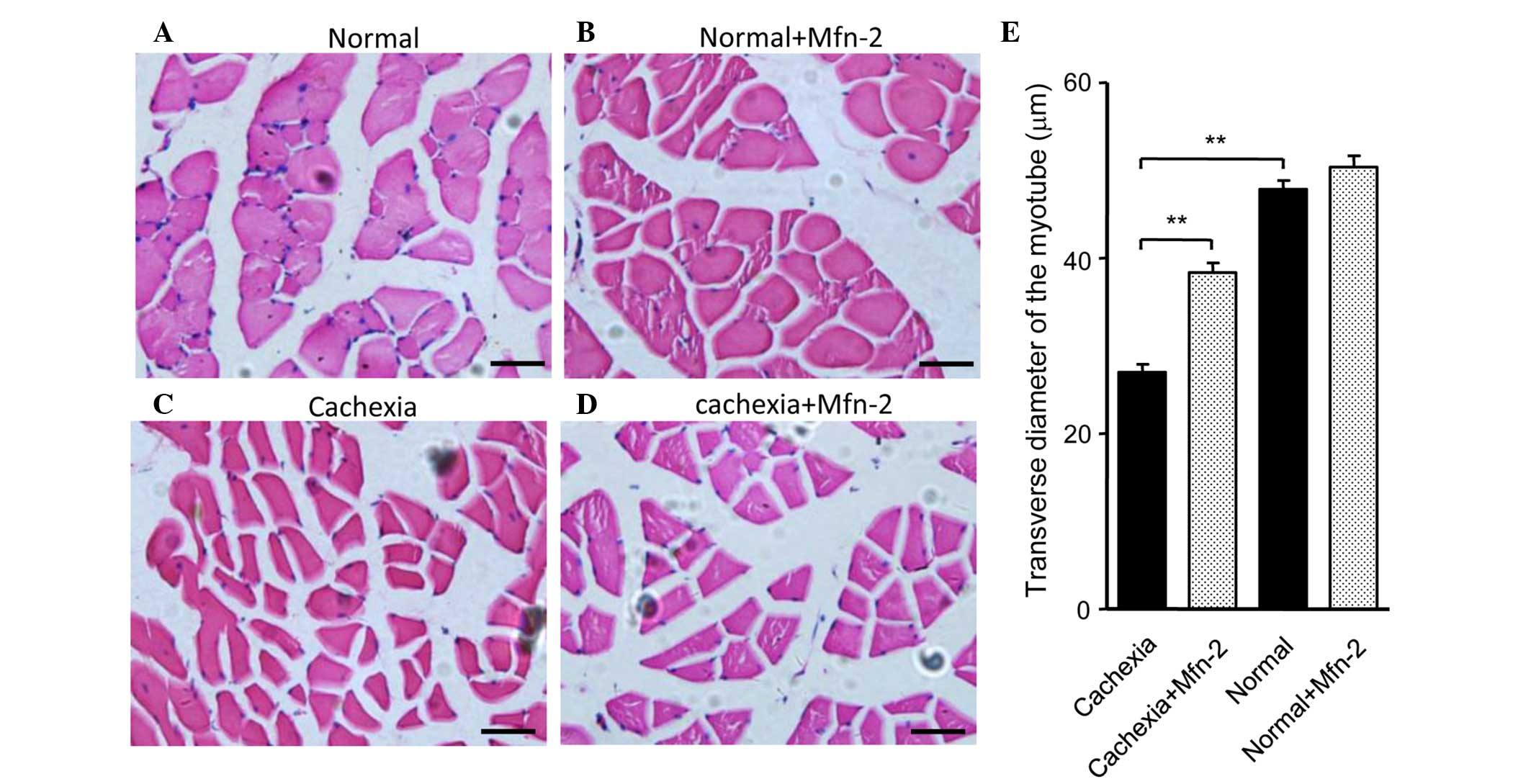
(P<0.05; Fig. 5C). HE staining of
gastrocnemius muscle tissue sections indicated that the morphology
of the control and control + Mfn2 gastrocnemius muscle was normal
(Fig. 6A and B). Conversely, staining
of the gastrocnemius muscle from mice with cachexia showed obvious
atrophy of the muscle (Fig. 6C).
Overexpression of Mfn2 partially ameliorated the
cachexia-associated morphological changes of the muscle (Fig. 6D). The transverse diameters of the
myotube were measured and are shown in Fig. 6E. It was observed that the transverse
diameters of the myotube of the gastrocnemius muscles in mice with
cachexia were significantly decreased when compared with the
control mice (P<0.01; Fig. 6E).
Notably, treatment of the cachexia mice with Mfn2 lentivirus
significantly increased the transverse diameters of the myotube
compared with the untreated cachexia mice (P<0.01; Fig. 6E).
Discussion
Cancer cachexia remains a leading cause of morbidity
and mortality worldwide, despite extensive research and clinical
trials, and is identified in ≤80% of upper gastrointestinal cancer
patients and 60% of lung cancer patients at the time of diagnosis
(5). The prominent clinical feature
of cancer cachexia is the continuous loss of skeletal muscle that
cannot be fully reversed by conventional nutritional support, and
which leads to progressive functional impairment (2). However, the underlying mechanism remains
largely unknown. The present study demonstrated that Mfn2 was
downregulated in the gastrocnemius muscles of patients with
gastrointestinal cancer, which was associated with weight loss.
In vitro cell tests and in vivo animal experiments
suggested that overexpression of Mfn2 was able to reverse
cachexia-induced muscle wasting. These results suggested that Mfn2
has an important role in skeletal muscle loss in patients with
cancer cachexia, and that Mfn2 may be a novel target for prevention
of skeletal muscle loss caused by cachexia.
Loss of skeletal muscle is the major symptom of
cancer cachexia, which leads to reduced mobility, a low quality of
life and a decreased life expectancy (5). The exact mechanism by which cancer
cachexia causes skeletal muscle loss is poorly understood. A
previous study suggested that inflammatory cytokines such as TNF-α,
interferon (IFN)-γ and interleukin (IL)-6 likely have a role in
muscle loss during cancer cachexia (25). In addition, dysfunction of the
mitochondria of skeletal muscle may also explain the muscle loss
(26). Mfn2 has an important role in
the regulation of mitochondrial function (14,15).
Therefore, we hypothesized that Mfn2 may have a role in muscle
wasting caused by cachexia. In order to investigate this, the
expression levels of Mfn2 in the rectus abdominis of cancer
patients with cachexia were determined. It was shown that Mfn2 was
downregulated in the rectus abdominis, which was associated with
the severity of muscle loss in these patients. These results
suggested that downregulation of Mfn2 expression is associated with
the progression of muscle wasting in patients with cancer cachexia.
The in vitro cell experiments showed that a reduction in
Mfn2 expression was observed in TNF-α-induced C2C12 myotube
atrophy, and in vivo animal tests indicated that loss of
Mfn2 was associated with gastrocnemius muscle mass loss in
tumor-bearing mice. Notably, it was demonstrated that
overexpression of Mfn2 in C2C12 cells was able to reverse
TNF-α-induced myotube atrophy, and re-introduction of this gene
into mice gastrocnemius could reverse cachexia-induced muscle
wasting. Together, these results suggested that Mfn2 has a critical
role in skeletal muscle wasting in cancer cachexia.
The results of the present study suggested that Mfn2
has a crucial role in muscle wasting during cachexia; however, the
underlying mechanism was unclear. The loss of fusion proteins may
predispose the mitochondria to undergo fragmentation (27). Mfn2 participates in mitochondrial
fusion and contributes to the maintenance and operation of the
mitochondrial network; thus it is possible that a decrease in the
expression of Mfn2 in muscle cells may lead to mitochondrial
dysfunction and thereby muscle wasting. In the present study, a
reduction in the expression levels of COX IV and Cyto C was
observed concomitantly with the downregulation of Mfn2. COX IV and
Cyto C are proteins of the mitochondrial respiratory chain and
their loss results in reduced ATP formation and mitochondrial
dysfunction (11). Therefore, it is
possible that cancer cachexia induces the downregulation of Mfn2 in
skeletal muscle cells, leading to mitochondrial dysfunction and
thereafter muscle mass loss.
Previous studies have reported that inflammatory
cytokines, such as TNF-α, IFNγ and IL-6, likely have a role in the
occurrence of muscle wasting in patients with cancer cachexia
(11,22,23,25). In
addition, increased levels of inflammatory cytokines have been
observed in patients with cancer cachexia (7,28).
Therefore, it may be hypothesized that inflammatory factors are
involved in the downregulation of Mfn2 expression in cancer
cachexia. In the present study, treatment of C2C12 cells with TNF-α
resulted in the downregulation of Mfn2 protein expression levels.
Additional studies are required to determine the underlying
molecular mechanisms.
In conclusion, the present study demonstrated that
downregulation of Mfn2 was associated with skeletal muscle loss in
cancer cachexia, and that overexpression of Mfn2 was able to
ameliorate this loss. These results suggested that Mfn2 protects
against cachexia-induced muscle loss, and that Mfn2 may be a novel
target for the treatment of cachexia in cancer patients.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81372197).
References
|
1
|
Fearon KC, Voss AC and Hustead DS: Cancer
Cachexia Study Group: Definition of cancer cachexia: Effect of
weight loss, reduced food intake, and systemic inflammation on
functional status and prognosis. Am J Clin Nutr. 83:1345–1350.
2006.PubMed/NCBI
|
|
2
|
Fearon K, Strasser F, Anker SD, Bosaeus I,
Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N,
Mantovani G, et al: Definition and classification of cancer
cachexia: An international consensus. Lancet Oncol. 12:489–495.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bachmann J, Heiligensetzer M,
Krakowski-Roosen H, Büchler MW, Friess H and Martignoni ME:
Cachexia worsens prognosis in patients with resectable pancreatic
cancer. J Gastrointest Surg. 12:1193–1201. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dewys WD, Begg C, Lavin PT, Band PR,
Bennett JM, Bertino JR, Cohen MH, Douglass HO Jr, Engstrom PF,
Ezdinli EZ, et al: Prognostic effect of weight loss prior to
chemotherapy in cancer patients. Eastern Cooperative Oncology
Group. Am J Med. 69:491–497. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Donohoe CL, Ryan AM and Reynolds JV:
Cancer cachexia: Mechanisms and clinical implications.
Gastroenterol Res Pract. 2011:6014342011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bozzetti F, Arends J, Lundholm K,
Micklewright A, Zurcher G and Muscaritoli M: ESPEN: ESPEN
guidelines on parenteral nutrition: Non-surgical oncology. Clin
Nutr. 28:445–454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Deans DA, Tan BH, Wigmore SJ, Ross JA, de
Beaux AC, Paterson-Brown S and Fearon KC: The influence of systemic
inflammation, dietary intake and stage of disease on rate of weight
loss in patients with gastro-oesophageal cancer. Br J Cancer.
100:63–69. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Romanello V, Guadagnin E, Gomes L, Roder
I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz
M, et al: Mitochondrial fission and remodelling contributes to
muscle atrophy. EMBO J. 29:1774–1785. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Soriano FX, Liesa M, Bach D, Chan DC,
Palacín M and Zorzano A: Evidence for a mitochondrial regulatory
pathway defined by peroxisome proliferator-activated receptor-gamma
coactivator-1alpha, estrogen-related receptor-alpha, and mitofusin
2. Diabetes. 55:1783–1791. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang P, Galloway CA and Yoon Y: Control
of mitochondrial morphology through differential interactions of
mitochondrial fusion and fission proteins. PLoS One. 6:e206552011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
White JP, Baltgalvis KA, Puppa MJ, Sato S,
Baynes JW and Carson JA: Muscle oxidative capacity during
IL-6-dependent cancer cachexia. Am J Physiol Regul Integr Comp
Physiol. 300:R201–R211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shen T, Zheng M, Cao C, Chen C, Tang J,
Zhang W, Cheng H, Chen KH and Xiao RP: Mitofusin-2 is a major
determinant of oxidative stress-mediated heart muscle cell
apoptosis. J Biol Chem. 282:23354–23361. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song Z, Ghochani M, McCaffery JM, Frey TG
and Chan DC: Mitofusins and OPA1 mediate sequential steps in
mitochondrial membrane fusion. Mol Biol Cell. 20:3525–3532. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ranieri M, Brajkovic S, Riboldi G, Ronchi
D, Rizzo F, Bresolin N, Corti S and Comi GP: Mitochondrial fusion
proteins and human diseases. Neurol Res Int. 2013:2938932013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kluge MA, Fetterman JL and Vita JA:
Mitochondria and endothelial function. Circ Res. 112:1171–1188.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bach D, Naon D, Pich S, Soriano FX, Vega
N, Rieusset J, Laville M, Guillet C, Boirie Y, Wallberg-Henriksson
H, et al: Expression of Mfn2, the Charcot-Marie-Tooth neuropathy
type 2A gene, in human skeletal muscle: Effects of type 2 diabetes,
obesity, weight loss, and the regulatory role of tumor necrosis
factor alpha and interleukin-6. Diabetes. 54:2685–2693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hernández-Alvarez MI, Thabit H, Burns N,
Shah S, Brema I, Hatunic M, Finucane F, Liesa M, Chiellini C, Naon
D, et al: Subjects with early-onset type 2 diabetes show defective
activation of the skeletal muscle PGC-1{alpha}/Mitofusin-2
regulatory pathway in response to physical activity. Diabetes Care.
33:645–651. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang HS, Chen Y, Fan L, Xi QL, Wu GH, Li
XX, Yuan TL, He SQ, Yu Y, Shao ML, Liu Y, et al: The endoplasmic
reticulum stress sensor IRE1α in intestinal epithelial cells is
essential for protecting against colitis. J Biol Chem.
290:15327–15336. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xue J, Li X, Jiao S, Wei Y, Wu G and Fang
J: Prolyl hydroxylase-3 is down-regulated in colorectal cancer
cells and inhibits IKKbeta independent of hydroxylase activity.
Gastroenterology. 138:606–615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eura Y, Ishihara N, Yokota S and Mihara K:
Two mitofusin proteins, mammalian homologues of FZO, with distinct
functions are both required for mitochondrial fusion. J Biochem.
134:333–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yaffe D and Saxel O: Serial passaging and
differentiation of myogenic cells isolated from dystrophic mouse
muscle. Nature. 270:725–727. 1977. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Reid MB and Li YP: Tumor necrosis
factor-alpha and muscle wasting: A cellular perspective. Respir
Res. 2:269–272. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
De Larichaudy J, Zufferli A, Serra F,
Isidori AM, Naro F, Dessalle K, Desgeorges M, Piraud M, Cheillan D,
Vidal H, et al: TNF-α- and tumor-induced skeletal muscle atrophy
involves sphingolipid metabolism. Skelet Muscle. 2:22012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu Z, Puigserver P, Andersson U, Zhang C,
Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC and
Spiegelman BM: Mechanisms controlling mitochondrial biogenesis and
respiration through the thermogenic coactivator PGC-1. Cell.
98:115–124. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tisdale MJ: Wasting in cancer. J Nutr.
129:(1S Suppl). S243–S246. 1999.
|
|
26
|
Argilés JM, Busquets S, Stemmler B and
López-Soriano FJ: Cancer cachexia: Understanding the molecular
basis. Nat Rev Cancer. 14:754–762. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sugioka R, Shimizu S and Tsujimoto Y:
Fzo1, A protein involved in mitochondrial fusion, inhibits
apoptosis. J Biol Chem. 279:52726–52734. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deans C and Wigmore SJ: Systemic
inflammation, cachexia and prognosis in patients with cancer. Curr
Opin Clin Nutr Metab Care. 8:265–269. 2005. View Article : Google Scholar : PubMed/NCBI
|