Introduction
The epidermal growth factor receptor (EGFR) family
consists of four homologous members, including: EGFR, also named
ErbB1 or human EGRF 1 (HER1); ErbB2 or HER2/Neu; ErbB3/HER3; and
ErbB4/HER4 (1). These receptors
contain an extracellular ligand-binding site, a hydrophobic
membrane-spanning region and an intracellular tyrosine
kinase-containing domain (2). Ligand
binding promotes EGFR dimerization and activates intrinsic protein
kinase activity, initiating cascades of cytoplasmic signal
transduction pathways, including the mitogen-activated protein
kinase/extracellular signal-regulated kinase (MAPK/Erk) and the
phosphoinositide-3-kinase/protein kinase B (PI3K/Akt) signaling
pathways. Activation of these pathways has been demonstrated to
regulate fundamental cellular processes, including cell
proliferation and survival (3).
Lung cancer is one of the most frequently diagnosed
types of cancer among males and females in China and in Western
countries, and it remains the leading cause of cancer mortality in
the USA, with estimated 222,520 new cases and 157,300 mortalities
in 2010 (4). Non-small cell lung
cancer (NSCLC) accounts for 85% of all lung cancer cases (5). Chemotherapy provides symptomatic control
and a moderate improvement in survival; however, prognosis is
relatively poor, with a median time for progression ranging between
3 and 5 months (6). Targeted
therapies, such as those based on the EGFR family, appear to
provide the most promising solution for lung cancer patients in the
future.
The EGFR family is implicated in the pathogenesis of
numerous malignant diseases, and its members are commonly
overexpressed in NSCLC, making them an attractive target for lung
cancer therapy. One of the representative agents in molecular
targeting is the tyrosine kinase inhibitor (TKI), an oral
nonpeptide anilinoquinazoline compound that competes with ATP
binding and blocks EGFR tyrosine kinases (7). Two large randomized phase II studies
(IDEAL1 and IDEAL2) in pretreated patients with advanced NSCLC have
demonstrated clinically significant benefits of gefitinib, an EGFR
TKI, in symptom improvement and tumor regression, as well as
tolerable toxicity mostly associated with skin rash and diarrhea
(8,9).
However, the emergence of mutations, such as T790M in the kinase
domain of EGFR, which is observed in ~50% of patients with acquired
resistance, has become a clinical concern (10).
Determining the prognosis of NSCLC patients is
challenging, thus identifying novel biological markers that can
predict the therapeutic outcome of gefitinib is urgent. Mutations
of the K-ras and EGFR have been shown to correlate with sensitivity
to EGFR TKIs in NSCLC (11,12). An intensive crosstalk exists among the
EGFR family members. However, in terms of HER2, there are
conflicting results regarding whether HER2 expression is correlated
with the susceptibility of tumor cells to gefitinib. Cappuzzo et
al demonstrated that increased HER2 gene copy number was
significantly associated with gefitinib sensitivity in a cohort of
EGFR-positive NSCLC patients (13).
By contrast, HER2 amplification was regarded as an unrecognized
mechanism of acquired resistance that occurs in a subset of tumors
lacking the EGFR T790M mutation (14). Therefore, the impact of HER2
dysregulation on gefitinib sensitivity in tumor cells harboring the
EGFR T790M mutation remains unclear.
The present study examined two human NSCLC cell
lines, H1975 and H1299, which harbor a low endogenous level of HER2
and are resistant to gefitinib treatment. Mutational analysis
revealed the presence of the L858R and T790M EGFR mutations in the
H1975 cell line, whereas no mutations were detected in the EGFR TK
domain and the K-ras gene in H1299 cells. In addition, the study
investigated whether an increase in HER2 expression was able to
alter cell sensitivity to gefitinib. To explore the underlying
mechanism, the phosphorylation of HER3 and the activation of HER2
downstream effectors, as observed by the phosphorylation of Akt and
Erk1/2, were also examined.
Materials and methods
Materials
Gefitinib was provided by AstraZeneca (Macclesfield,
UK). Antibodies against EGFR (ab52894; 1:1,000), HER2 (ab134182;
1:10,000), HER3 (ab32121; 1:1,000), Erk1/2 (ab196883; 1:1,000), Akt
(ab32505; 1:5,000); phospho-Akt (Ser473) (ab81283; 1:5,000),
phospho-HER3 (ab5470; 1:500) and β-actin (ab6276; 1:5,000) were
obtained from Epitomics (Abcam, Cambridge, MA, USA).
Anti-phospho-Erk1/2 (Thr202/Tyr204) (9101S; 1:1,000) was purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Cell culture and stable
transfections
The cell lines H1975 and H1299 (Cell Bank of the
Chinese Academy of Sciences, Shanghai, China) were cultured in
RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS;
both from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
HER2-transfected H1975 and H1299 cells were cultured in RPMI-1640
medium supplemented with 10% FBS and G418 (250 and 200 µg/ml,
respectively). Cells were maintained in a humidified chamber at
37°C with 5% carbon dioxide.
A HER2 expression plasmid pcDNA3.1 was provided by
Dr Yikang Shi (National Glycoengineering Research Center, Shandong
University, Jinan, China). The full-length human HER2 cDNA (GenBank
no., M11730.1) was sequence confirmed, digested by restriction
enzymes NheI and XhoI, and ligated into the
expression vector pIRES2-EGFP (Clontech; Takara Bio, Inc., Mountain
View, CA, USA). Stable transfections were performed using
Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Subsequent to transfection, the vector-transfected and
HER2-expressing H1975 and H1299 cell lines were exposed for 2–3
weeks in G418-containing medium (with 250 and 200 µg/ml G418,
respectively). The pIRES2-EGFP-HER2 transfectant, mock vector
transfectant and the parent cells of the two cell lines were named
as follows: H1975/HER2, H1975/mock and H1975/parent, respectively;
H1299/HER2, H1299/mock and H1299/parent, respectively. HER2 protein
expression was determined by western blot analysis.
MTT assay
Following hemocytometer counting, the cells were
plated in triplicate at a density of 3,500 cells/well in 96-well
plates. The cells were allowed to attach overnight on the dishes
and were treated the following day with serially diluted
concentrations of gefitinib. The concentrations of gefitinib were
serially diluted (40, 20, 10, 5, 2.5 and 1.25 µM). Following 2 days
of incubation with gefitinib, 20 µl of 5 mg/ml MTT solution [also
known as 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide; Sigma-Aldrich, Oakville, ON, USA] in phosphate-buffered
saline (PBS) was added to each well and incubated under standard
cell culture conditions for 4 h. At the end of the assay, the
crystals were dissolved in 150 µl dimethyl sulfoxide (Sangon
Biotech Co., Ltd., Shanghai, China) and vibrated slightly for 10
min. The optical density (OD) of each well was measured with an
ELISA plate reader at 570 nm, and the half maximal inhibitory
concentration (IC50) was determined. The results are
presented as the mean ± standard deviation of triplicate
experiments and are shown as a percentage of untreated control
cells.
Molecular analyses of the status of
EGFR and K-ras genes
The mutation status of EGFR (exons 18–21) and K-ras
(exons 12 and 13) were analyzed by polymerase chain reaction (PCR)
amplification of genomic DNA and direct sequencing. Genomic DNA was
extracted from the H1975 and H1299 cell lines using a DNA
extraction kit (catalog no. #D0063; Beyotime Institute of
Biotechnology, Hangzhou, China), according to the manufacturer's
protocol. Amplification of EGFR and K-ras was performed using the
primers listed in Table I. PCR was
performed in a total volume of 50 µl, in a mixture containing 5 µl
Takara 10X Ex Taq buffer (Mg2+ Plus), 4 µl Takara dNTP
mixture (2.5 mM each), 0.25 µl Takara Ex Taq DNA polymerase (1.25
U), 0.5 µg DNA, and 1 µl of each primer pair for EGFR and K-ras.
Initial denaturation was conducted at 95°C for 2 min, followed by
30 cycles of PCR program, which consisted of denaturation at 94°C
for 30 sec, specific annealing temperature (50–61°C) for 30 sec,
and extension at 72°C for 1 min. A final extension step was
performed at 72°C for 10 min. The samples were subjected to
sequence analysis using a cycle sequencing kit (Applied Biosystems;
Thermo Fisher Scientific, Inc.) and the 3730XL DNA Analyzer
(Applied Biosystems; Thermo Fisher Scientific, Inc.).
 | Table I.Primers used for amplification of
genomic DNA sequences of EGFR and K-ras exons. |
Table I.
Primers used for amplification of
genomic DNA sequences of EGFR and K-ras exons.
| Gene | Forward | Reverse |
|---|
| K-ras |
|
|
| Exon
2 |
5′-CTTAAGCGTCGATGGAGGAG-3′ |
5′-CCCTGACATACTCCCAAGGA-3′ |
| Exon
3 |
5′-TGGGTATGTGGTAGCATCTCA-3′ |
5′-AATCCCAGCACCACCACTAC-3′ |
| EGFR tyrosine
kinase |
|
|
| Exon
18 |
5′-CAAATGAGCTGGCAAGTGCCGTGT-3′ |
5′-GAGTTTCCCAAACACTCAGTGAAA-3′ |
| Exon
19 |
5′-GCAATATCAGCCTTAGGTGCGGCTC-3′ |
5′-CATAGAAAGTGAACATTTAGGATGTG-3′ |
| Exon
20 |
5′-ACTTCACAGCCCTGCGTAAAC-3′ |
5′-ATGGGACAGGCACTGATTTGT-3′ |
| Exon
21 |
5′-CTAACGTTCGCCAGCCATAAGTCC-3′ |
5′-GCTGCGAGCTCACCCAGAATGTCTGG-3′ |
Western blot analysis
The monolayer culture cells were maintained on 100
mm dishes in medium supplemented with 10% FBS. Cells were exposed
to gefitinib at increasing concentrations (0, 1 and 10 µm) for 1 or
24 h at 37°C. Subsequently, the cells were rinsed with ice-cold PBS
and lysed in mammalian protein extraction reagent (Pierce; Thermo
Fisher Scientific, Inc.) containing phosphatase inhibitor cocktail
(Pierce; Thermo Fisher Scientific, Inc.) for 10–20 min on ice.
Protein concentration was determined using a BCA reagent (Pierce;
Thermo Fisher Scientific, Inc.). Equal amounts of each protein
sample (30 µg) were separated by electrophoresis on 8–12%
SDS-polyacrylamide gels and transferred to polyvinylidene
difluoride Hybond-P membranes (GE Healthcare, Chicago, IL, USA).
Following transfer, the blots were incubated with the blocking
solution and probed with antibodies against EGFR, HER2, HER3,
Erk1/2, Akt, phospho-Akt (Ser473), phospho-Erk1/2 (Thr202/Tyr204),
phospho-HER3 and β-actin, followed by successive washes with
Tris-buffered saline. An enhanced chemiluminescence kit (catalog
no. KGP1121-KGP1126; KeyGen Biotech Co., Ltd., Nanjing, China) was
used for detection. All experiments were performed in duplicate and
provided similar results.
Results
Sensitivity of lung cancer cell lines
to gefitinib
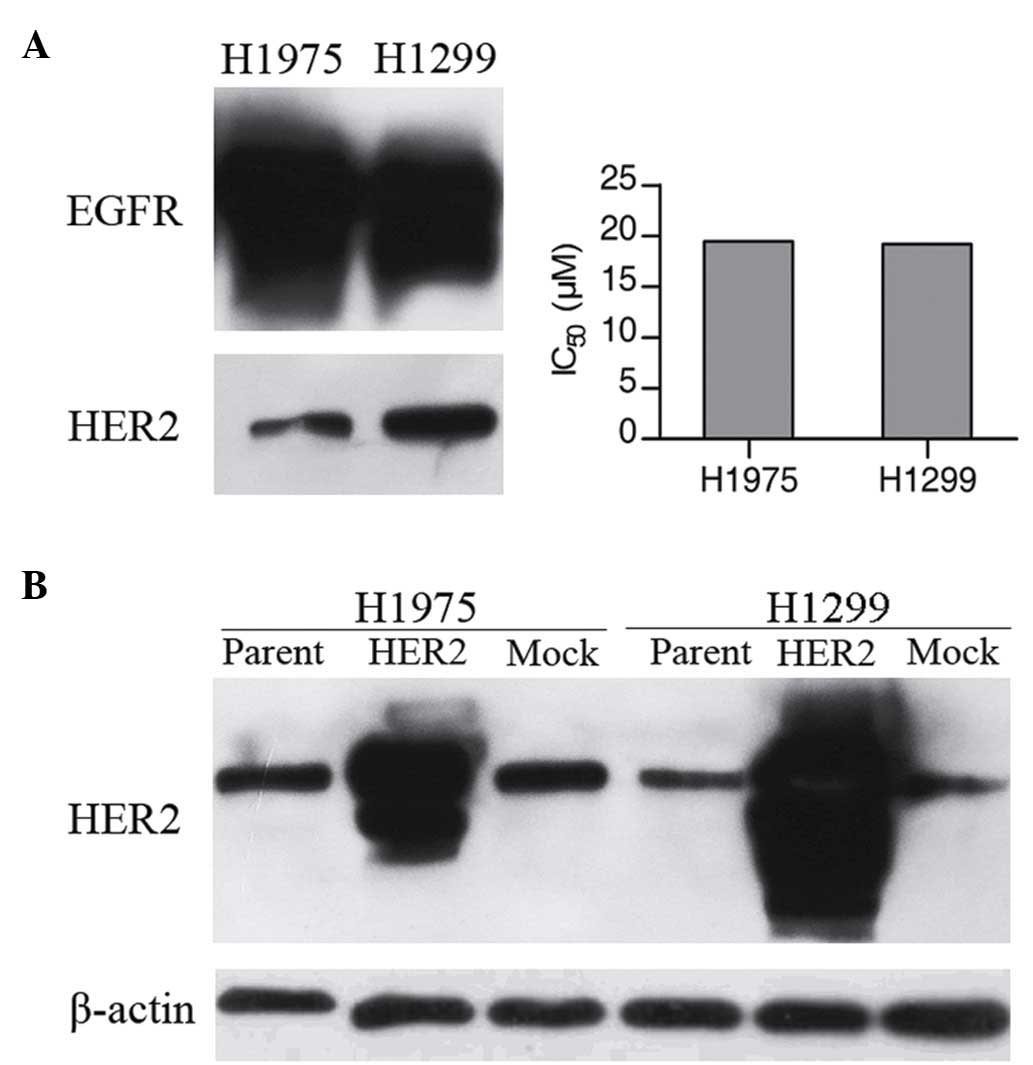
To determine the sensitivity of H1975 and H1299
cells to gefitinib, the cell viability upon treatment with
different concentrations of gefitinib was examined by MTT assay
(Fig. 1A). IC50 values
were calculated using GraphPad Prism software version 3.0 (GraphPad
Software, Inc., La Jolla, CA, USA). The two cell lines were
observed to be resistant of gefitinib, with IC50 values
of ~20 µm. The results of western blot analysis verified the
comparable expression levels of EGFR and HER2 in these two cell
lines (Fig. 1A).
Molecular analysis
Mutations in EGFR and K-ras have been identified in
lung adenocarcinomas and these mutations are correlated with
sensitivity of gefitinib (15). Since
resistance to gefitinib in the two cell lines investigated in the
present study was observed, the EGFR (exons 18–21) and K-ras (exons
12 and 13) mutation status was examined by direct sequencing.
Mutational analysis revealed the presence of the L858R and T790M
EGFR mutation in the H1975 cell line. However, no mutations were
detected in the EGFR TK domain and the K-ras gene in H1299 cells
(Table II).
| Table II.Characteristics of non-small cell
lung cancer cell lines. |
Table II.
Characteristics of non-small cell
lung cancer cell lines.
| Cell line | EGFR status | K-ras status |
|---|
| H1975 | L858R/T790M | Wild type |
| H1299 | Wild type | Wild type |
Overexpression of the HER2 enhances
the sensitivity of the H1975 cell line to gefitinib
The present study next investigated whether
increased expression of HER2 was able to alter the sensitivity of
cells to gefitinib. Therefore, HER2 expression plasmids were
introduced into the two lung cancer cell lines. The HER2 stable
transfectants (H1975/HER2 and H1299/HER2) exhibited higher HER2
protein expression levels compared with the corresponding parental
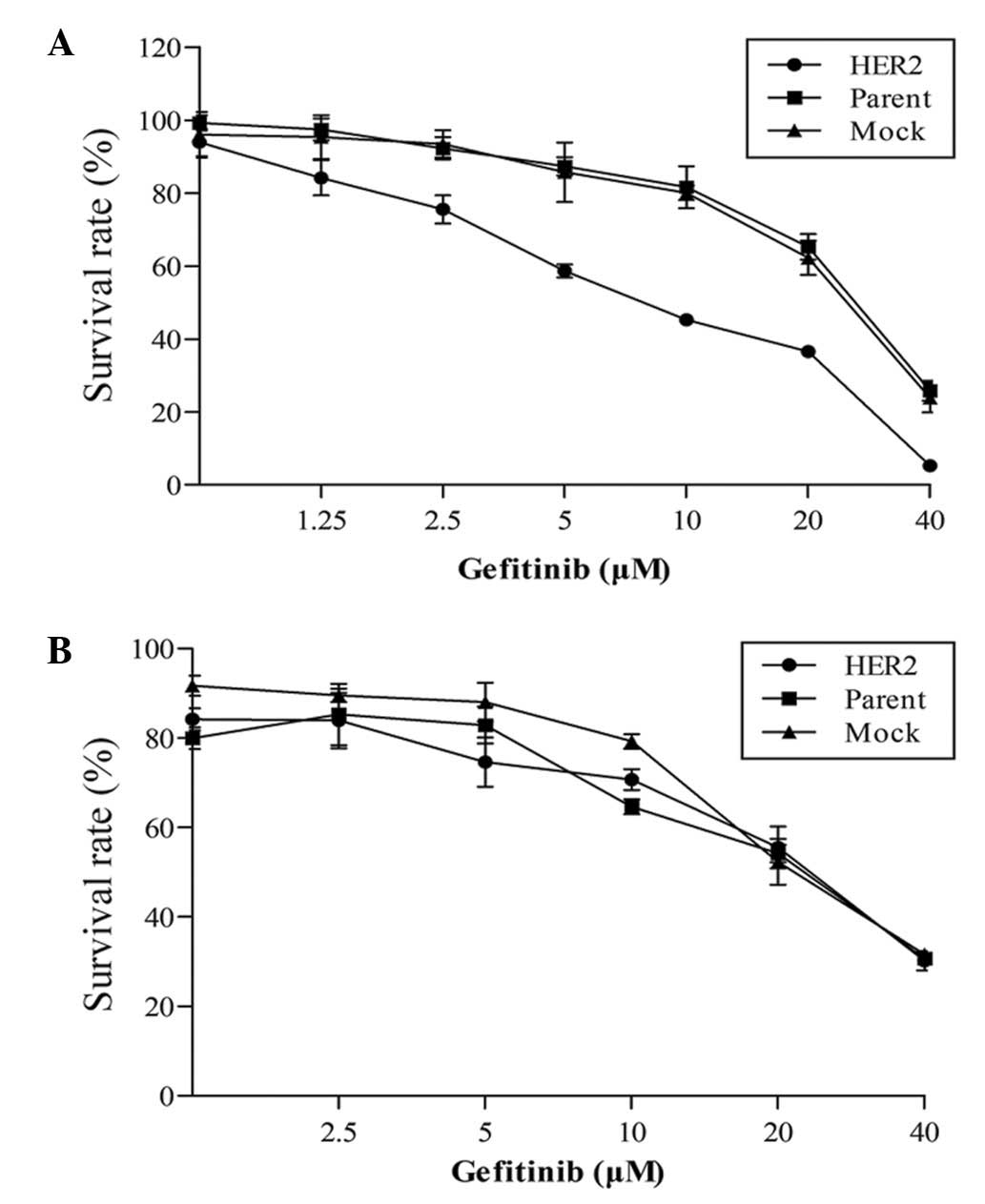
cell lines or with the mock transfectants (Fig. 1B). An MTT assay was then performed in
the presence of increasing doses of gefitinib. Upon HER2
overexpression, H1975/HER2 cells displayed a 3-4-fold higher
sensitivity to gefitinib (IC50 at 6.19 µm compared with
~20 µm in mock or parental cells). On the contrary, HER2
overexpression in H1299 cells did not significantly affect
gefitinib sensitivity (Fig. 2).
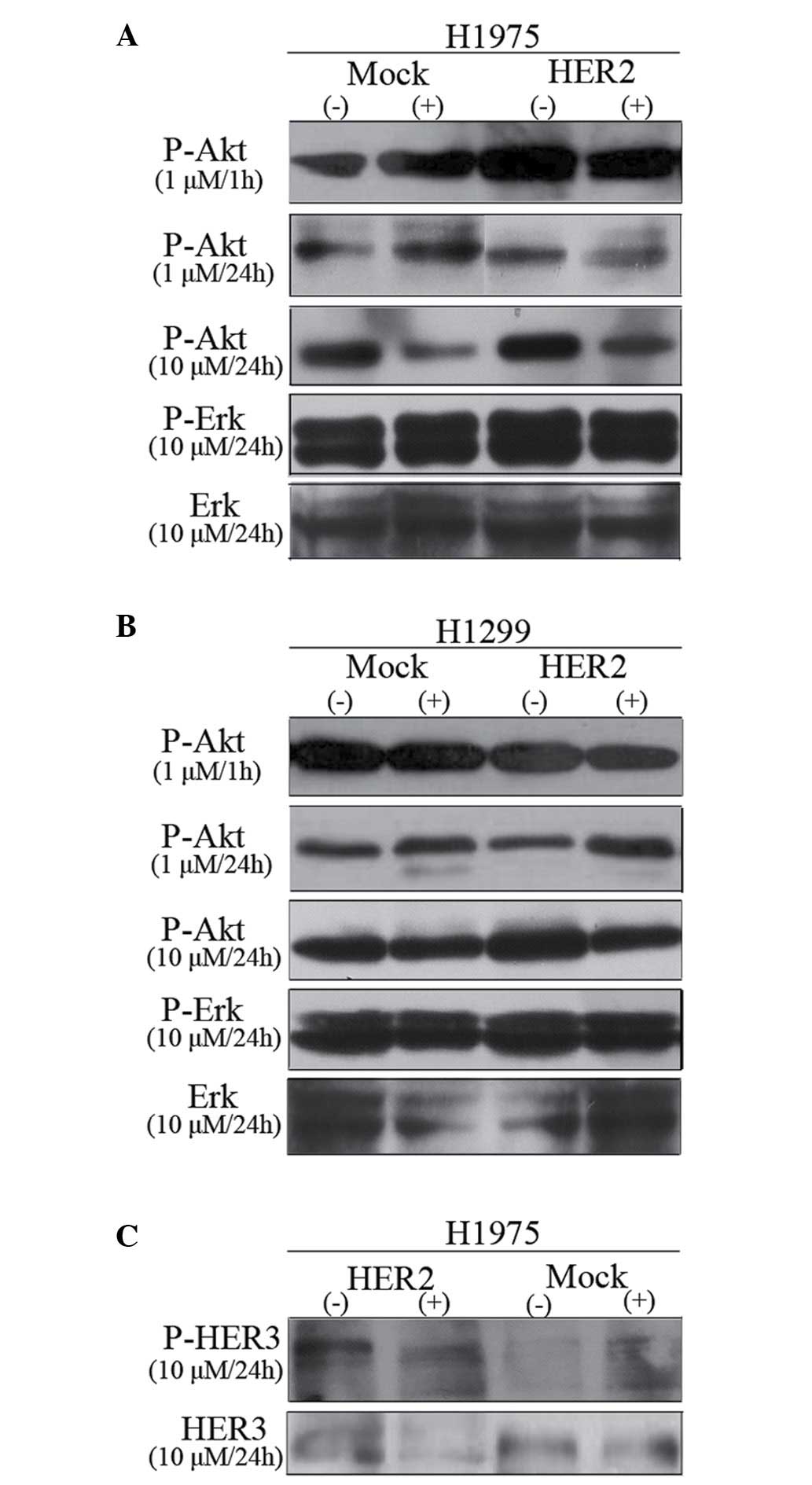
Effect of gefitinib on phospho-Akt
(Ser473), phospho-Erk1/2 (Thr202/Tyr204) and phospho-HER3
Fig. 3A and B shows
the inhibitory dose kinetics of gefitinib for phospho-Akt and
phospho-Erk in H1975 and H1299 cells, with the phosphorylation
level in untreated cells normalized to 100%. The results of the
present study identified that gefitinib treatment effectively
decreased Akt phosphorylation in a dose- and time-dependent manner
in EGFR-mutant H1975 cell line. In addition, gefitinib inhibited
Akt phosphorylation equally in H1299/HER2 or H1299/mock cells. By
contrast, Erk1/2 phosphorylation was not altered by gefitinib
treatment up to 10 µm in both H1975 and H1299 cells.
The effect of gefitinib on phospho-HER3 in H1975
cell lines in the presence of 10% FBS was also assayed (Fig. 3C). Phospho-HER3 was increased by
~2-fold in H1975/HER2 in the absence of gefitinib treatment.
However, treatment with 10 µm gefitinib for 24 h evidently reduced
HER3 phosphorylation. In conclusion, HER2 overexpression was able
to sensitize H1975 cells to gefitinib, which stimulated HER2-driven
signaling cascades accompanied by the activation of Akt.
Discussion
EGFR and/or HER2 are expressed at high levels in a
wide spectrum of malignancies, including lung cancer, breast
cancer, esophageal carcinoma, ovary cancer and gliomas (16,17). Their
overexpression usually correlates with a more aggressive disease
course and poor clinical prognosis (18). Gefitinib, a tyrosine kinase inhibitor,
can compete with ATP, as well as inhibit EGFR and HER2 kinase
activities in vitro with an IC50 of 27–33 nmol/l
and >3.7 µmol/l, respectively (19). Although gefitinib exerts its antitumor
activity selectively through the inhibition of EGFR, recent reports
have demonstrated that the antitumor activity of gefitinib does not
directly depend on EGFR (12,20). In addition, no apparent difference in
sensitivity to gefitinib was observed between the human lung cancer
line LK2 and its EGFR derivatives (LK2/EGFR) (21). On the contrary, according to several
retrospective studies of patients with advanced NSCLC, increased
HER2 gene copy number was statistically significantly associated
with response to gefitinib treatment (13,22).
Furthermore, the impact of HER2 expression level on gefitinib
sensitivity has also been reported in a series of human breast
cancer and epithelial cell lines (19,23). Taken
together, these results suggested that HER2 levels appeared to be
correlated with the susceptibility of tumor cells to gefitinib.
However, a study by Takezawa et al indicated
that HER2 amplification is a mechanism of acquired resistance to
EGFR TKI in EGFR-mutant tumor cell lines that lack the second-site
EGFR T790M mutation (14). The impact
of HER2 dysregulation on gefitinib sensitivity in tumor cells
harboring the T790M mutation remains unclear. In the present study,
mutational analysis revealed the presence of the L858R (T→G
mutation in exon 21) and T790M (C→T mutation in exon 20) EGFR
mutations in the H1975 cell line, whereas no mutation was detected
in both the EGFR TK domain and the K-ras gene in H1299 cells. The
H1975 and H1299 cell lines were selected as cell models to examine
whether an increase in HER2 expression level was able to alter the
cell sensitivity to gefitinib in cells harboring the EGFR T790M
mutation. The NSCLC cell line H1975 with HER2 overexpression
presented increased sensitivity to gefitinib with an
IC50 of ~6 µm, compared with the parental cell line that
had an IC50 of 20 µm. The discrepancies between these
values and the maximum plasma concentration of gefitinib observed
in patients (~1 µm) may be due to the different exposure time to
gefitinib (24). The results of the
present study strongly suggest that overexpression of HER2 may
overcome T790M-mediated resistance. Notably, the relatively
resistance of lung cancer cell line H1975 to gefitinib can be
reversed by the overexpression of HER2. Therefore, the expression
of HER2 may also be employed as a prognostic marker for lung cancer
patients, particularly those harboring the T790M mutation in the
kinase domain of EGFR.
HER2 mutations within the tyrosine kinase domain are
also considered to be associated with responsiveness of EGFR-TKIs
in NSCLC and squamous cell carcinoma of the head and neck (25,26).
Prospective studies are required to confirm the role of HER2 in
NSCLC and other tumor types as a prognostic marker for tumor
responsiveness, in addition to mutations of the EGFR and K-ras.
It has been suggested that breast cancer cells with
HER2 amplification were selectively dependent on Akt signaling and
downregulation of Akt activity was a marker of the antitumor effect
of gefitinib (23). In a study by
Noro et al, cancer cell lines showing sensitivity to
gefitinib also exhibited greater phospho-Akt levels in a panel of
lung cancer cell lines (24). Data
presented in the current study indicated that overexpression of
HER2 in H1975 cells resulted in a marked inhibitory effect of
gefitinib on Akt phosphorylation, but not on phospho-Erk1/2. Akt
activity was demonstrated to promote resistance to chemotherapy and
radiotherapy (27). This may provide
an explanation for the combination of gefitinib with chemotherapy
or radiotherapy as an effective strategy in phospho-Akt positive
patients who are refractory to standard chemotherapy regimens
(28). Noro et al also assumed
that phosphorylation of Akt may be just a hallmark of sensing
activation of other upstream molecules, such as HER2 (24). Therefore, phospho-Akt is a potential
surrogate marker of the response to gefitinib, and precise
assessment of clinical markers of Akt activation may help for
efficient prognosis of gefitinib treatment.
The data of the current study revealed that a
significant reduction in the levels of activation of Akt was
required for gefitinib to induce cell death in the EGFR-mutant
H1975 cell line. PI3K docking sites are absent on EGFR or HER2,
however, docking sites for PI3K are highly prevalent on HER3
(29). It is noteworthy that HER2 is
an orphan receptor for which no ligand has yet been identified,
while the kinase activity of HER3 is defective (30). In this regard, EGFR and HER2 are not
able to directly activate the PI3K/Akt pathway (20). In these tumors, activation of the
PI3K/Akt pathway occurs through the transphosphorylation of HER3.
Furthermore, HER2 has been identified as the preferred binding
partner of the other ErbB receptors (31). Thus, HER2 and HER3 can form
heterodimeric complexes and act in a complementary manner, and the
HER2/HER3 heterodimer is considered the most transforming and
mitogenic receptor complex (32).
Treatment with gefitinib can most significantly reduce the activity
of the HER2/HER3 heterodimer and block the downstream signaling
(20). In the present study, it was
observed that phospho-HER3 was 2-fold higher in H1975/HER2 compared
with that in the H1975/mock cells in the absence of drug.
Furthermore, HER2 overexpression leads to more HER2 heterodimers,
which increase their ligand binding affinity and result in more
prolonged signals (33). These
findings can thus account for the increase of HER2/HER3 heterodimer
and HER2/HER2 homodimer levels in the HER2-overexpressing H1975
cell line. Therefore, gefitinib may downregulate the phospho-Akt
more effectively in H1975/HER2 cells due to an increased number of
available target sites. Chandarlapaty et al reported that
PI3K/Akt inhibitors caused a feedback activation of receptor
tyrosine kinase signaling, and simultaneous inhibition of Akt and
HER kinase activity is more effective compared with single
treatment (34). Hence, combined
inhibition of Akt and EGFR family members may warrant further
evaluation in clinical trials of NSCLC patients.
Restoration of p53 to p53-defective H1299 cells can
enhance the sensitivity to gefitinib (35). In the present study, the resistant of
H1299 cells to gefitinib could not be reversed by the ectopic
overexpression of HER2. The present study observations were
consistent with the hypothesis of Strobel et al (36) that an intact apoptotic pathway was
necessary for chemotherapy-induced cytotoxicity. Furthermore, the
lack of p53 gene affected the ability of a cell to undergo
apoptosis and finally led to the development of chemotherapy
resistance (36). Thus, the
hypothesis that HER2 overexpression per se is sufficient to
determine sensitivity to gefitinib may be over-simplistic.
The objective response rate of gefitinib was ~15%
when administered to lung cancer patients (37). However, the underlying mechanism of
different drug sensitivity to gefitinib is not fully understood. In
the present study, stable lung cancer cell lines of H1975 and H1299
overexpressing HER2 have been established. The data indicated that
the sensitivity of H1975 cells to gefitinib could be restored by
overexpression of HER2, which stimulated HER2-driven signaling
cascades, accompanied by Akt activation. By contrast, ectopic HER2
overexpression in H1299 cells did not significantly alter the
sensitivity to gefitinib. The present study demonstrated that a
more comprehensive analysis is required to maximize the clinical
benefits of gefitinib. Future studies should include potential
clinical markers, such as p53, HER2, EGFR, the phosphorylation
state of HER3 and Akt, along with other molecules that are
associated with gefitinib sensitivity.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81201811).
References
|
1
|
Jorissen RN, Walker F, Pouliot N, Garrett
TP, Ward CW and Burgess AW: Epidermal growth factor receptor:
Mechanisms of activation and signalling. Exp Cell Res. 284:31–53.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hynes NE and Lane HA: ERBB receptors and
cancer: The complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arora A and Scholar EM: Role of tyrosine
kinase inhibitors in cancer therapy. J Pharmacol Exp Ther.
315:971–979. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xue C, Hu Z, Jiang W, Zhao Y, Xu F, Huang
Y, Zhao H, Wu J, Zhang Y, Zhao L, et al: National survey of the
medical treatment status for non-small cell lung cancer (NSCLC) in
China. Lung Cancer. 77:371–375. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shepherd FA, Pereira J Rodrigues, Ciuleanu
T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S,
Smylie M, Martins R, et al: Erlotinib in previously treated
non-small-cell lung cancer. N Engl J Med. 353:123–132. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anido J, Matar P, Albanell J, Guzmán M,
Rojo F, Arribas J, Averbuch S and Baselga J: ZD1839, a specific
epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor,
induces the formation of inactive EGFR/HER2 and EGFR/HER3
heterodimers and prevents heregulin signaling in
HER2-overexpressing breast cancer cells. Clin Cancer Res.
9:1274–1283. 2003.PubMed/NCBI
|
|
8
|
Fukuoka M, Yano S, Giaccone G, Tamura T,
Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S,
Rischin D, et al: Multi-institutional randomized phase II trial of
gefitinib for previously treated patients with advanced
non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin
Oncol. 21:2237–2246. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kris MG, Natale RB, Herbst RS, Lynch TJ
Jr, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H,
Sandler A, et al: Efficacy of gefitinib, an inhibitor of the
epidermal growth factor receptor tyrosine kinase, in symptomatic
patients with non-small cell lung cancer: A randomized trial. JAMA.
290:2149–2158. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arcila ME, Oxnard GR, Nafa K, Riely GJ,
Solomon SB, Zakowski MF, Kris MG, Pao W, Miller VA and Ladanyi M:
Rebiopsy of lung cancer patients with acquired resistance to EGFR
inhibitors and enhanced detection of the T790M mutation using a
locked nucleic acid-based assay. Clin Cancer Res. 17:1169–1180.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Landi L, Tiseo M, Chiari R, Ricciardi S,
Rossi E, Galetta D, Novello S, Milella M, D'Incecco A, Minuti G, et
al: Activity of the EGFR-HER2 dual inhibitor afatinib in
EGFR-mutant lung cancer patients with acquired resistance to
reversible EGFR tyrosine kinase inhibitors. Clin Lung Cancer.
15:411–417.e4. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Han SW, Kim TY, Jeon YK, Hwang PG, Im SA,
Lee KH, Kim JH, Kim DW, Heo DS, Kim NK, et al: Optimization of
patient selection for gefitinib in non-small cell lung cancer by
combined analysis of epidermal growth factor receptor mutation,
K-ras mutation, and Akt phosphorylation. Clin Cancer Res.
12:2538–2544. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cappuzzo F, Varella-Garcia M, Shigematsu
H, Domenichini I, Bartolini S, Ceresoli GL, Rossi E, Ludovini V,
Gregorc V, Toschi L, et al: Increased HER2 gene copy number is
associated with response to gefitinib therapy in epidermal growth
factor receptor-positive non-small-cell lung cancer patients. J
Clin Oncol. 23:5007–5018. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Takezawa K, Pirazzoli V, Arcila ME, Nebhan
CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ,
Melnick MA, et al: HER2 amplification: A potential mechanism of
acquired resistance to EGFR inhibition in EGFR-mutant lung cancers
that lack the second-site EGFRT790M mutation. Cancer Discov.
2:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pao W, Wang TY, Riely GJ, Miller VA, Pan
Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG and Varmus HE: KRAS
mutations and primary resistance of lung adenocarcinomas to
gefitinib or erlotinib. PLoS Med. 2:e172005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wei Q, Chen L, Sheng L, Nordgren H, Wester
K and Carlsson J: EGFR, HER2 and HER3 expression in esophageal
primary tumours and corresponding metastases. Int J Oncol.
31:493–499. 2007.PubMed/NCBI
|
|
17
|
Hirsch FR, Varella-Garcia M, Cappuzzo F,
McCoy J, Bemis L, Xavier AC, Dziadziuszko R, Gumerlock P, Chansky
K, West H, et al: Combination of EGFR gene copy number and protein
expression predicts outcome for advanced non-small-cell lung cancer
patients treated with gefitinib. Ann Oncol. 18:752–760. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mendelsohn J and Baselga J: The EGF
receptor family as targets for cancer therapy. Oncogene.
19:6550–6565. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moulder SL, Yakes FM, Muthuswamy SK,
Bianco R, Simpson JF and Arteaga CL: Epidermal growth factor
receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits
HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in
vivo. Cancer Res. 61:8887–8895. 2001.PubMed/NCBI
|
|
20
|
Normanno N, Maiello MR and De Luca A:
Epidermal growth factor receptor tyrosine kinase inhibitors
(EGFR-TKIs): Simple drugs with a complex mechanism of action? J
Cell Physiol. 194:13–19. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ono M, Hirata A, Kometani T, Miyagawa M,
Ueda S, Kinoshita H, Fujii T and Kuwano M: Sensitivity to gefitinib
(Iressa, ZD1839) in non-small cell lung cancer cell lines
correlates with dependence on the epidermal growth factor (EGF)
receptor/extracellular signal-regulated kinase 1/2 and EGF
receptor/Akt pathway for proliferation. Mol Cancer Ther. 3:465–472.
2004.PubMed/NCBI
|
|
22
|
Soh J, Toyooka S, Ichihara S, Fujiwara Y,
Hotta K, Suehisa H, Kobayashi N, Ichimura K, Aoe K, Aoe M, et al:
Impact of HER2 and EGFR gene status on gefitinib-treated patients
with nonsmall-cell lung cancer. Int J Cancer. 121:1162–1167. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moasser MM, Basso A, Averbuch SD and Rosen
N: The tyrosine kinase inhibitor ZD1839 (‘Iressa’) inhibits
HER2-driven signaling and suppresses the growth of
HER2-overexpressing tumor cells. Cancer Res. 61:7184–7188.
2001.PubMed/NCBI
|
|
24
|
Noro R, Gemma A, Kosaihira S, Kokubo Y,
Chen M, Seike M, Kataoka K, Matsuda K, Okano T, Minegishi Y, et al:
Gefitinib (IRESSA) sensitive lung cancer cell lines show
phosphorylation of Akt without ligand stimulation. BMC Cancer.
6:2772006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shigematsu H, Takahashi T, Nomura M,
Majmudar K, Suzuki M, Lee H, Wistuba II, Fong KM, Toyooka S,
Shimizu N, et al: Somatic mutations of the HER2 kinase domain in
lung adenocarcinomas. Cancer Res. 65:1642–1646. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cohen EE, Lingen MW, Martin LE, Harris PL,
Brannigan BW, Haserlat SM, Okimoto RA, Sgroi DC, Dahiya S, Muir B,
et al: Response of some head and neck cancers to epidermal growth
factor receptor tyrosine kinase inhibitors may be linked to
mutation of ERBB2 rather than EGFR. Clin Cancer Res. 11:8105–8108.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brognard J, Clark AS, Ni Y and Dennis PA:
Akt/protein kinase B is constitutively active in non-small cell
lung cancer cells and promotes cellular survival and resistance to
chemotherapy and radiation. Cancer Res. 61:3986–3997.
2001.PubMed/NCBI
|
|
28
|
Han SW, Hwang PG, Chung DH, Kim DW, Im SA,
Kim YT, Kim TY, Heo DS, Bang YJ and Kim NK: Epidermal growth factor
receptor (EGFR) downstream molecules as response predictive markers
for gefitinib (Iressa, ZD1839) in chemotherapy-resistant non-small
cell lung cancer. Int J Cancer. 113:109–115. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lurje G and Lenz HJ: EGFR signaling and
drug discovery. Oncology. 77:400–410. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ricciardi GR, Russo A, Franchina T,
Ferraro G, Zanghì M, Picone A, Scimone A and Adamo V: NSCLC and
HER2: Between lights and shadows. J Thorac Oncol. 9:1750–1762.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Garrido-Castro AC and Felip E: HER2 driven
non-small cell lung cancer (NSCLC): Potential therapeutic
approaches. Transl Lung Cancer Res. 2:122–127. 2013.PubMed/NCBI
|
|
32
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fuller SJ, Sivarajah K and Sugden PH: ErbB
receptors, their ligands, and the consequences of their activation
and inhibition in the myocardium. J Mol Cell Cardiol. 44:831–854.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chandarlapaty S, Sawai A, Scaltriti M,
Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK,
Baselga J and Rosen N: AKT inhibition relieves feedback suppression
of receptor tyrosine kinase expression and activity. Cancer Cell.
19:58–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rho JK, Choi YJ, Ryoo BY, Na II, Yang SH,
Kim CH and Lee JC: p53 enhances gefitinib-induced growth inhibition
and apoptosis by regulation of Fas in non-small cell lung cancer.
Cancer Res. 67:1163–1169. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Strobel T, Swanson L, Korsmeyer S and
Cannistra SA: BAX enhances paclitaxel-induced apoptosis through a
p53-independent pathway. Proc Natl Acad Sci USA. 93:14094–14099.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dudek AZ, Kmak KL, Koopmeiners J and
Keshtgarpour M: Skin rash and bronchoalveolar histology correlates
with clinical benefit in patients treated with gefitinib as a
therapy for previously treated advanced or metastatic non-small
cell lung cancer. Lung Cancer. 51:89–96. 2006. View Article : Google Scholar : PubMed/NCBI
|