Introduction
Colorectal cancer (CRC) has been considered the
third most common malignant tumor in the world, with an increasing
incidence rate (1). As the
development of resection treatment, chemotherapy and radiotherapy
in previous years has improved, the prognosis of patients diagnosed
at an early stage has been enhanced to a certain degree. However,
the prognosis of those with advanced tumors remains poor (2,3). Molecular
biology research has increasingly become a popular avenue for
cancer therapies. Therefore, to additionally understand the
molecular mechanisms of cancer occurrence and progression and to
find effective molecular treatment targets is important for basic
and clinical researchers (4,5).
Tumor necrosis factor receptor (TNFR) is composed of
two different subtypes, TNFR1 and TNFR2, which exhibit 30% homology
at amino-acid level in extracellular, cysteine-rich, and
ligand-binding regions and are encoded by different genes (6). TNFR1 is ubiquitously expressed in
various cells and mediates apoptosis induced by tumor necrosis
factor α (TNF-α) (7). However, TNFR2
is mainly expressed in hematopoietic and endothelial cells and has
been found to be required for anti-inflammation, immunoregulation
(8), protection against LPS-induced
lung damage (9) and bone fracture
healing (10). However, studies about
TNFR2 in tumors are limited and the functional properties of TNFR2
in CRC remain unknown.
In the present study, immunohistochemistry (IHC)
found that TNFR2 was positively associated with Ki67 expression in
CRC tissues; western blot analysis found that TNFR2 promoted Ki67
expression in CRC cells via the phosphoinositide 3-kinase
(PI3K)/protein kinase B (AKT) signaling pathway; methyl thiazolyl
tetrazolium (MTT) assay and clone formation assay also showed that
TNFR2 successfully promoted CRC cell growth and clone formation
abilities via the PI3K/AKT signaling pathway. All the results
confirmed the pro-growth function of TNFR2 in CRC and provided a
new target for CRC treatment.
Materials and methods
Cell lines and culture
Colorectal cancer cell lines SW1116 and HT29 were
purchased from American Type Culture Collection (Manassas, VA,
USA). SW1116 and HT29 cells were cultured in RPMI 1640 medium
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and
McCoy's 5A Medium (Invitrogen; Thermo Fisher Scientific, Inc.),
respectively. The two cell lines were supplemented with 10% fetal
bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.).
Tissue samples
Approved by the review board and ethics committee of
the Second People's Hospital of Dongying (Dongying, China), the
present study selected 90 primary CRC specimens from CRC patients
who had undergone surgery between January 2009 and December 2015 in
The Second People's Hospital of Dongying (Dongying, China). No
patients received chemotherapy, radiotherapy or immunomodulatory
therapy prior to surgery.
IHC
Tissue samples were cut into 4 µm thickness and
deparaffinized in xylene and rehydrated in graded ethanol, then
boiled in 10 mmol/l citrate buffer (pH 6.0) for 3 min at 100°C for
antigen unmasking. The sections were then immersed in 3%
H2O2 for 10 min to block the endogenous
peroxidase and in goat serum blocking solution (ZLI-9022; ZsBio,
Beijing, China) for 15 min to block non-specific antigens at room
temperature. Subsequent to being incubated at room temperature for
2 h in rabbit anti-human polyclonal primary antibodies against
TNFR2 (catalog no. 19272-1-AP; dilution, 1:400; Proteintech Group
Inc., Chicago, IL, USA) and Ki67 (working solution; catalog no.
RB-9043; Maixin Biotech Co., Ltd., Fuzhou, China), the sections
were washed with phosphate-buffered saline (PBS) and incubated in
goat anti-rabbit/mouse IgG H&L (HRP) polymer (working solution)
(catalog no. KIT-9921; Maixin Biotech Co., Ltd.) at room
temperature for 30 min. Finally, slides were stained with
3,3′-diaminobenzidine and counterstained with hematoxylin (catalog
no. DAB-0031; Maixin Biotech Co., Ltd.). Two independent
pathologists, who were blind to clinical parameters and clinical
outcomes of patients, performed the staining analysis. The
percentage of stained cells was recorded at ×400 magnification in
at least 5 random fields. The proportion score represented the
estimated fraction of positive staining tumor cells (0, ≤25%; 1,
26–50%; 2, 51–75%; 3, >75%). The intensity score represented the
estimated average staining intensity of positive tumor cells (0,
negative; 1, weak; 2, moderate; 3, strong). The expression level of
TNFR2 was evaluated using the product of proportion score and
intensity score at five fields and mean value was obtained (≤4 as
low expression, >4 as high expression); the expression level of
Ki67 was directly evaluated using the proportion score.
Transfection
pEZ-M61/green fluorescent protein (GFP)/TNFR2 or
vector control (Shanghai GenePharma Co., Ltd., Shanghai, China) was
transfected into SW1116 cells using HP X-treme GENE HP Reagents
(Roche Diagnostics, Basel, Switzerland) to upregulate TNFR2
expression; p-GPU6/GFP/Neo/TNFR2, including sequence 1 (TNFR2-sh1)
and sequence 2 (TNFR2-sh2), or small hairpin RNA control (Shanghai
GenePharma Co., Ltd.) was transfected into HT29 cells to silence
TNFR2 expression. All procedures were assessed according to the
manufacturer's protocol. Cells were collected for additional study
48 h later.
Western blot analysis
Cells were washed twice in PBS and lysed in
radioimmunoprecipitation assay buffer containing 1% protease
inhibitor (Beijing ComWin Biotech Co., Ltd., Beijing, China).
Protein concentration was determined by spectrophotometer ND-1000
(NanoDrop Technologies; Thermo Fisher Scientific, Inc.). Equal
total protein was separated by sodium dodecyl sulfate
polyacrylamide gel electrophoresis and transferred to 0.45 µm
polyvinylidene fluoride membrane. Subsequent to being blocked in
Tris-buffered saline and Tween 20 containing 5% non-fat dry milk
for 1 h at room temperature, the membrane was incubated in primary
antibodies at 4°C overnight and in goat anti-rabbit polyclonal
horseradish peroxidase (HRP)-conjugated secondary antibody (catalog
no. SA00001-2; dilution, 1:5,000; Proteintech Group, Inc., Chicago,
IL, USA) at room temperature for 1 h. The proteins on the membrane
were then detected by chemiluminescence solution (ratio of HRP
substrate luminol reagent and HRP substrate peroxide solution was
1:1; EMD Millipore, Billerica, MA, USA); the band intensity was
determined by ImageJ software (National Institutes of Health,
Bethesda, MA, USA). The primary rabbit anti-human antibodies were
as follows: TNFR2 (polyclonal; catalog no. 19272-1-AP; dilution,
1:1,000; Proteintech Group, Inc.); Ki67 (polyclonal; catalog no.
ab15580; dilution, 1:1,000; Abcam, Cambridge, MA, USA);
phospho-extracellular signal-related kinases (pERK) 1/2
(polyclonal; catalog no. ~2219-1; dilution, 1:1,000; Epitomics,
Burlingame, CA, USA); ERK1/2 (monoclonal; catalog no. 4695;
dilution, 1:1,000; Cell Signaling Technology, Inc., Danvers, MA,
USA); phospho-AKT (monoclonal; catalog no. 9611; dilution, 1:1,000;
Cell Signaling Technology, Inc.); AKT (monoclonal; catalog no.
4685; dilution, 1:1,000; Cell Signaling Technology, Inc.);
glyceraldehyde 3-phosphate dehydrogenase (polyclonal; catalog no.
10494-1-AP; dilution, 1:5,000; Proteintech Group, Inc.).
MTT assay
Cells were counted and plated in 96-well plates in
triplicate at 3×103 cells in 100 µl medium per well.
Cell viability was determined at 24, 48, 72, 96 and 120 h using MTT
assay, according to the manufacturers protocol (Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China). Optical
density (OD) values were read at 490 nm using enzyme-labeled
instrument (Bio-Rad Model 680; Bio-Rad Laboratories, Beijing,
China) and growth curves were drawn. The OD values at 120 h were
compared. The experiment was repeated three times.
Clone formation assay
Cells were counted and plated in 6-well plates at a
density of 1×103 cells in 2 ml medium per well. Medium
was refreshed every 3 days. Subsequent to 12 days, cells were fixed
using methanol for 30 min and stained using Giemsa, according to
the manufacturer's protocol (Beijing Solarbio Science &
Technology Co., Ltd.), then clones were counted by eye by two
independent researchers and images were captured. The experiment
was repeated 3 times.
Statistical analysis
SPSS 13.0 software (SPSS Inc., Chicago, IL, USA) was
used for statistical analysis. All data were expressed as the mean
± standard deviation. The differences between the two groups were
analyzed using Student's two-tailed t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
TNFR2 promoted Ki67 expression in
CRC
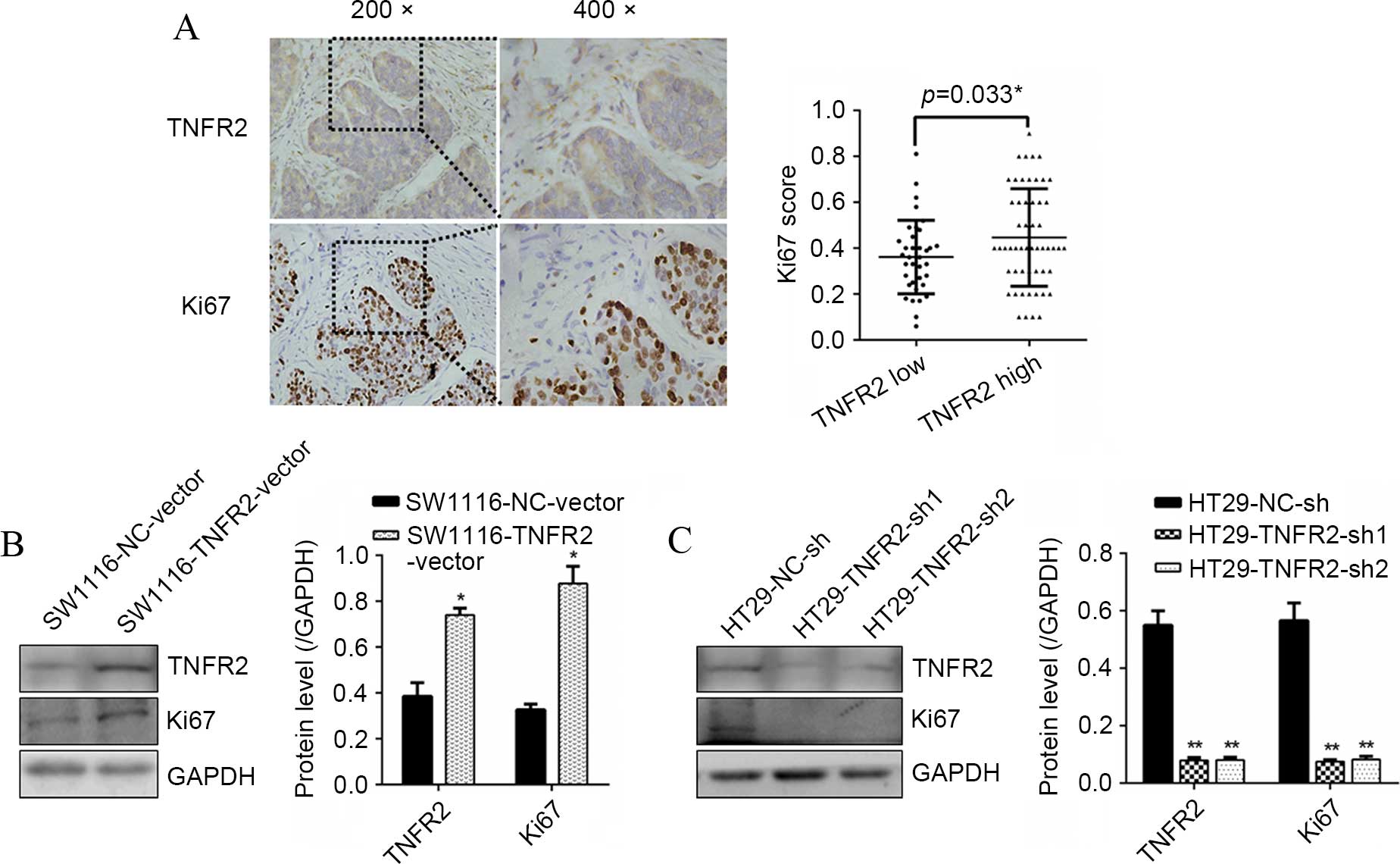
To confirm the association of TNFR2 with
proliferation marker Ki67 in CRC tissues, IHC was assessed. It was
found that diffused strong staining of TNFR2 was localized in CRC
cell cytoplasm, sporadic staining was also found in the mesenchyme.
A positive association between TNFR2 and Ki67 was confirmed
(P=0.033; Fig. 1A). At the cellular
level, western blot analysis found that subsequent to
overexpressing TNFR2 in SW1116 cells, Ki67 was significantly
upregulated (P=0.021; Fig. 1B).
Additionally, subsequent to silencing TNFR2 in HT29 cells, Ki67 was
significantly downregulated (P=0.009 and P=0.008; Fig. 1C). These results suggest that TNFR2
can promote Ki67 expression in CRC, indicating pro-growth
potential.
TNFR2 promoted growth and
proliferation of CRC cells
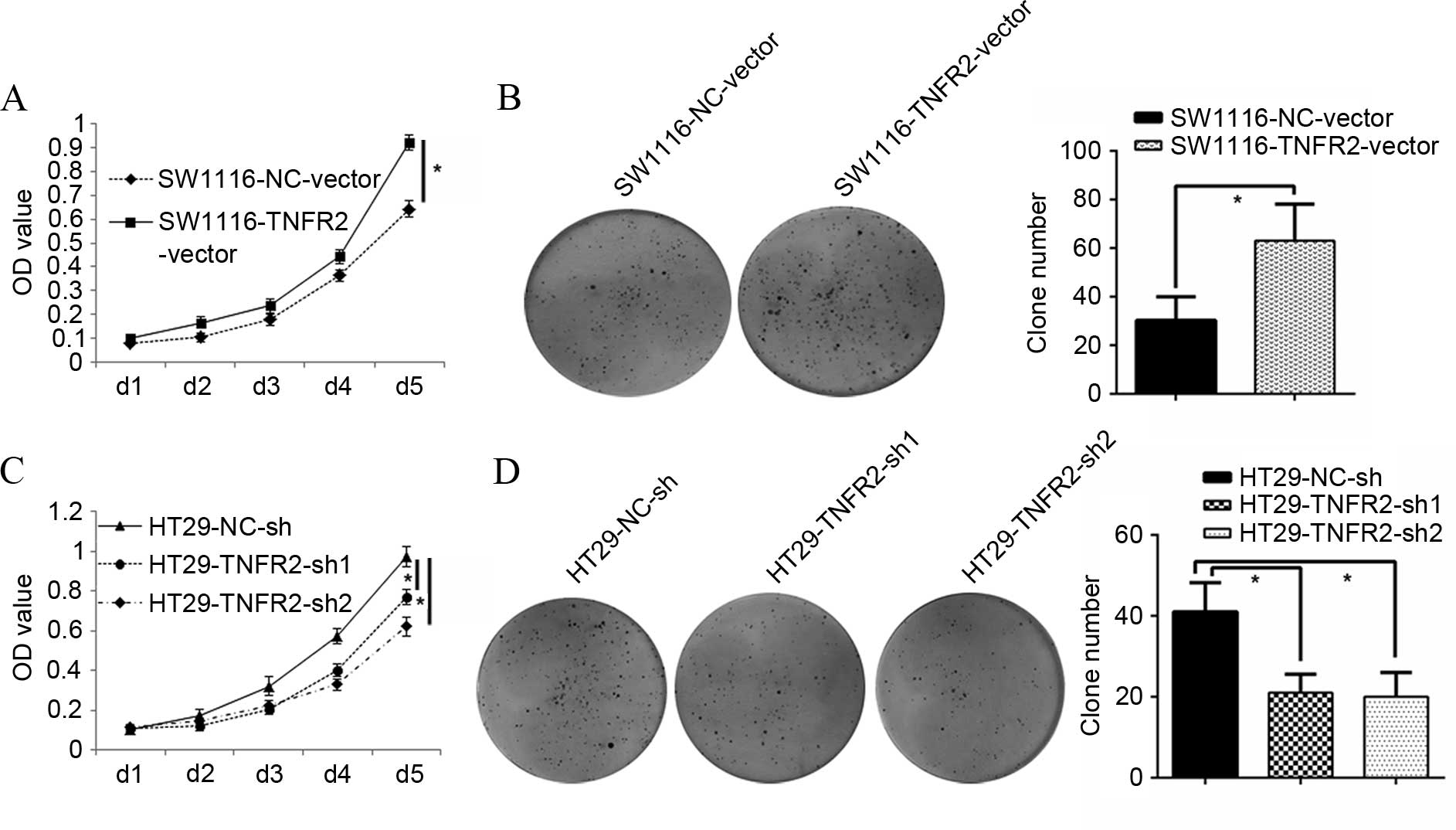
To additionally identify whether TNFR2 could
regulate CRC growth, an MTT assay was assessed. It was found that
SW1116 cells overexpressing TNFR2 (SW1116-TNFR2-vector) grew much
faster than the control group, with a greater OD value at 120 h in
the MTT assay (P=0.016; Fig. 2A). The
clone forming assay also showed that the clone number of
SW1116-TNFR2-vector was 63.00±15.10, almost twice that of the
control group (30.33±9.61) (P=0.031; Fig.
2B). Additionally, HT29 cells with silenced TNFR2
(HT29-TNFR2-sh1 and HT29-TNFR2-sh2) grew much slower than the
control group, with a weaker OD value at end time (120 h; P=0.036
and P=0.021; Fig. 2C). The clone
forming assay also showed that the clone number of HT29-TNFR2-sh1
and HT29-TNFR2-sh2 was 21.00±4.58 and 20±6.18, respectively, much
less than the control group (41±7.21; P=0.027 and P=0.028; Fig. 2D). All the data suggest that TNFR2 can
successfully promote growth and proliferation of CRC cells.
PI3K/AKT was required for growth and
proliferation of CRC cells promoted by TNFR2
To additionally confirm the molecular mechanism
responsible for regulation of TNFR2 on CRC growth, activation of
candidate signal targets was detected by western blot analysis.
Overexpressing TNFR2 in SW1116 cells significantly stimulated the
phosphorylation of AKT, without change of total AKT, whereas
phosphorylation of ERK and total levels of ERK did not alter. In
addition, subsequent to silencing TNFR2 in HT29 cells, only the
phosphorylation of AKT was inhibited. This indicated that AKT may
contribute to regulation of TNFR2 on CRC growth (Fig. 3A and B). To test this hypothesis, the
specific AKT inhibitor LY294002 was used. Subsequent to treatment
with LY294002, the upregulation of Ki67 induced by TNFR2 was
significantly abrogated (P=0.017; Fig.
3C). Additionally, the growth (P=0.012; Fig. 3D) and clone formation ability
(P=0.033; Fig. 3E) of the
SW1116-TNFR2-vector was also significantly inhibited. The present
data confirmed the hypothesis that AKT could contribute to
regulation of CRC growth induced by TNFR2.
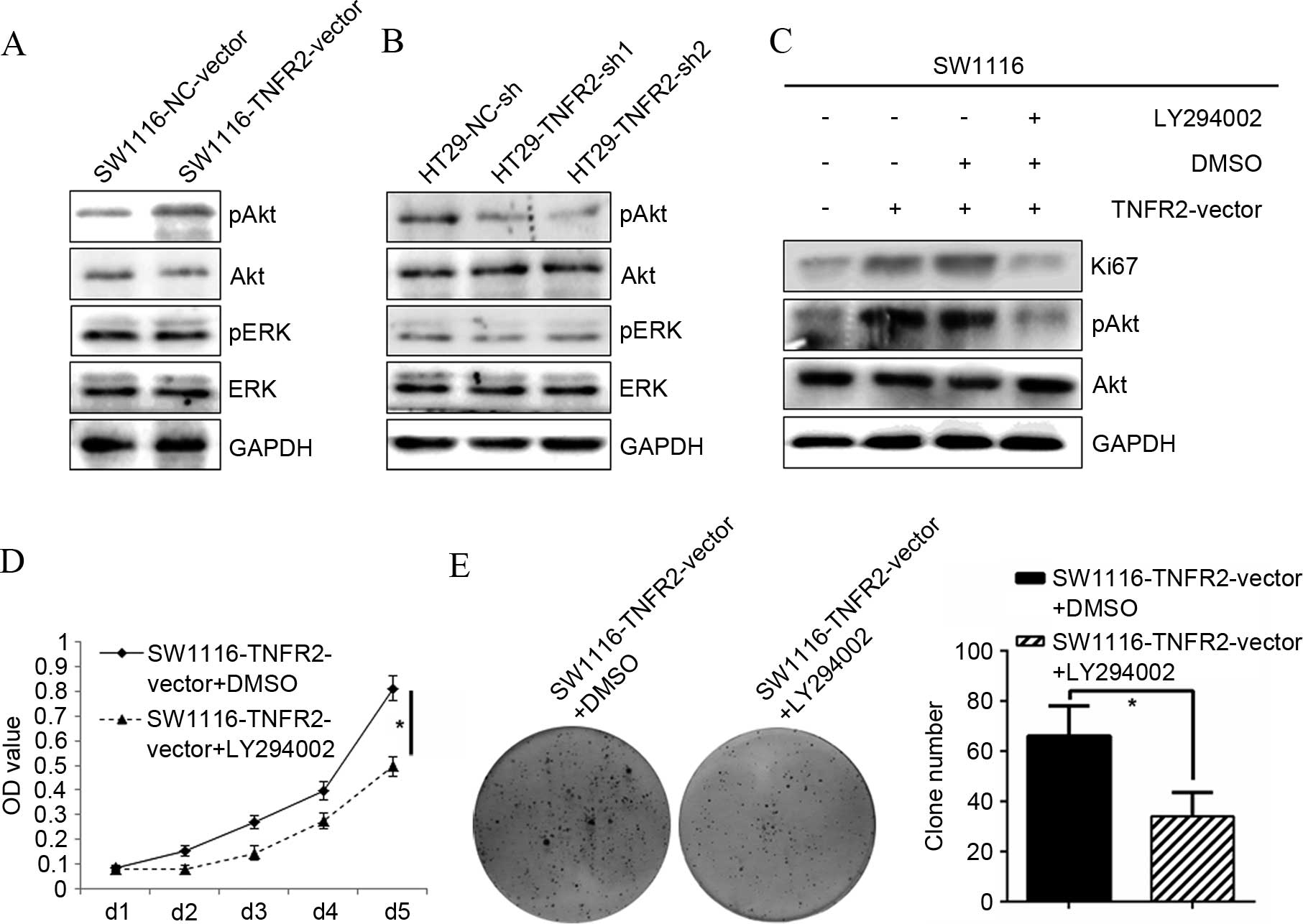 | Figure 3.PI3K/AKT was required for the growth
and proliferation of CRC cells induced by TNFR2. (A) pAKT, AKT,
pERK, ERK expression in SW1116-NC-vector and SW1116-TNFR2-vector
cells, as determined by western blot analysis. (B) pAKT, AKT, pERK,
ERK expression in HT29-NC-sh, HT29-TNFR2-sh1 and HT29-TNFR2-sh2
cells, as determined by western blot analysis. (C) Ki67 and pAKT
upregulation induced by TNFR2 in SW1116 cells was abrogated
subsequent to treatment using LY294002. (D) Growth promotion of
SW1116 cells induced by TNFR2 was inhibited following treatment
using LY294002. (E) Clone formation promotion of SW1116 cells
induced by TNFR2 was inhibited subsequent to treatment using
LY294002 and quantitative histogram of clone numbers. *P<0.05.
PI3K, phosphoinositide 3-kinase; (p)AKT, (phospho)-protein kinase
B; (p)ERK, (phospho)-extracellular signal-related kinases; TNFR2,
tumor necrosis factor receptor 2; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; DMSO, dimethyl sulfoxide; OD, optical density. |
Discussion
TNFR2 is the receptor for TNF-α. Unlike TNFR1 it
does not contain the death domain (DD), and its functions are less
understood (11). It is reported that
activation of TNFR2 in skin tumors could promote proliferation of
tumor cells, not induce apoptosis (12). Tanimura et al (11) reported that TNF-α could promote
invasiveness of cholangiocarcinoma cells via TNFR2 and Jöhrer et
al (13) reported that TNFR2 was
required for migration of myeloma cells induced by TNF-α. However,
the studies by Arnott et al, Baud and Karin, and Grell et
al (14–16) reported that, regardless of DD, TNFR2
could also activate death signals and induce cell death by ligand
passing to TNFR1. Due to the specific functional properties of
TNFR2 in different tumors, the contribution of TNFR2 in CRC
remained unknown. In the present study, IHC found that TNFR2 was
highly expressed in CRC cells and significantly positively
associated with Ki67 expression. Western blot analysis also showed
that TNFR2 successfully regulated Ki67 expression, revealing the
potential proliferative ability of TNFR2. To additionally verify
whether TNFR2 could regulate CRC growth, MTT assay was performed
and it was found that upregulating TNFR2 significantly promoted the
growth of SW1116 cells. In addition, silencing TNFR2 significantly
inhibited the growth of HT29 cells. To test the effect of TNFR2 on
cell proliferation ability, clone formation assay was performed and
the same trends were identified as found in the MTT assay. This
indicates that TNFR2 can promote CRC growth partly through
regulating proliferation ability of CRC cells. This is consistent
with the hypothesis that TNFR2 is required for cell
proliferation.
Signaling pathways play important roles in the
occurrence and progression of tumors and thorough knowledge of
signal transduction may provide new targets for tumor therapy.
PI3K/AKT and mitogen-activated protein kinase/ERK are important
signaling pathways for numerous cellular functions, including
proliferation, survival, adhesion and migration (17–22). It
has been reported that pretreatment of cells with TNFR2
neutralizing antibody could effectively block the activation of
PI3K/AKT and ERK signal pathway in cholangiocarcinoma cells
(11). To investigate whether AKT
and/or ERK are associated in CRC growth, they were selected as
targets for western blot analysis. It was found that TNFR2
upregulation activated phosphorylation of AKT, but not ERK.
Furthermore, following treatment using the AKT-specific inhibitor
LY294002, not only the upregulation of Ki67 induced by TNFR2 was
significantly abrogated, but the growth and clone formation
abilities of CRC cells were also inhibited. This was not completely
consistent with previous studies in which PI3K/AKT and ERK worked
in cholangiocarcinoma. It is possible that the downstream target of
TNFR2 in different tumors is type-dependent.
In conclusion, the present study confirmed the
contribution of TNFR2 in CRC growth for the first time and
additionally elucidated the molecular mechanism. This enriched the
understanding of functional properties of TNFR2 and provided
evidential support for finding a new promising target for CRC
treatment.
References
|
1
|
Becker C, Fantini MC, Wirtz S, Nikolaev A,
Lehr HA, Galle PR, Rose-John S and Neurath MF: IL-6 signaling
promotes tumor growth in colorectal cancer. Cell Cycle. 4:217–220.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smith AR, Marquez RT, Tsao WC, Pathak S,
Roy A, Ping J, Wilkerson B, Lan L, Meng W, Neufeld KL, et al: Tumor
suppressive microRNA-137 negatively regulates Musashi-1 and
colorectal cancer progression. Oncotarget. 6:12558–12573. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
National Research Council (US) Committee
on A Framwework for Developing a New Taxonomy of Disease, . Toward
Precision Medicine: Building a Knowledge Network for Biomedical
Research and a New Taxonomy of Disease. National Academies Press
(US); Washington (DC): 2011
|
|
5
|
Mirnezami R, Nicholson J and Darzi A:
Preparing for precision medicine. N Engl J Med. 366:489–491. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hosono K, Yamada E, Endo H, Takahashi H,
Inamori M, Hippo Y, Nakagama H and Nakajima A: Increased tumor
necrosis factor receptor 1 expression in human colorectal adenomas.
World J Gastroenterol. 18:5360–5368. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seitz C, Muller P, Krieg RC, Mannel DN and
Hehlgans T: A novel p75TNF receptor isoform mediating NFkappa B
activation. J Biol Chem. 276:19390–19395. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ,
Liu GY, Syed NM, Lai Y, Lin EA, Kong L, et al: The growth factor
progranulin binds to TNF receptors and is therapeutic against
inflammatory arthritis in mice. Science. 332:478–484. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guo Z, Li Q, Han Y, Liang Y, Xu Z and Ren
T: Prevention of LPS-induced acute lung injury in mice by
progranulin. Mediators Inflamm. 2012:5407942012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao YP, Tian QY, Frenkel S and Liu CJ:
The promotion of bone healing by progranulin, a downstream molecule
of BMP-2, through interacting with TNF/TNFR signaling.
Biomaterials. 34:6412–6421. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tanimura Y, Kokuryo T, Tsunoda N, Yamazaki
Y, Oda K, Nimura Y, Mon N Naing, Huang P, Nakanuma Y, Chen MF, et
al: Tumor necrosis factor alpha promotes invasiveness of
cholangiocarcinoma cells via its receptor, TNFR2. Cancer Lett.
219:205–213. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Szlosarek PW and Balkwill FR: Tumour
necrosis factor alpha: A potential target for the therapy of solid
tumours. Lancet Oncol. 4:565–573. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jöhrer K, Janke K, Krugmann J, Fiegl M and
Greil R: Transendothelial migration of myeloma cells is increased
by tumor necrosis factor (TNF)-alpha via TNF Receptor 2 and
autocrine Up-regulation of MCP-1. Clin Cancer Res. 10:1901–1910.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Arnott CH, Scott KA, Moore RJ, Robinson
SC, Thompson RG and Balkwill FR: Expression of both TNF-alpha
receptor subtypes is essential for optimal skin tumour development.
Oncogene. 23:1902–1910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baud V and Karin M: Signal transduction by
tumor necrosis factor and its relatives. Trends Cell Biol.
11:372–377. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Grell M, Zimmermann G, Hülser D,
Pfizenmaier K and Scheurich P: TNF receptors TR60 and TR80 can
mediate apoptosis via induction of distinct signal pathways. J
Immunol. 153:1963–1972. 1994.PubMed/NCBI
|
|
17
|
Dahlmann M, Okhrimenko A, Marcinkowski P,
Osterland M, Herrmann P, Smith J, Heizmann CW, Schlag PM and Stein
U: RAGE mediates S100A4-induced cell motility via MAPK/ERK and
hypoxia signaling and is a prognostic biomarker for human
colorectal cancer metastasis. Oncotarget. 5:3220–3233. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu SY, Tai CC, Li YH and Wu JL:
Progranulin compensates for blocked IGF-1 signaling to promote
myotube hypertrophy in C2C12 myoblasts via the PI3K/Akt/mTOR
pathway. FEBS Lett. 586:3485–3492. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bai T, Lian LH, Wu YL, Wan Y and Nan JX:
Thymoquinone attenuates liver fibrosis via PI3K and TLR4 signaling
pathways in activated hepatic stellate cells. Int Immunopharmacol.
15:275–281. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li N, Cui J, Duan X, Chen H and Fan F:
Suppression of type I collagen expression by miR-29b via PI3K, Akt,
and Sp1 pathway in human Tenon's fibroblasts. Invest Ophthalmol Vis
Sci. 53:1670–1678. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miao B and Degterev A: Targeting
phospshatidylinositol 3-kinase signaling with novel
phosphatidylinositol 3,4,5-triphosphate antagonists. Autophagy.
7:650–651. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gentilini A, Marra F, Gentilini P and
Pinzani M: Phosphatidylinositol-3 kinase and extracellular
signal-regulated kinase mediate the chemotactic and mitogenic
effects of insulin-like growth factor-I in human hepatic stellate
cells. J Hepatol. 32:227–234. 2000. View Article : Google Scholar : PubMed/NCBI
|