Introduction
B-cell precursor acute lymphoblastic leukemia (BCP
ALL) is the most common type of cancer that occurs during
childhood. Although the majority of patients may be cured by the
currently available therapeutic regimens, novel treatments are
urgently required for the 20% of patients who experience relapse
(1). The prevalence of BCP ALL may
result, in part, from the fact that transformation generally occurs
in cells arrested at the B-cell receptor checkpoint, a stage of
differentiation associated with a balance between proliferation and
recombination-activating gene-mediated DNA rearrangement (2–4). The
stromal cytokine interleukin (IL)-7 serves an important role during
this highly ordered process of differentiation, proliferation and
somatic recombination (5–7). IL-7 binds to a receptor (IL-7R) that is
composed of cluster of differentiation (CD) 127 (IL-7Rα) and CD132
(γc), which is found on lymphoid progenitor cells, BCP cells and T
cells. It has been demonstrated that IL-7 facilitates the
proliferation and survival of leukemia cells (8–10),
contributes to the progression and relapse of leukemia (11,12) and
induces resistance to chemotoxins (13), thus contributing to the pathogenesis
of ALL. As a result, identifying the leukemia-specific mediators of
these IL-7-driven effects may enable the therapeutic targeting of
this pathway.
Y-box-binding protein 1 (YB-1) regulates
differentiation, proliferation and survival in a broad range of
cells (14). In the cytoplasm, this
highly conserved protein acts as a translational inhibitor to
repress mRNA involved in survival and proliferation (15), whereas activation by stress and other
factors leads to YB-1 phosphorylation and translocation to the
nucleus where it functions as a transcription factor (16–18).
Aberrant YB-1 expression may contribute to the development and
maintenance of malignancy in several ways, including dysregulation
of hormone/growth factor receptors (19–21),
enhancement of survival during stress, and induction of drug
resistance (22–25). Increased expression of YB-1 has been
implicated as a poor prognostic factor in various different types
of malignancy, including B-cell lymphoma (26–28).
However, YB-1 expression has not yet been investigated in ALL.
YB-1 functions downstream of growth factor receptors
in other malignancies (29);
therefore, it was hypothesized that YB-1 is involved in
IL-7-mediated survival pathways in ALL. The present report
describes a comparative analysis of YB-1 and IL-7Rα expression in
pediatric BCP ALL and normal precursor B cells, in order to
determine whether crosstalk occurs between these two key regulators
of proliferation and survival and whether YB-1 is involved in the
IL-7-mediated protection of BCP ALL cells against rapamycin. The
present study aimed to determine whether the inhibition of YB-1 may
represent a novel strategy to increase the susceptibility of BCP
ALL to mammalian target of rapamycin (mTOR) inhibitors.
Materials and methods
Cells, tissue samples and
antibodies
Cell lines derived from relapsed human BCP ALL were
obtained from commercial vendors: 697, 380, RCH-ACV and MHH-CALL-2
were purchased from DSMZ (Braunschweig, Germany); RS4;11 was
acquired from ATCC (Manassas, VA, USA). All cell lines were
cultured at 37°C in 5% CO2 in RPMI 1640 medium
supplemented with 10% heat-inactivated fetal bovine solution (FBS),
100 U/ml penicillin/streptomycin, and 2 mM glutamine (all from
Thermo Fisher Scientific Inc., Waltham, MA, USA), with the
exception of MHH-CALL-2, which was cultured in RPMI with 20% FBS.
The phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 (30 µM)
and the Janus kinase (JAK) 1/2 inhibitor ruxolitinib (100 nM) were
purchased from Selleck Chemicals (Houston, TX, USA), and the MEK1/2
inhibitor U0126 (10 µM) was acquired from Cell Signaling
Technology, Inc. (Danvers, MA, USA). The inhibitors in dimethyl
sulfoxide (DMSO) were added 2 h prior to IL-7 stimulation. Cells
were then incubated for a further 30 min after the addition of
recombinant 25 ng/ml IL-7 (BioLegend, Inc., San Diego, CA,
USA).
The use of human tissue samples was approved by the
ethics boards of the British Columbia Children's and Women's
Hospital (Vancouver, BC, Canada) and the Alberta Children's
Hospital (Calgary, AB, Canada), in accordance with the Declaration
of Helsinki. BCP ALL samples, obtained from surplus clinical tissue
banked between November 1998 and July 2002, were of varied or
unknown risk group classifications; the median ages of the patients
at diagnosis (n=20) and relapse (n=7) were 6 years (range, 1–16
years) and 13 years (range, 1–17 years), respectively. One adult
BCP ALL relapse sample was included in the current study. Normal
precursor and mature B cells were purchased as bone marrow
mononuclear cells from Lonza Inc. (Allendale, NJ, USA) or obtained
from fresh cord blood or bone marrow samples from pediatric
non-hematological malignancy patients without bone marrow
involvement. Xenograft-mediated expansion of primary BCP ALL
samples in NOD.Cg-Prkdcscid/IL2rgtm1Wjl/SzJ mice (NSG
mice™; Jackson Laboratory, Bar Harbor, ME, USA) was performed as
previously described (30). All
animal experiments were performed in accordance with a University
of British Columbia Animal Care Committee-approved protocol
(A11-0130).
Western blotting
Cell lines were harvested and lysed for 20 min on
ice in the presence of a protease and phosphatase inhibitor
cocktail (Roche Life Science, Benzberg, Germany) using 1X Cell
lysis buffer from Cell Signaling Technology, Inc. (catalog no.
9803). After spinning down (18,000 × g for 10 min at 4°C),
the amount of protein in the supernatant (lysate) was determined
using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific,
Inc.), and an equal amount of protein was collected and mixed with
Laemmli sample buffer and denatured for 5 min at 95°C followed by 1
min on ice. Proteins were separated on a 10% sodium dodecyl sulfate
gel followed by electro-transfer blotting onto polyvinylidene
difluoride membranes (BioRad, Hercules, CA, USA) using the Mini
Trans-Blot Electrophoretic Transfer Cell device (BioRad). The
membranes were then blocked using protease-free bovine serum
albumin (catalog no. A7030; Sigma-Aldrich) blocking buffer (5% w/v
in Tris-buffered saline). The following primary antibodies were
used: PhosphoAKTS473 (catalog no. 5012), phospho-signal
transducer and activator of transcription (STAT) 5Y694
(catalog no. 9351), phosphoYB-1S102 (catalog no. 2900),
phospho42/44 MAPK [extracellular signal-regulated kinase (ERK) 1/2]
T202/Y204 (catalog no. 4370), total p90 ribosomal S6
kinase (RSK) 1 (catalog no. 8408), total AKT (catalog no. 4691),
total STAT5 (catalog no. 9363) and total p42/44 (catalog no. 4695).
Anti-total YB-1 was purchased from Abcam (Cambridge, UK; catalog
no. ab12148) and anti-pRSK1/2S221/227/218/232 was
obtained from Invitrogen (catalog no. PA5-37829; Thermo Fisher
Scientific, Inc.). Anti-rabbit or -mouse immunoglobulin
(Ig)G-horseradish peroxidase (HRP) was used as a secondary antibody
(Biorad catalog no. 1721019). All antibodies were purchased from
Cell Signaling Technology, Inc., unless otherwise stated and were
used at a dilution of 1:1,000. Signals were detected using the
Novex ECL HRP Chemiluminiscent Substrate Reagent kit, and captured
on X-ray film (both from Invitrogen; Thermo Fisher Scientific,
Inc.).
Flow cytometry
For detection of total YB-1 in hematogones and BCL
ALL primary cells, frozen cells were thawed, washed and counted. A
total of 1–2×106 peripheral blood mononuclear cells were
stained with a 1:25 dilution of the following antibodies:
CD19-peridinin chlorophyll (PerCP), CD10-allophycocyanin (APC),
CD20-Brilliant™ Violet 421, CD45-APC-Cy7 and CD127-phycoerythrin
(PE) antibodies (BioLegend, Inc.), and incubated for 15 min on ice.
The cells were then fixed using BD Biosciences Fix buffer (BD
Biosciences, Franklin Lakes, NJ, USA) for 15 min on ice and then
permeabilized using Perm II buffer (BD Biosciences) for 10 min at
room temperature. All cell washes were in 2% FBS in phosphate
buffered-saline (PBS) and the spun at 4°C and 500 × g for 5
min. For detection of total intracellular YB-1, the cells were
stained with a rabbit monoclonal antibody against YB-1 (Abcam;
catalog no. ab76540) followed by anti-rabbit IgG-fluorescein
isothiocyanate (BioLegend, Inc.; catalog no. 406403). Hematogones
were identified as CD45dim, CD19+,
CD20dim to negative and CD10+ cells. Flow
cytometry data was acquired using the BD LSRII with FACSDiva
software™ V8 (BD Biosciences) and analyzed using FlowJo v9.2 (Tree
Star, Inc., Ashland, OR, USA). Detection of phosphoYB-1 in primary
cells or cell lines after IL-7 treatment was performed as
aforementioned for total YB-1 but substituting the
pYB-1S102 antibody (catalog no. ab74162; Abcam). Primary
transfected B-cells were stained at 24 h post-transfection for
surface markers using CD19-PB (catalog no. 302223), CD127-PerCP
(catalog no. 351322) (all 1/25 dilution; BioLegend, Inc.), then
fixed/permeabilized and stained for intracellular YB-1 as
aforementioned, with the exception that anti-rabbit IgG-PE
(Biolegend, Inc.; catalog no. 406421) was used.
Knockdown and overexpression of
YB-1
A total of 1×106 MHH-CALL-2 cells in PBS
were transfected with 200 nM small interfering RNA (siRNA), or 2–4
µg plasmid, using electroporation (Amaxa™ nucleofection, Lonza,
Inc.) and the proprietary program C005. Silencer® Select
stealth siRNA (Invitrogen; Thermo Fisher Scientific, Inc.),
modified to extend cell half-life, was used for the targeted
knockdown of YB-1. The universal negative control MEDGC
(Invitrogen; Thermo Fisher Scientific, Inc.) served as a control
for non-specific interactions. The siRNA sequences used for YB-1
knockdown were as follows: siRNA YB-1-1,
5′-CAAGGUAGACCAGUGAGGCAGAAUA-3′; siRNA YB-1-2,
5′-GGUCCUCCACGCAAUUACCAGCAAA-3′. Transcript and protein levels were
determined by quantitative polymerase chain reaction (qPCR) or flow
cytometry three days after transfection. RNA was extracted from the
cells using the Qiagen RNAeasy Mini kit (Qiagen, Mississauga, ON,
Canada) and cDNA synthesized with a first-strand cDNA synthesis kit
(GE Healthcare Life Sciences, Chalfont, UK) using random primers.
The TaqMan® Universal PCR Master Mix was used for qPCR
with the TaqMan gene expression assay for human YB-1
(Hs00898625_g1), with human TATA-box binding protein
(Hs99999910_m1) used as an endogenous control. Amplification was
performed using a GeneAmp® 5700 Sequence Detection
system (40 cycles; 95.0°C for 15 sec and 60.0°C for 60 sec). All
qPCR reagents were purchased from Applied Biosystems (Thermo Fisher
Scientific, Inc.). Quantification of RNA was performed using the
2−ΔΔCq method (31).
For overexpression of YB-1, CD19+ B cells
were isolated from cord or peripheral blood using a RosetteSep™
Human B Cell Enrichment Cocktail (catalog no. 15064; Stemcell
Technologies, Inc., Vancouver, BC, Canada) and 1×106
cells were transfected with the plasmid as described above; a
pEGFP-based vector expressing the green fluorescent protein
(GFP)/YB-1 fusion protein, which was generously provided by Dr
Michel Lebel (Cancer Research Center of Laval University, QC,
Canada), was used for overexpression (32), and pEGFP-C1 (Clontech Laboratories,
Inc., Mountain View, CA, USA) served as a negative control.
Cytotoxicity assays
MHH-CALL-2 cells were incubated overnight with 25
ng/ml IL-7. IL-7 treatment was repeated 30 min prior to the
addition of 140 µM rapamycin (LC Laboratories, Woburn, MA, USA).
After 8 h, cells were harvested and the percentage of dead cells
was determined by flow cytometry using the Fixable Viability Dye
eFluor® 450 (eBioscience Inc., San Diego, CA, USA).
For experiments combining rapamycin and U0126, cells
were incubated for 2 h with 10 mM U0126 or 0.15% DMSO and
subsequently overnight with 25 ng/ml IL-7. The following morning,
cultures were administered a second dose of U0126 and IL-7 for 30
min prior to the addition of 34 µM rapamycin for a further 18 h.
The percentage of dead cells was subsequently determined. The
extent of apoptosis (A) induced by rapamycin with or without IL-7
and U0126 was calculated as previously described (28). The percent rescue with IL-7 was
calculated as follows: Percent rescue = [1 - (Awith IL-7
/ Awithout IL-7)] × 100.
Statistical analysis
To compare levels of YB-1 and IL-7 in different
populations, selected data sets were analyzed by one-way analysis
of variance with corrections for multiple comparisons. All other
data sets were compared using an unpaired Student's t-test unless
otherwise indicated. All tests were two-tailed. P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression of IL-7Rα and YB-1 in
normal and malignant BCP cells
YB-1 is well established as a risk factor for
various types of malignancy (14),
and IL-7Rα mediates proliferation and survival signaling in BCP ALL
cells (33); therefore, the levels of
these two proteins in normal BCP cells and unmatched BCP ALL cells
obtained at diagnosis or relapse were compared. As shown in
Fig. 1A, levels of YB-1 in BCP ALL at
diagnosis and relapse were significantly higher than those of cells
within the broader BCP cell population (defined as
CD45dimCD10+CD19+) obtained from
healthy donors (P=0.0018). Furthermore, levels of YB-1 expression
were higher in relapsed BCP ALL than in diagnostic samples
(P=0.0019). Similarly, IL-7Rα expression levels were significantly
higher in BCP ALL cells compared with normal BCP cells (Fig. 1B; P=0.0286); however, given the broad
range of expression levels in the different patient samples, IL-7Rα
expression did not differ significantly between diagnostic and
relapse samples. These data indicate that IL-7Rα and YB-1 may serve
a potential role in the onset and/or progression of BCP ALL.
Therefore, it was investigated whether YB-1 and IL-7R co-expression
was fully restricted to leukemic BCP cells.
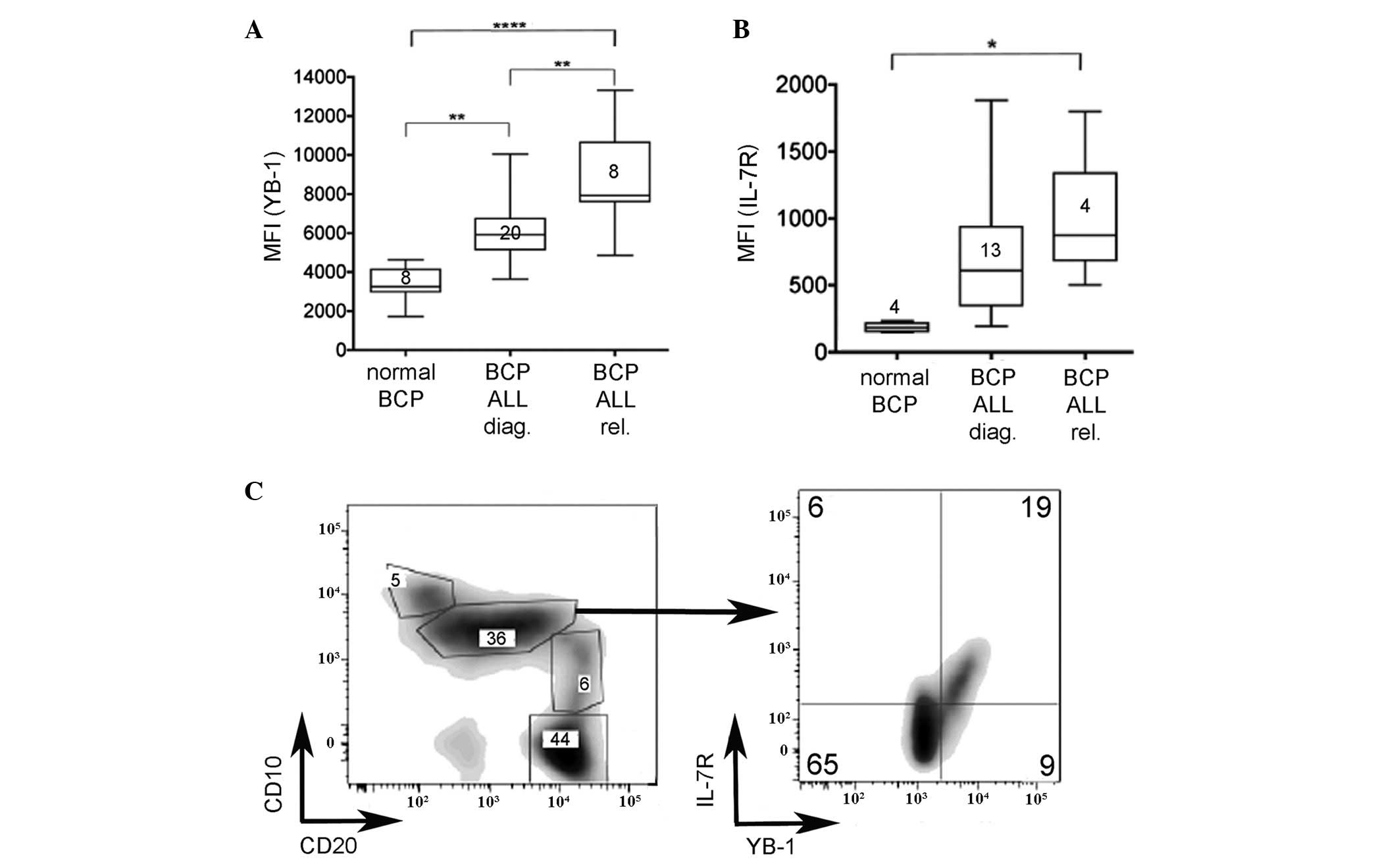 | Figure 1.YB-1 is significantly higher in BCP
ALL, at diagnosis and relapse, compared with normal counterparts.
Flow cytometric analyses of (A) YB-1 and (B) IL-7Rα expression in
BCP cells from the bone marrow of healthy individuals and pediatric
BCP ALL patients at diagnosis and at relapse. Normal BCP cells were
defined by a CD45dim/CD19+/CD10+
phenotype. The number of samples evaluated in each case is
indicated on each plot. Results are presented as a box and whisker
plot (Tukey method). (C) Evaluation of normal BCP subsets
(percentage of total BCP population is indicated in each box)
revealed an IL-7Rα/YB-1-expressing subpopulation within the
CD10+/CD20dim subset. *P<0.05,
**P<0.005, ****P<0.0001. YB-1, Y-box-binding protein-1;
IL-7Rα, interleukin-7 receptor α; BCP, B-cell precursor; ALL, acute
lymphoblastic leukemia; diag., diagnosis; rel., relapse; CD,
cluster of differentiation; MFI, mean fluorescence intensity. |
As 90% of the primary ALL samples were
CD10+/CD20dim, the phenotypically equivalent
normal BCP population was evaluated for YB-1 and IL-7R expression
(Fig. 1C). This analysis demonstrated
that a particular subset of cells, previously hidden in the more
broadly defined CD45dimCD10+CD19+
BCP cell population, expressed YB-1 and IL-7R. In this cell subset,
the mean fluorescence intensity for YB-1 staining of cells
expressing IL-7R was not significantly different from that observed
on diagnostic BCP ALL cells (data not shown). This result indicates
that YB-1 is co-expressed with IL-7R in a small subset of normal
B-cell progenitors, and therefore not restricted to leukemic
cells.
Surface levels of IL-7Rα increase with
overexpression of YB-1
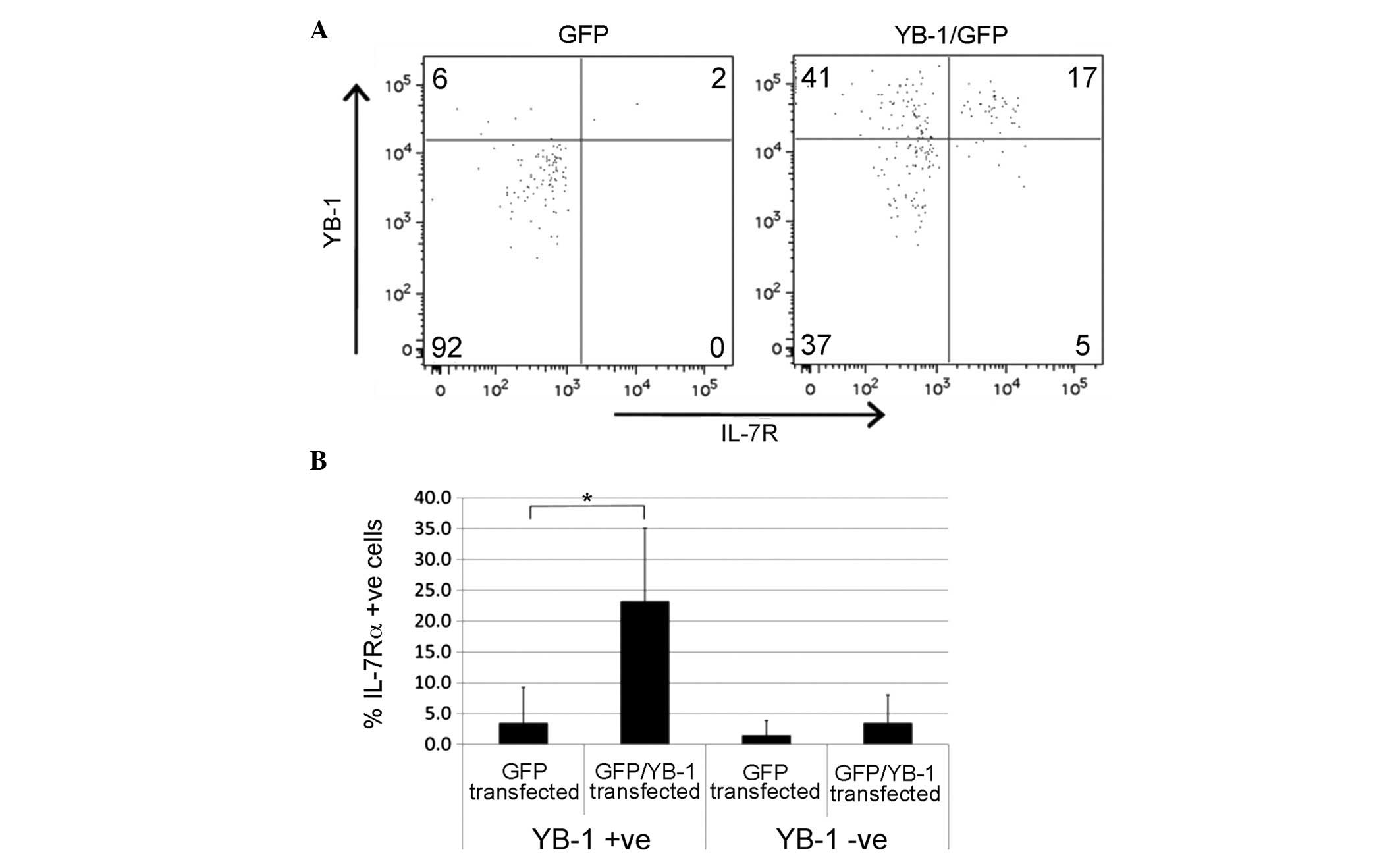
Having established that YB-1 and IL-7Rα are highly
expressed in BCP ALL and a subset of normal BCP cells, it was
investigated whether the co-expression of the two proteins was
functionally related. Previously, YB-1 has been demonstrated to
regulate the expression of hormone and growth factor receptors in
cancer (19,20); therefore the effect of YB-1
overexpression on IL-7 receptor levels was evaluated. Peripheral
blood- or cord blood-derived CD19+ B cells were
transiently transfected with a YB-1-GFP fusion construct or a GFP
control, and subsequently analyzed for IL-7Rα surface expression. A
comparison of GFP-positive cells in the two groups indicated that
the marked increase in YB-1 expression in B cells containing the
GFP/YB-1 plasmid was accompanied by an increase in the proportion
of IL-7Rα-positive cells. IL-7Rα expression did not change in the
YB-1-negative fraction or in cells rendered GFP-positive by the
control plasmid (Fig. 2A and B).
While YB-1 expression alone was not always sufficient to increase
the expression of IL-7Rα (transfection of cells with YB-1-GFP
increased the proportion of YB-1+/IL-7Rα−
cells), this result supports the concept that surface expression of
IL-7Rα is influenced by YB-1 expression in B-lineage cells.
IL-7 induces phosphorylation of YB-1
in BCP ALL cell lines and patient samples
In the cytoplasmic compartment, YB-1 forms an
inhibitory complex with mRNA. It has previously been demonstrated
that phosphorylation at Ser102 activates YB-1, inducing the release
and translation of mRNA and enabling nuclear localization for
transcriptional regulation (16,34,35). As
pYB-1S102 is induced by epidermal growth factor
stimulation in breast cancer cell lines (36), the ability of IL-7 to induce the
phosphorylation of YB-1 in BCP ALL cells was examined. Several
different BCP ALL cell lines (380, RS;4:11, 697, RCH-ACV and
MHH-CALL-2) were stimulated with PBS or 25 ng/ml IL-7 for 5–60 min.
Western blotting was subsequently performed to analyze the
expression of phosphorylated and total YB-1 (Fig. 3A). Within 15 min of stimulation with
IL-7, all of the cell lines exhibited YB-1 phosphorylation. Similar
results were obtained for pYB-1 following intracellular flow
cytometric analyses of the cell lines (data not shown).
Furthermore, analysis of primary BCP ALL diagnostic samples
(deidentified code nos. A193 and A199) and the primary BCP ALL
relapse sample (deidentified code no. A247) by flow cytometry
indicated an increase in pYB-1S102 within 5 min of IL-7
stimulation (Fig. 3B). Thus, IL-7 has
the ability to activate YB-1 by S102 phosphorylation in BCP ALL
cell lines and in primary samples of pediatric BCP ALL at diagnosis
and relapse.
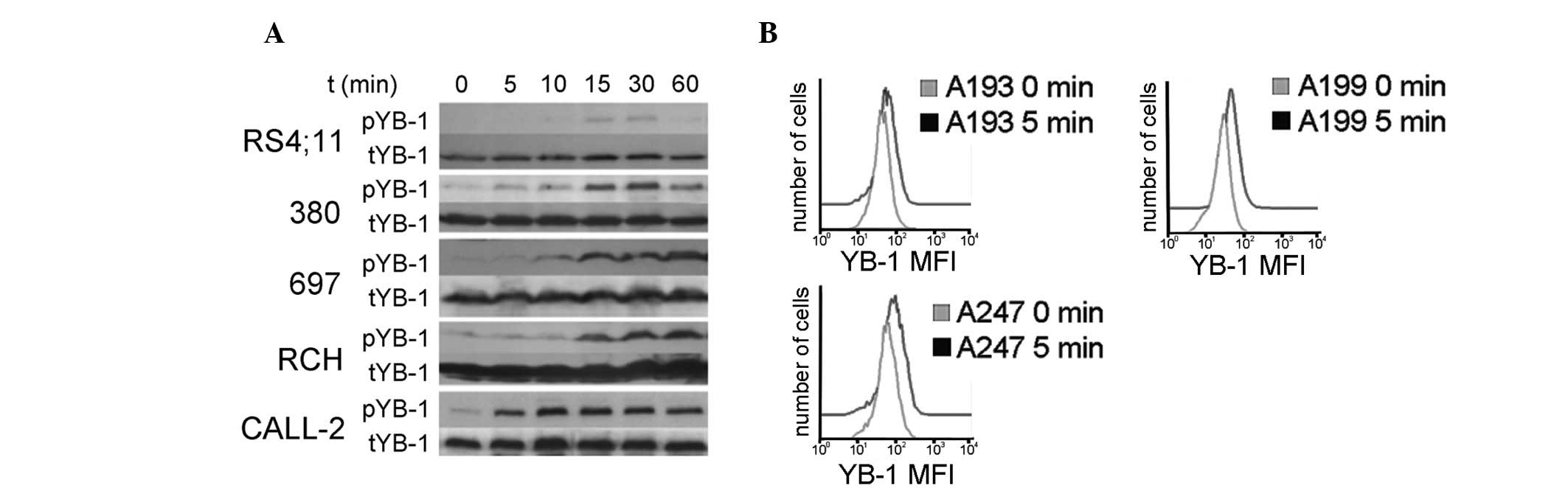 | Figure 3.IL-7 induces YB-1 phosphorylation in
BCP ALL cell lines and in primary patient samples. (A) Western blot
analysis of BCP ALL cell lines (RS4;11, 380, 697, RCH and CALL-2)
for pYB-1. Cell lines were serum-starved for 24 h, stimulated with
25 ng/ml IL-7 for 0–60 min and sequentially probed with anti-pYB-1
(S102) and anti-tYB-1 antibodies. (B) Flow cytometric determination
of pYB-1 following IL-7 stimulation of BCP ALL samples. Primary
samples, expanded in NOD.Cg-Prkdcscid/IL2rgtm1Wjl/SzJ
mice, were thawed, rested for 1.5 h in serum-free medium and
stimulated with 25 ng/ml IL-7 for the indicated times. Histograms
of pYB-1 from each expanded patient sample prior to and following
stimulation with IL-7 at the time of maximum phosphorylation are
presented. IL-7, interleukin-7; YB-1, Y-box-binding protein-1;
pYB-1, phosphorylated YB-1; tYB-1, total YB-1; BCP, B-cell
precursor; ALL, acute lymphoblastic leukemia. |
IL-7-mediated phosphorylation of YB-1
is MEK1-dependent and PI3K/Akt-independent
The binding of IL-7 to its receptor on BCP cells
triggers a distinct set of responses including differentiation,
proliferation and survival. JAK1/3 phosphorylates tyrosine residues
on the IL-7Rα chain allowing recruitment and activation of STAT1, 3
and 5 proteins (37), PI3K (38) and the Ras/Raf signaling cascade
(39,40). To identify which of these signaling
pathways are involved in the IL-7-mediated phosphorylation of YB-1,
the induction of these pathways in a BCP ALL cell line by IL-7 was
assessed. Stimulation of CALL-2 cells with 25 ng/ml IL-7 induced
the phosphorylation of STAT5Y694, AktS473 and
ERK1/2T202/Y204 (Fig. 4A). It has been demonstrated that
RSK1/2, downstream of ERK1/2, is able to phosphorylate YB-1
(36); therefore, the induction of
pRSKS221/S227 was investigated. RSK1 was strongly and
constitutively phosphorylated; however, RSK2 was only transiently
induced by IL-7 (Fig. 4A). To
determine which signaling components were required for YB-1
phosphorylation, CALL-2 cells were treated with pharmacological
inhibitors of the JAK/STAT, PI3K and Raf/ERK signaling pathways
prior to IL-7 stimulation, and the phosphorylation of YB-1 was then
examined (Fig. 4B). Inhibition of
signaling downstream of PI3K with Ly294002 blocked phosphorylation
of Akt completely; however, phosphorylation of YB-1 was only
slightly reduced. Phosphorylation of YB-1 downstream of IL-7 was
strongly inhibited by the MEK1/2 inhibitor U0126. Likewise, the
JAK1/2 inhibitor ruxolitinib blocked all IL-7-induced signaling and
thus phosphorylation of YB-1. Consistent with this finding, U0126
blocked the increase in levels of phosphorylated YB-1 in
IL-7-stimulated ALL patient samples (Fig.
4C). These results indicate that IL-7-mediated activation of
YB-1, in cell lines and patient-derived BCP ALL, requires
MEK1/2.
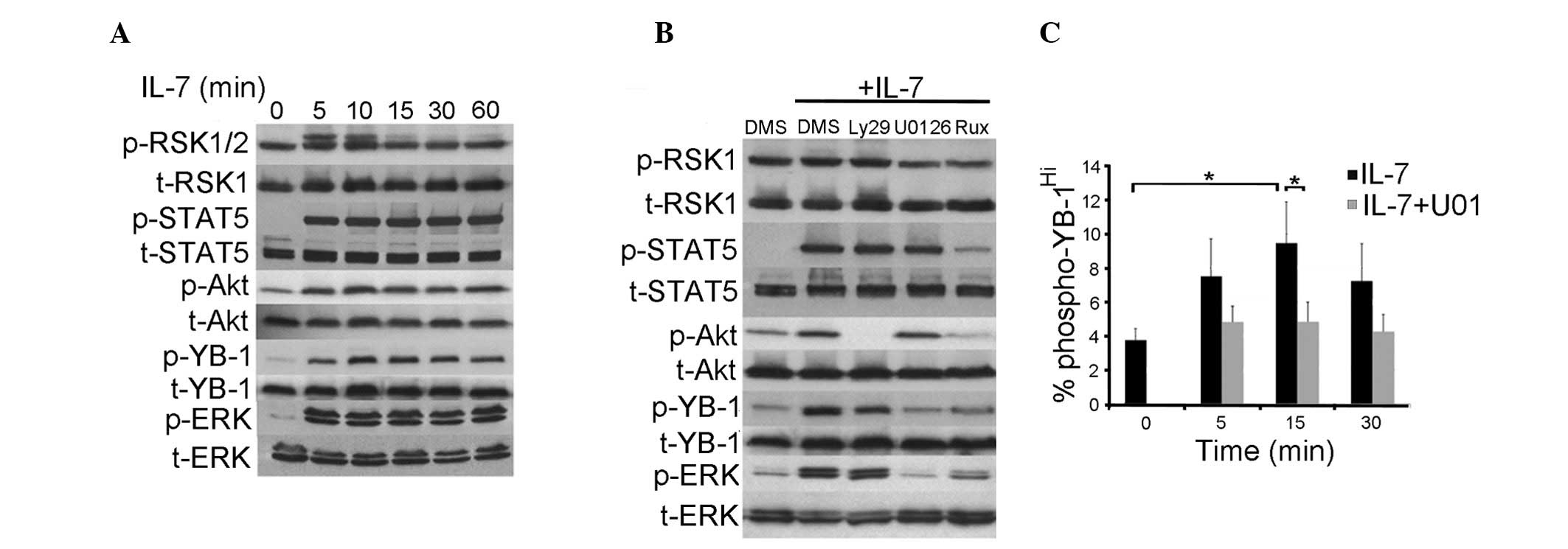 | Figure 4.IL-7-mediated phosphorylation of YB-1
is MEK1-dependent but PI3K/AKT independent.
Cells were serum-starved for 24 h prior to IL-7 stimulation.
Lysates were then probed with antibodies specific for
phosphorylated (phosphoRSK S221, phosphoSTAT5 Y694, phosphoAkt
S473, phosphoYB-1 S102, phosphoERK1/2 T202/Y204) and total
proteins. (A) A time course of phosphorylation responses to IL-7
added for the indicated durations. (B) Inhibition of signaling
pathways in CALL-2 cells pretreated with 0.15% DMSO or the
indicated inhibitor (30 µM LY294002, 10 µM U0126 or 100 nM
ruxolitinib) for 2 h prior to the addition of IL-7 for 30 min.
Blots are representative of at ≥3 different experiments. (C)
Inhibition of YB-1 phosphorylation with U0126 in expanded patient
samples. An arbitrary gate was used to define
phospho-YB-1hi and applied to all samples; the
percentage of phospho-YB-1hi cells prior to and
following stimulation with 25 ng/ml IL-7 with and without prior
treatment with 10 µM U0126 are presented. The graph shows the
averages from three NSG-expanded cell lines (A193, A247 and A199),
each repeated three times. *P<0.05. NSG mice,
NOD.Cg-Prkdcscid/IL2rgtm1Wjl/SzJ mice; IL-7,
interleukin 7; YB-1, Y-box-binding protein-1; p-YB-1,
phosphorylated Y-box-binding protein 1; STAT5, signal transducer
and activator of transcription 5; DMSO/DMS, dimethyl sulfoxide;
U0126, MEK1/2 inhibitor; Ly29, LY294002. |
YB-1 contributes to IL-7-mediated
inhibition of rapamycin-induced cell death
Having identified YB-1 as an IL-7 signaling
intermediate in BCP ALL, the functional significance of this
relationship was examined. YB-1 is a prognostic marker for
multi-drug resistance in many different types of cancer (22–25); thus,
the finding that YB-1 expression is highest in relapsed BCP ALL
suggests that YB-1 serves a potential role in contributing to
chemotherapy resistance. The ability of IL-7 to protect BCP ALL
cells against cell death has previously been demonstrated with the
mTOR inhibitor rapamycin (13). In
the present study, the role of YB-1 in this type of protection was
assessed using two approaches: Targeted knockdown of YB-1 and
pharmacological inhibition of YB-1 phosphorylation. Determination
of YB-1 levels in CALL-2 cells 72 h following transfection with
siRNA YB-1-1 or YB-1-2 demonstrated respective reductions of 88 and
68% in mRNA, and 67 and 37% in protein levels compared with control
cells transfected with a non-specific control siRNA (Fig. 5A). Knockdown of YB-1 alone did not
cause a significant decrease in cell viability in the absence or
presence of IL-7 in the 24 h assay (Fig.
5B). To evaluate the effect of YB-1 knockdown on rapamycin
sensitivity, 48 h after siRNA transfection, cells were treated
overnight with IL-7 or vehicle control and then challenged with
rapamycin for a further 8 h. Even in the absence of IL-7, cells
transfected with siRNA exhibited increased susceptibility to
rapamycin (91 and 86% cell death for siRNA YB-1-1 and YB-1-2,
respectively), compared with cells transfected with control siRNA
(71% cell death; Fig. 5B; P<0.05).
In cells transfected with control siRNA, treatment with IL-7
reduced the proportion of dead cells following rapamycin challenge
from 71 to 36%, corresponding to 49% cell rescue. In cells
transfected with siRNA targeting YB-1, IL-7 reduced the percentage
of cell death following rapamycin treatment to 66% (for siRNA
YB-1-1) and 58% (for siRNA YB-1-2); this corresponded to rescue
rates of 27 and 33%, respectively, which were significantly lower
than the rescue rate obtained by IL-7 treatment of control
siRNA-transfected cells (Fig. 5B;
P<0.005 and P<0.01, respectively). This result indicates that
YB-1 knockdown reduces the ability of IL-7 to protect BCP ALL cells
against rapamycin.
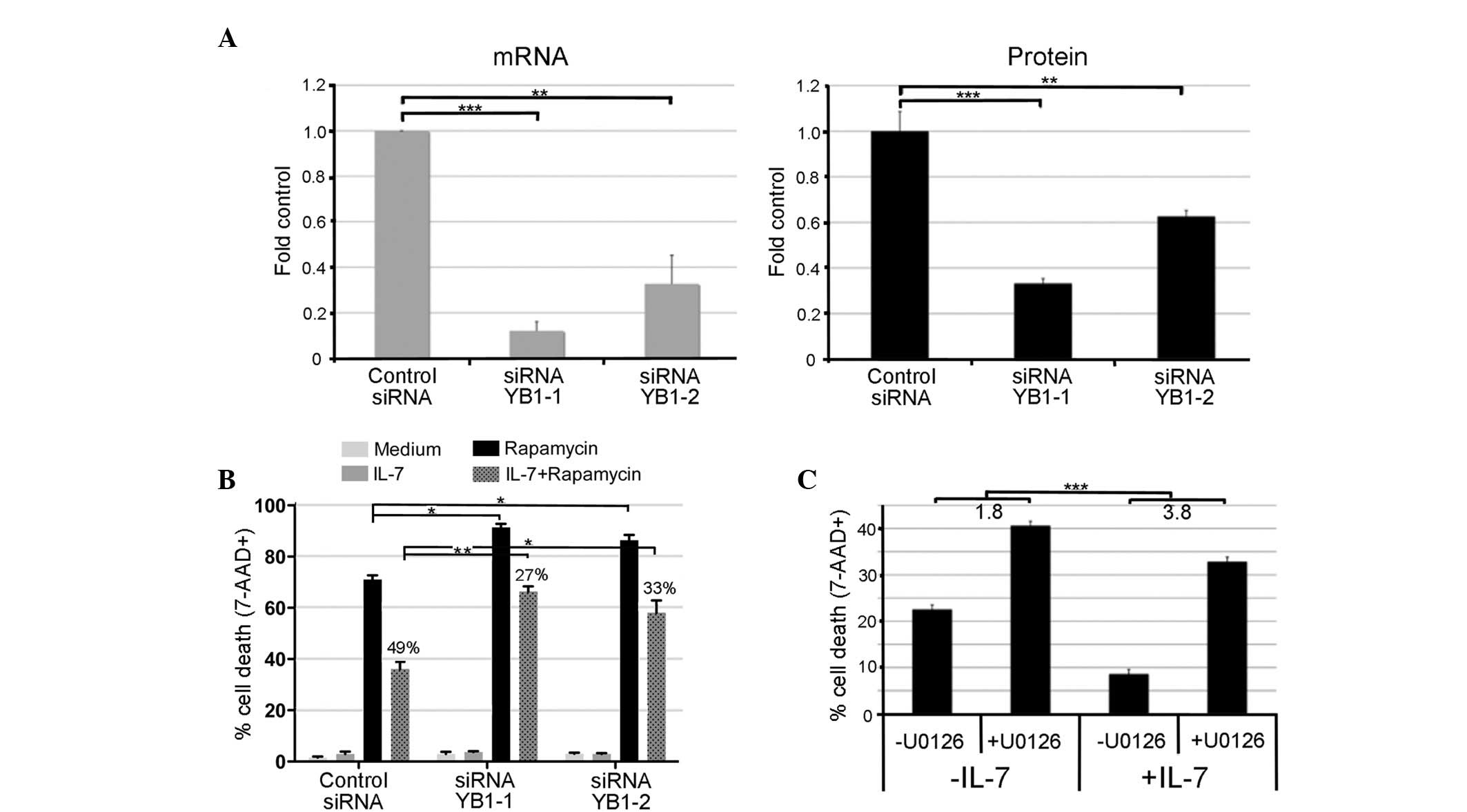 | Figure 5.YB-1 contributes to IL-7-mediated
protection against rapamycin-induced cell death. CALL-2 cells were
electroporated with 200 nM non-specific control siRNA, siRNA YB-1-1
or siRNA YB-1-2. Figures are compiled from a minimum of three
experiments. (A) YB-1 siRNA-mediated knockdown of mRNA (left panel)
and protein (right panel) 72 h after transfection. Results are
expressed as the fold of the negative control + standard deviation.
(B) YB-1 involvement in IL-7 protection against rapamycin
challenge. CALL-2 cells were transfected with siRNA against YB-1 or
control siRNA, and 48 h later they were treated overnight with
medium or 25 ng/ml IL-7, and then for further 8 h with or without
140 µM rapamycin. The average percentage of dead cells + standard
error of the mean compiled from three experiments are presented and
the numbers indicate the percentage of rescue following IL-7
treatment compared with cells transfected with control siRNA. (C)
The effect of a combination of U0126 and rapamycin in the absence
and presence of IL-7. CALL-2 cells were treated with 10 µM U0126 or
vehicle for 2 h prior to overnight stimulation with PBS or 25 ng/ml
IL-7. U0126 and PBS or IL-7 treatment was repeated the next day.
Cells were incubated for a further 18 h with 34 µM rapamycin 30 min
after the second addition of IL-7 and the percentage of cell death
was determined by 7-AAD exclusion. Results from three independent
experiments are presented. Numbers indicate the fold difference
with and without IL-7 treatment. *P<0.01, **P<0.005,
***P<0.001. siRNA, small interfering RNA; IL-7, interleukin 7;
YB-1, Y-box-binding protein-1; PBS, phosphate-buffered saline;
STAT5, signal transducer and activator of transcription 5; DMSO,
dimethyl sulfoxide; U0126, MEK1/2 inhibitor; 7-AAD,
7-aminoactinomycin D. |
To confirm the validity of the knockdown experiment
results, the ability of MEK1/2 inhibition by U0126 (which was shown
to completely inhibit IL-7-induced YB-1 phosphorylation) to augment
cell death in response to rapamycin in cells with and without IL-7
pretreatment was evaluated. Rapamycin was added to U0126-treated
cells which had been exposed to IL-7 or medium, and the percentage
of dead cells was determined 18 h later (Fig. 5C). In the presence of IL-7, MEK
inhibition increased the rate of rapamycin-induced apoptosis from
9–33% (P<0.001). Additionally, MEK inhibition increased
rapamycin-mediated apoptosis in cells not treated with IL-7 (from
24–41%, P<0.001) indicating downstream involvement of
additional, IL-7-independent survival pathways. However, the
U0126-mediated increase in cell death was greater in IL-7-treated
cells than untreated cells (3.8 fold vs. 1.8 fold, P<0.001),
indicating a significant contribution from IL-7-mediated survival
pathways. These results identify YB-1 as a significant factor in
the IL-7-mediated protection of BCP ALL against rapamycin.
Discussion
To the best of our knowledge, the present study
provides the first evidence for the involvement of YB-1 in IL-7
signaling in B cells. The results suggest that crosstalk occurs
between these proteins, with increased YB-1 levels contributing to
IL-7Rα upregulation, and IL-7 signaling leading to YB-1
phosphorylation. While the full implications of this interaction
remain unknown, the crosstalk mediates a clear survival signal for
BCP ALL cells. Although the current study is limited by the lack of
matched samples, the increase in YB-1 expression between diagnosis
and relapse ALL samples is in accordance with its role in
chemotherapy resistance described for other malignancies (24,25) and
suggests the physiological relevance of this pathway for survival
during therapy. Although YB-1 has not been identified in RNA
microarray-based studies comparing BCP ALL samples at diagnosis and
relapse, evidence is emerging that indicates that this protein is
largely regulated at a post-transcriptional level (41).
The IL-7 receptor and YB-1 are overexpressed in
malignant compared with normal BCP cells. It has been demonstrated
that YB-1 increases the expression of growth factor receptors in
prostate and breast cancer (19,20).
Transfection of YB-1 into normal B cells led to the upregulation of
YB-1 expression in the majority of transfected cells; however, the
induction of IL-7Rα expression occurred in only a proportion of
these, suggesting that this relationship is influenced by
additional factors. These data support the hypothesis that YB-1 may
contribute to IL-7Rα overexpression, which in turn may contribute
to the development or maintenance of malignant BCP cells by binding
IL-7 or thymic stromal lymphopoietin (TSLP) (42), a cytokine which signals through a
dimer composed of cytokine receptor-like factor 2 and IL-7Rα. The
pro-leukemic potential of such signaling is suggested by the gain
of function mutations in IL-7Rα or associated pathways that occur
in the context of TSLP signaling and have been described in BCP ALL
(43,44).
Addition of IL-7 to BCP ALL cells induces YB-1
phosphorylation at S102. In previous studies using breast cancer
cells, Akt (16,18) and RSK (34) have been implicated as the serine
kinase responsible for YB-1 phosphorylation; the signaling pathway
may vary according to cell type and environmental context. In the
current study, the pharmacological blockade of ERK1/2 signaling by
MEK1/2 inhibition, but not of Akt signaling by PI3K inhibition,
completely abolished the IL-7-mediated phosphorylation of S102.
Downstream targets of ERK remain to be identified; however, RSK2,
which phosphorylates YB-1 in breast cancer cells (34) and is induced by IL-7 in BCP ALL, is a
primary candidate. Phosphorylation of YB-1S102 activates
the translation of mRNA involved in growth and survival, while
direct phosphorylation or subsequent stress induces nuclear
localization enabling YB-1 to exert transcriptional control on a
further subset of genes, including those involved in drug
resistance (16,45). In contrast to results from studies on
squamous carcinoma cells (46),
reduced leukemia cell viability was not observed following YB-1
knockdown alone. The current study, however, implicates the
activation of YB-1 by IL-7 as a mechanism contributing to the
survival of BCP ALL in the presence of cytotoxic stressors.
IL-7 inhibits the induction of apoptosis by the mTOR
inhibitor rapamycin (13).
Curtailment of YB-1 activity, through targeted knockdown or
pharmacological inhibition, reduced the IL-7-mediated protection
against rapamycin. mTOR is a translational regulator of mRNA which
is involved in cell cycle progression, proliferation and survival.
IL-7, mTOR and YB-1 may work in synergy to augment survival: IL-7
can induce mTOR downstream of Akt (47), YB-1 is involved in the mTOR-mediated
increase of translation downstream of growth factor-induced
PI3K/Akt (15), and mTOR may
influence the translation of YB-1 (41). IL-7-mediated induction of YB-1
requires ERK rather than Akt in BCP ALL, and the induced survival
pathway is effective against inhibitors of mTOR; therefore, it is
assumed that this pathway is at least partly mTOR-independent.
Factors downstream of IL-7 and YB-1 that contribute to increased
survival remain to be determined. One potential explanation is
increased expression of the ATP-binding cassette transporter
multi-drug resistance 1, which is known to influence YB-1-mediated
drug resistance (25,27). When activated downstream of IL-7, YB-1
may also contribute to survival by stimulating a STAT5-mediated
increase in B-cell lymphoma 2 expression (38,48).
Finally, it must be noted that the ability of IL-7 to protect cells
from cytotoxins may be difficult to observe in cell monocultures as
it is likely to depend on interaction with stromal cells (49).
In conclusion, although the functional analysis of
the present study is limited to BCP ALL cells, the observation that
YB-1 and IL-7Rα expression are closely correlated in a
non-malignant subset of BCP cells, and that augmented YB-1
expression drives increased IL-7R levels on normal CD19+
B cells, suggests that this novel interaction may also have
relevance to B cell biology. The similarly elevated levels of YB-1
in BCP ALL cells at diagnosis and in the normal BCP cell subset is
intriguing; however, further studies are required to determine
whether this reflects the fact that leukemia frequently arises from
this subset of BCP cells or that there is selective pressure for
ALL cells to recapitulate the pathway. YB-1 directs
differentiation, proliferation and survival programs in a broad
range of cells (25); therefore, it
is conceivable that YB-1 pathways mediate the expression of factors
that facilitate transformation by increasing the potential for
self-renewal and drug resistance, as has been reported for breast
cancer cells (50). Overall, the
results of the present study suggest that inhibition of YB-1 may
represent a novel strategy to increase the susceptibility of BCP
ALL to mTOR inhibitors.
Acknowledgements
The authors would like to thank the following
CFRI-affiliated researchers: Mr. Johan VanMeerlo for providing
technical support, Dr Laura Sly and Dr Chinten James Lim for useful
discussions, Dr Pascal Lavoie for arranging cord blood, and from
Life Technologies, Dr. John Verberg for providing technical
support. The current study was supported by the Michael Cuccione
Foundation at British Columbia Children's Hospital and by a grant
from the Canadian Institute for Health Research (no.
MOP-97905).
Glossary
Abbreviations
Abbreviations:
|
YB-1
|
Y-box-binding protein-1
|
|
BCP
|
B-cell precursor
|
|
ALL
|
acute lymphoblastic leukemia
|
|
IL-7R
|
interleukin-7 receptor
|
References
|
1
|
Pui CH, Relling MV and Downing JR: Acute
lymphoblastic leukemia. N Engl J Med. 350:1535–1548. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsai AG, Lu H, Raghavan SC, Muschen M,
Hsieh CL and Lieber MR: Human chromosomal translocations at CpG
sites and a theoretical basis for their lineage and stage
specificity. Cell. 135:1130–1142. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Papaemmanuil E, Rapado I, Li Y, Potter NE,
Wedge DC, Tubio J, Alexandrov LB, Van Loo P, Cooke SL, Marshall J,
et al: RAG-mediated recombination is the predominant driver of
oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia.
Nat Genet. 46:116–125. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Swaminathan S, Klemm L, Park E,
Papaemmanuil E, Ford A, Kweon SM, Trageser D, Hasselfeld B, Henke
N, Mooster J, et al: Mechanisms of clonal evolution in childhood
acute lymphoblastic leukemia. Nat Immunol. 16:766–774. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mandal M, Powers SE, Ochiai K,
Georgopoulos K, Kee BL, Singh H and Clark MR: Ras orchestrates exit
from the cell cycle and light-chain recombination during early B
cell development. Nat Immunol. 10:1110–1117. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Corcoran AE, Smart FM, Cowling RJ,
Crompton T, Owen MJ and Venkitaraman AR: The interleukin-7 receptor
alpha chain transmits distinct signals for proliferation and
differentiation during B lymphopoiesis. EMBO J. 15:1924–1932.
1996.PubMed/NCBI
|
|
7
|
Milne CD and Paige CJ: IL-7: A key
regulator of B lymphopoiesis. Semin Immunol. 18:20–30. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Touw I, Pouwels K, van Agthoven T, van
Gurp R, Budel L, Hoogerbrugge H, Delwel R, Goodwin R, Namen A and
Löwenberg B: Interleukin-7 is a growth factor of precursor B and T
acute lymphoblastic leukemia. Blood. 75:2097–2101. 1990.PubMed/NCBI
|
|
9
|
Nishii K, Katayama N, Miwa H, Shikami M,
Masuya M, Shiku H and Kita K: Survival of human leukaemic B-cell
precursors is supported by stromal cells and cytokines: Association
with the expression of bcl-2 protein. Br J Haematol. 105:701–710.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barata JT, Keenan TD, Silva A, Nadler LM,
Boussiotis VA and Cardoso AA: Common gamma chain-signaling
cytokines promote proliferation of T-cell lymphoblastic leukemia.
Haematologica. 89:1459–1467. 2004.PubMed/NCBI
|
|
11
|
Silva A, Laranjeira AB, Martins LR,
Cardoso BA, Demengeot J, Yunes JA, Seddon B and Barata JT: IL-7
contributes to the progression of human T-cell acute lymphoblastic
leukemias. Cancer Res. 71:4780–4789. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thiant S, Yakoub-Agha I, Magro L, Trauet
J, Coiteux V, Jouet JP, Dessaint JP and Labalette M: Plasma levels
of IL-7 and IL-15 in the first month after myeloablative BMT are
predictive biomarkers of both acute GVHD and relapse. Bone Marrow
Transplant. 45:1546–1552. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brown VI, Fang J, Alcorn K, Barr R, Kim
JM, Wasserman R and Grupp SA: Rapamycin is active against
B-precursor leukemia in vitro and in vivo, an effect that is
modulated by IL-7-mediated signaling. Proc Natl Acad Sci USA.
100:15113–15118. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kosnopfel C, Sinnberg T and Schittek B:
Y-box binding protein 1-A prognostic marker and target in tumour
therapy. Eur J Cell Biol. 93:61–70. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Evdokimova V, Ovchinnikov LP and Sorensen
PH: Y-box binding protein 1: Providing a new angle on translational
regulation. Cell Cycle. 5:1143–1147. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Evdokimova V, Ruzanov P, Anglesio MS,
Sorokin AV, Ovchinnikov LP, Buckley J, Triche TJ, Sonenberg N and
Sorensen PH: Akt-mediated YB-1 phosphorylation activates
translation of silent mRNA species. Mol Cell Biol. 26:277–292.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koike K, Uchiumi T, Ohga T, Toh S, Wada M,
Kohno K and Kuwano M: Nuclear translocation of the Y-box binding
protein by ultraviolet irradiation. FEBS Lett. 417:390–394. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sutherland BW, Kucab J, Wu J, Lee C,
Cheang MC, Yorida E, Turbin D, Dedhar S, Nelson C, Pollak M, et al:
Akt phosphorylates the Y-box binding protein 1 at Ser102 located in
the cold shock domain and affects the anchorage-independent growth
of breast cancer cells. Oncogene. 24:4281–4292. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shiota M, Takeuchi A, Song Y, Yokomizo A,
Kashiwagi E, Uchiumi T, Kuroiwa K, Tatsugami K, Fujimoto N, Oda Y
and Naito S: Y-box binding protein-1 promotes castration-resistant
prostate cancer growth via androgen receptor expression. Endocr
Relat Cancer. 18:505–517. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stratford AL, Habibi G, Astanehe A, Jiang
H, Hu K, Park E, Shadeo A, Buys TP, Lam W, Pugh T, et al: Epidermal
growth factor receptor (EGFR) is transcriptionally induced by the
Y-box binding protein-1 (YB-1) and can be inhibited with Iressa in
basal-like breast cancer, providing a potential target for therapy.
Breast Cancer Res. 9:R612007. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Davies AH and Dunn SE: YB-1 drives
preneoplastic progression: Insight into opportunities for cancer
prevention. Oncotarget. 2:401–406. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim ER, Selyutina AA, Buldakov IA,
Evdokimova V, Ovchinnikov LP and Sorokin AV: The proteolytic YB-1
fragment interacts with DNA repair machinery and enhances survival
during DNA damaging stress. Cell Cycle. 12:3791–3803. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bargou RC, Jürchott K, Wagener C, Bergmann
S, Metzner S, Bommert K, Mapara MY, Winzer KJ, Dietel M, Dörken B
and Royer HD: Nuclear localization and increased levels of
transcription factor YB-1 in primary human breast cancers are
associated with intrinsic MDR1 gene expression. Nat Med. 3:447–450.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chatterjee M, Rancso C, Stühmer T,
Eckstein N, Andrulis M, Gerecke C, Lorentz H, Royer HD and Bargou
RC: The Y-box binding protein YB-1 is associated with progressive
disease and mediates survival and drug resistance in multiple
myeloma. Blood. 111:3714–3722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lasham A, Print CG, Woolley AG, Dunn SE
and Braithwaite AW: YB-1: Oncoprotein, prognostic marker and
therapeutic target? Biochem J. 449:11–23. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Szczuraszek K, Halon A, Materna V, Mazur
G, Wrobel T, Kuliczkowski K, Donizy P, Holm PS, Lage H and Surowiak
P: Elevated YB-1 expression is a new unfavorable prognostic factor
in non-Hodgkin's lymphomas. Anticancer Res. 31:2963–2970.
2011.PubMed/NCBI
|
|
27
|
Shen H, Xu W, Luo W, Zhou L, Yong W, Chen
F, Wu C, Chen Q and Han X: Upregulation of mdr1 gene is related to
activation of the MAPK/ERK signal transduction pathway and YB-1
nuclear translocation in B-cell lymphoma. Exp Hematol. 39:558–569.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hanzawa K, Momose S, Higashi M, Tokuhira
M, Watanabe R, Kajino K, Hino O, Itoyama S, Kizaki M and Tamaru J:
Y-box binding protein-1 expression in diffuse large B-cell
lymphoma: An impact on prognosis in the rituximab era. Leuk
Lymphoma. 51:2054–2062. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Castellana B, Aasen T, Moreno-Bueno G,
Dunn SE and Ramón y Cajal S: Interplay between YB-1 and IL-6
promotes the metastatic phenotype in breast cancer cells.
Oncotarget. 6:38239–38256. 2015.PubMed/NCBI
|
|
30
|
Barrett DM, Seif AE, Carpenito C, Teachey
DT, Fish JD, June CH, Grupp SA and Reid GS: Noninvasive
bioluminescent imaging of primary patient acute lymphoblastic
leukemia: A strategy for preclinical modeling. Blood.
118:e112–e117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guay D, Evoy AA, Paquet E, Garand C,
Bachvarova M, Bachvarov D and Lebel M: The strand separation and
nuclease activities associated with YB-1 are dispensable for
cisplatin resistance but overexpression of YB-1 in MCF7 and
MDA-MB-231 breast tumor cells generates several chemoresistance
signatures. Int J Biochem Cell Biol. 40:2492–2507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brown VI, Fang J, Alcorn K, Barr R, Kim
JM, Wasserman R and Grupp SA: Rapamycin is active against
B-precursor leukemia in vitro and in vivo, an effect that is
modulated by IL-7-mediated signaling. Proc Natl Acad Sci USA.
100:15113–15118. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fujii T, Kawahara A, Basaki Y, Hattori S,
Nakashima K, Nakano K, Shirouzu K, Kohno K, Yanagawa T, Yamana H,
et al: Expression of HER2 and estrogen receptor alpha depends upon
nuclear localization of Y-box binding protein-1 in human breast
cancers. Cancer Res. 68:1504–1512. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Basaki Y, Hosoi F, Oda Y, Fotovati A,
Maruyama Y, Oie S, Ono M, Izumi H, Kohno K, Sakai K, et al:
Akt-dependent nuclear localization of Y-box-binding protein 1 in
acquisition of malignant characteristics by human ovarian cancer
cells. Oncogene. 26:2736–2746. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stratford AL, Fry CJ, Desilets C, Davies
AH, Cho YY, Li Y, Dong Z, Berquin IM, Roux PP and Dunn SE: Y-box
binding protein-1 serine 102 is a downstream target of p90
ribosomal S6 kinase in basal-like breast cancer cells. Breast
Cancer Res. 10:R992008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
van der Plas DC, Smiers F, Pouwels K,
Hoefsloot LH, Löwenberg B and Touw IP: Interleukin-7 signaling in
human B cell precursor acute lymphoblastic leukemia cells and
murine BAF3 cells involves activation of STAT1 and STAT5 mediated
via the interleukin-7 receptor alpha chain. Leukemia. 10:1317–1325.
1996.PubMed/NCBI
|
|
38
|
Venkitaraman AR and Cowling RJ:
Interleukin-7 induces the association of phosphatidylinositol
3-kinase with the alpha chain of the interleukin-7 receptor. Eur J
Immunol. 24:2168–2174. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Steelman LS, Abrams SL, Whelan J, Bertrand
FE, Ludwig DE, Bäsecke J, Libra M, Stivala F, Milella M, Tafuri A,
et al: Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and
Jak/STAT pathways to leukemia. Leukemia. 22:686–707. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fleming HE and Paige CJ: Pre-B cell
receptor signaling mediates selective response to IL-7 at the Pro-B
to Pre-B cell transition via an ERK/MAP kinase-dependent pathway.
Immunity. 15:521–531. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lyabin DN, Eliseeva IA and Ovchinnikov LP:
YB-1 synthesis is regulated by mTOR signaling pathway. PLoS One.
7:e525272012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Brown VI, Hulitt J, Fish J, Sheen C, Bruno
M, Xu Q, Carroll M, Fang J, Teachey D and Grupp SA: Thymic
stromal-derived lymphopoietin induces proliferation of pre-B
leukemia and antagonizes mTOR inhibitors, suggesting a role for
interleukin-7Ralpha signaling. Cancer Res. 67:9963–9970. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shochat C, Tal N, Bandapalli OR, Palmi C,
Ganmore I, te Kronnie G, Cario G, Cazzaniga G, Kulozik AE, Stanulla
M, et al: Gain-of-function mutations in interleukin-7 receptor-α
(IL7R) in childhood acute lymphoblastic leukemias. J Exp Med.
208:901–908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zenatti PP, Ribeiro D, Li W, Zuurbier L,
Silva MC, Paganin M, Tritapoe J, Hixon JA, Silveira AB, Cardoso BA,
et al: Oncogenic IL7R gain-of-function mutations in childhood
T-cell acute lymphoblastic leukemia. Nat Genet. 43:932–939. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Somasekharan SP, Stoynov N, Rotblat B,
Leprivier G, Galpin JD, Ahern CA, Foster LJ and Sorensen PH:
Identification and quantification of newly synthesized proteins
translationally regulated by YB-1 using a novel Click-SILAC
approach. J Proteomics. 77:e1–e10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Troiano A, Lomoriello IS, di Martino O,
Fusco S, Pollice A, Vivo M, La Mantia G and Calabrò V: Y-box
binding Protein-1 Is part of a complex molecular network linking
ΔNp63α to the PI3K/akt pathway in cutaneous squamous cell
carcinoma. J Cell Physiol. 230:2067–2074. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Silva A, Cebola I, Santos CI, Antunes F
and Barata JT: Intracellular reactive oxygen species are essential
for PI3K/Akt/mTOR-dependent IL-7-mediated viability of T-cell acute
lymphoblastic leukemia cells. Leukemia. 25:960–967. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Winston LA and Hunter T: JAK2, Ras, and
Raf are required for activation of extracellular signal-regulated
kinase/mitogen-activated protein kinase by growth hormone. J Biol
Chem. 270:30837–30840. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Johnson SE, Shah N, Panoskaltsis-Mortari A
and LeBien TW: Murine and human IL-7 activate STAT5 and induce
proliferation of normal human pro-B cells. J Immunol.
175:7325–7331. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
To K, Fotovati A, Reipas KM, Law JH, Hu K,
Wang J, Astanehe A, Davies AH, Lee L, Stratford AL, et al: Y-box
binding protein-1 induces the expression of CD44 and CD49f leading
to enhanced self-renewal, mammosphere growth, and drug resistance.
Cancer Res. 70:2840–2851. 2010. View Article : Google Scholar : PubMed/NCBI
|