Introduction
Polyphenols, such as epigallocatechin gallate,
resveratrol and flavonoids, exist in nature (1). Flavonoids are a group of polyphenols
that share similar chemical features and are abundant in
vegetables, fruits and tea, amongst other products (2). Humans absorb a large amount of
flavonoids orally, and epidemiological studies have revealed that
the risk of certain types of cancer, particularly cancers of the
breast, digestive tract, skin, prostate and certain hematological
malignancies, is inversely correlated with intake of flavonoids
(3,4).
Flavonoids possess antioxidative, anti-inflammatory and antitumor
properties (5). A number of flavonoid
compounds have previously been demonstrated to enhance the effects
of conventional chemotherapeutic drugs in cancer cells, prompting
increased attention (6–8).
Apigenin (4′,5,7-trihydroxyflavone) is a promising
chemopreventive agent that is abundantly present in fruits,
vegetables, and teas (3). This
compound has anticarcinogenic properties via diverse mechanisms and
is suggested to have a tumor preventative effect (3,9). The
mechanism of action of apigenin appears to involve p53, as apigenin
(15–60 µM) has previously been
reported to induce the necrosis and apoptosis of neuroblastoma
cells expressing wild-type, but not mutant, p53. Apigenin was
suggested to elevate the levels of p53, and p53 effector gene
expression in these neuroblastoma cells (10). Apigenin also causes cell cycle arrest
and sensitizes leukemia cells to vincristine (11). Apigenin has additionally been reported
to induce apoptosis of prostate and colon cancer cells through
induction of death receptor 5, and to act synergistically with
exogenous tumor necrosis factor-related apoptosis-inducing ligand
to induce cell apoptosis (12).
Furthermore, apigenin enhances the anticancer activity or minimizes
the resistance of cancer cells to a number chemotherapeutics,
including gemcitabine, paclitaxel, 5-fluorouracil and doxorubicin
by inducing apoptosis (13–17). It was reported that apigenin
sensitizes neuroblastoma cells to the anticancer drug etoposide by
retention of p53 in cell nuclei (18).
The present study aimed to determine the effects of
apigenin on cisplatin cytotoxic activity using several tumor cell
lines, and to elucidate the potential mechanisms of this. Cisplatin
is a member of a class of platinum-containing anticancer drugs,
which has demonstrated therapeutic properties against a broad range
of cancers (19,20). Cisplatin exerts a DNA-binding effect
by cross-linking DNA in several different ways, interfering with
mitotic cell division. The damaged DNA prompts induction of DNA
repair mechanisms, which activates apoptotic mechanisms when repair
proves impossible (21). Many tumors
demonstrate good responsiveness to this drug, but drug resistance
eventually develops, particularly in patients with lung,
colorectal, prostate and ovarian cancer (22). Several approaches have been proposed
to maintain cisplatin efficacy, including increased accumulation or
stabilization of p53, upregulating reactive oxygen species
production and inhibiting NF-κB and antiapoptotic proteins
(23–26). The current study investigated the
effects of apigenin on p53 accumulation, and its effect on
cisplatin cytotoxic activity in several tumor cell lines. The
present results demonstrated that apigenin enhances the inhibition
of proliferation observed following cisplatin treatment in a
p53-dependent manner, potentially enhancing the antitumor effects
of cisplatin.
Materials and methods
Cell culture and inhibition of cell
growth
The HeLa, A549, HCT 116, H1299, and MCF-7 cells were
obtained from the Cell Bank of the Chinese Academy of Sciences and
cultured in high-glucose Dulbecco's Modified Eagle's medium (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). H1299 cells were
cultured in RPMI 1640 medium (Thermo Fisher Scientific, Inc.).
Apigenin, U0126 (a mitogen-activated protein kinase kinase
inhibitor), MTT (all Sigma-Aldrich; Merck Millipore, Darmstadt,
Germany) and cisplatin (Jiangsu Hansoh Pharm. Co., Ltd.,
Lianyungang, China) were obtained commercially. To determine the
effect of the compounds on cell viability, MTT assays were
performed. Briefly, ~5×103 cells per well were seeded in
96-well plates (Corning Incorporated, Corning, NY, USA) for 24 h at
37°C. The cells were treated in triplicate with apigenin (in DMSO;
final concentration, ≤0.1%) or cisplatin (in PBS). These treatments
were 30 µM apigenin for 2 h, followed by different doses of
cisplatin (2.5, 5.0 and 10 µM) for another 48 h. At the end of
treatment, MTT was added to determine cell viability as previously
reported (27).
Western blotting
Western blotting was performed to monitor protein
expression and modifications using a standard protocol (28). Briefly, total protein was extracted
from the cultured cells using lysis buffer (Sigma-Aldrich; Merck
Millipore). The proteins were separated by 10% SDS-PAGE and then
transferred onto polyvinylidene difluoride membranes. The membranes
were blocked with 2% bovine serum albumin (Roche Diagnostics,
Basel, Switzerland) at room temperature for 1 h and incubated
overnight at 4°C with primary antibodies. The primary antibodies
were against caspase-3 (catalog no. 9662S; dilution, 1:1,000),
caspase-9 (catalog no. 9502S; dilution, 1:1,000), poly ADP ribose
polymerase (PARP; catalog no. 9542S; dilution, 1:2,000), c-Jun
N-terminal kinase (JNK1/2; catalog no. 9252S; dilution, 1:1,000),
p-JNK (catalog no. 9251S; dilution, 1:1,000), p38 (catalog no.
9212S; dilution, 1:1,000), p-p38 (catalog no. 9211S; dilution,
1:1,000), cleaved caspase-9 (catalog no. 9501S; dilution, 1:1,000),
cleaved caspase-3 (catalog no. 9661S; dilution, 1:2,000) (all Cell
Signaling Technology, Inc., Danvers, MA, USA), extracellular
signal-regulated kinase (Erk)2 (catalog no. sc-154; dilution,
1:4,000), p-Erk (catalog no. sc-7383; dilution, 1:1,000), caspase-8
(catalog no. sc-56070, dilution, 1:1,000), Bax (catalog no. sc-493;
dilution, 1:200), p53 (catalog no. sc-47698; dilution, 1:1,000),
p-p53/Ser15 (catalog no. sc-101762; dilution, 1:1,000), mouse
double minute 2 homolog (MDM2; catalog no. sc-5304; dilution,
1:1,000) (all Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
cleaved caspase-8 (catalog no. PA5-35558; dilution, 1:1,000;
Invitrogen; Thermo Fisher Scientific, Inc.) and GAPDH (catalog no.
AG019; 1:500; Beyotime Institute of Biotechnology, Haimen, China).
After washing, the membranes were incubated with the horseradish
peroxidase-conjugated secondary antibody (anti-mouse; catalog no.
A3682-1ML; dilution, 1:80,000; and anti-rabbit; catalog no.
A0545-1ML; dilution, 1:80,000; Abcam, Cambridge, UK) overnight at
4°C. The images were captured using a FluorChem HD2 imaging system
(Protein Simple, San Jose, CA, USA) following incubation with an
enhanced chemiluminescence reagent kit (Pierce; Thermo Fisher
Scientific, Inc.). Protein expression levels were compared with
GAPDH expression. The blots were quantified using Adobe Photoshop
CS6 (Adobe Inc., San Jose, CA, USA).
DAPI staining
Cell nuclei were stained with 4′,
6-diamidino-2-phenylindole (DAPI) to reveal morphological changes.
Briefly, cells (1×105/ml) were grown for 24 h at 37°C on
sterilized, fibronectin-coated coverslips to allow cells to attach
and spread. The cells were treated with the apigenin and cisplatin
(50, 100, 150 or 200 mg/ml) for 48 h, fixed using 4%
paraformaldehyde and stained with 50 µl/well of DAPI (1:2,000
dilution in TBST) for 5 min. The cells were then imaged under a
BX-53 fluorescence microscope (Olympus Corporation, Tokyo, Japan)
equipped with the QImaging system (Surrey, BC, Canada).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR was performed to determine p53 gene
expression in apigenin and apigenin-cisplatin treated cells. Total
RNA was extracted from the cultured cells using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. RT was performed using 5X AMV buffer, AMV
enzyme, 10 mM dNTP and R-primer (Takara Bio Inc., Otsu, Japan) at
16°C for 15 min, 42°C for 1 h and 85°C for 5 min. p53 levels were
determined with RT-qPCR. PCR was perfomed using 10X PCR buffer, 10
mM dNTP, Taq enzyme and primers from Takara Inc., and SYBR Green
from Applied Biosystems (Thermo Fisher Scientific, Inc.). The
following primers were used to amplify the p53 gene: Forward
primer, 5′-AACGGTACTCCGCCACC and reverse primer,
5′-CGTGTCACCGTCGTGGA. The following primers were used for the GAPDH
gene: Forward, 5′-ACCACAGTCCATGCCATCAC-3′ and reverse,
5′-TCCACCACCCTGTTGCTGTA-3′. The cycling conditions were as follows:
95°C for 5 min, 95°C for 15 sec and 60°C for 1 min, for 40 cycles.
The relative amount of gene normalized to control was calculated
using the 2−ΔΔCq method (29). The mean Cq was calculated from
triplicate PCRs.
Statistical analysis
All data were performed at least in triplicate. The
results were presented as the mean ± standard deviation of at least
three independent experiments. Statistical analysis was performed
using Student's t-test by utilizing IBM SPSS Statistics, version
20.0 (IBM, Armonk, NY, USA). P<0.05 was used to indicate a
statistically significant difference.
Results and Discussion
Apigenin enhances the cytotoxic effect
of cisplatin
To study the effect of apigenin on cisplatin-induced
cell death, A549, MCF-7, HCT 116 and HeLa cells that possess
wild-type p53 and H1299 cells, which are p53-null due to homozygous
deletion of the TP53 gene (30), were
treated with apigenin for 2 h, followed by different doses of
cisplatin for 48 h, as indicated. The cells were visually inspected
under an inverted fluorescence microscope at 24 and 48 h after
cisplatin treatment. Cisplatin treatment caused cell death at 48 h.
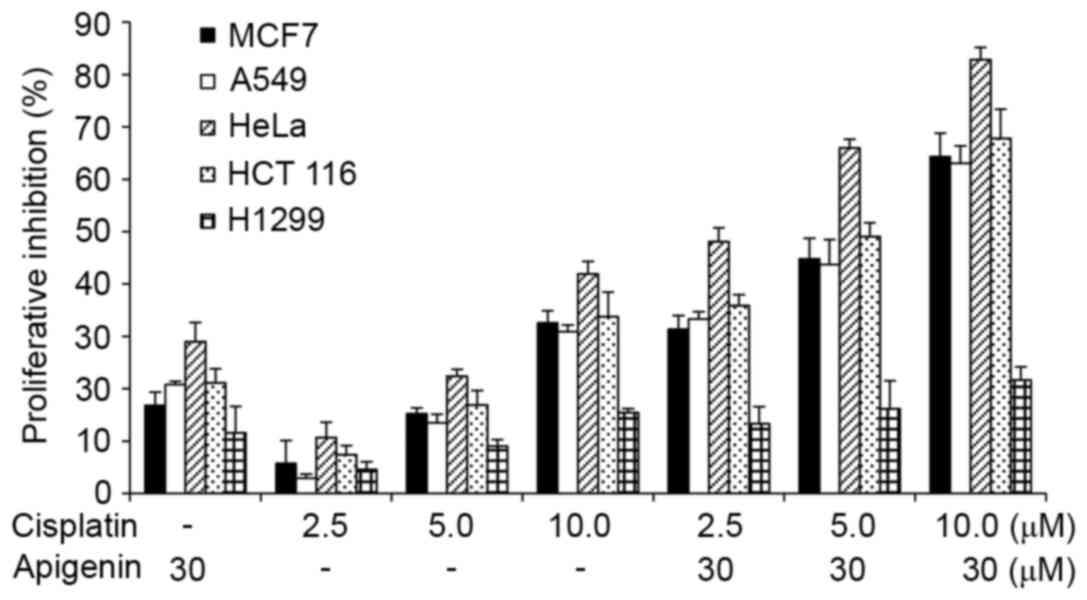
The effect was quantitatively measured using an MTT assay. As
reported in Fig. 1, apigenin
treatment alone caused a low viablity of cells to 10–30% in all
cell lines, while cisplatin treatment alone caused a viability of
3% at low doses to 42% at higher concentrations in HeLa cells.
Co-treatment of apigenin at 30 µM with cisplatin at 2.5, 5.0, and
10.0 µM increased the inhibitory effect of cisplatin in all cell
lines tested, except the H1299 cells.
Apigenin enhances cisplatin-induced
apoptosis
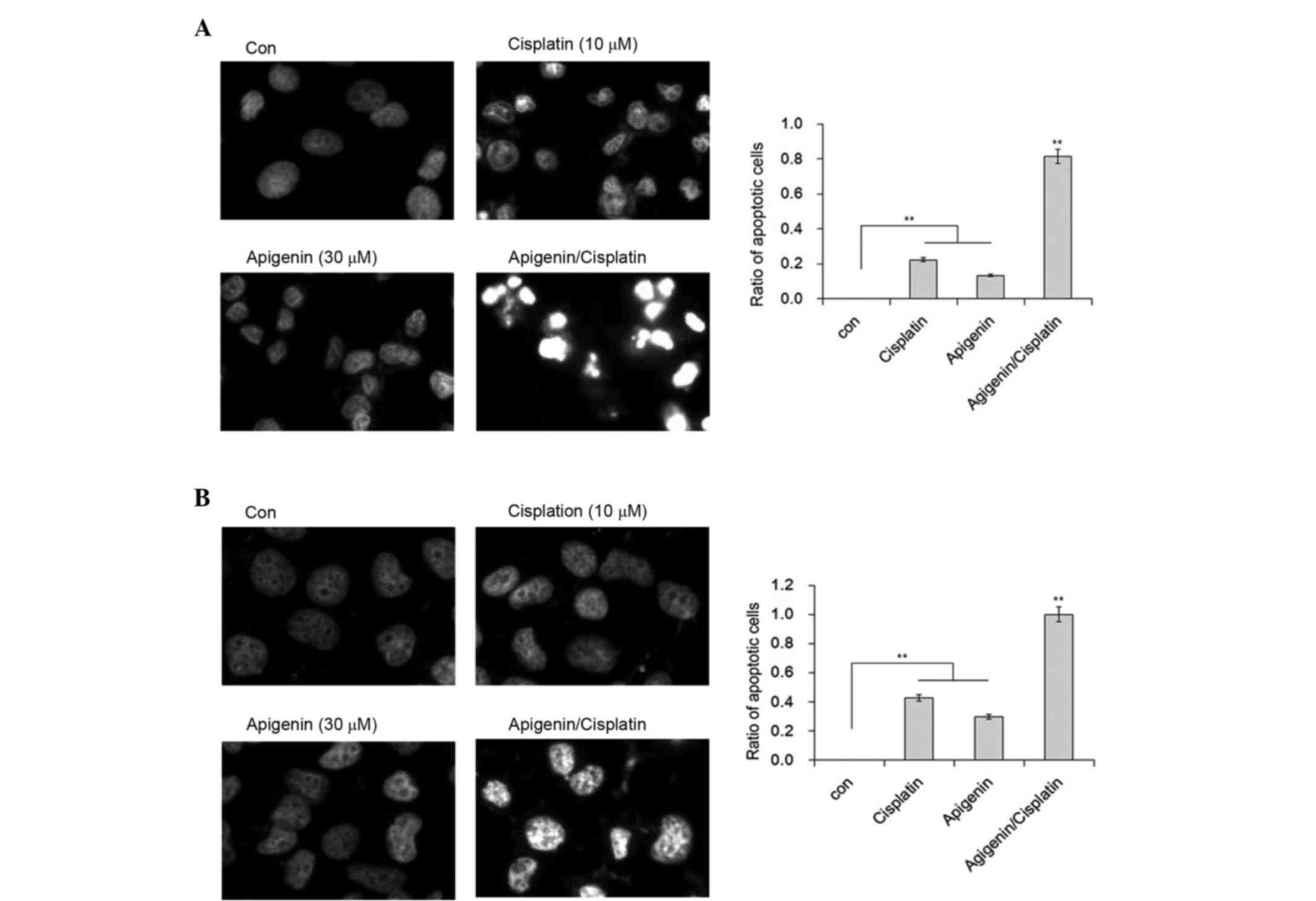
Cisplatin appeared to exert a cytotoxic effect via
the induction of apoptosis. Marked cell shrinkage resembling cell
apoptosis was observed in apigenin plus cisplatin-treated samples.
To confirm whether this shrinkage was due to apoptosis, DAPI
staining was performed to illustrate the nuclear morphology of A549
and H1299 cells. Although a proportion of A549 cells demonstrated
chromatin condensation in cells treated with cisplatin, inclusion
of apigenin resulted in marked changes to chromatin condensation,
nuclear shrinkage and the formation of apoptotic bodies in A549
cells (Fig. 2A). In comparison, no
nuclear fragmentation was observed in the H1299 cells under the
same conditions, although the formation of heterochromatin foci was
apparent in these cells (Fig. 2B).
These results indicate that apigenin is likely responsible for the
apoptotic effect of cisplatin in A549 cells.
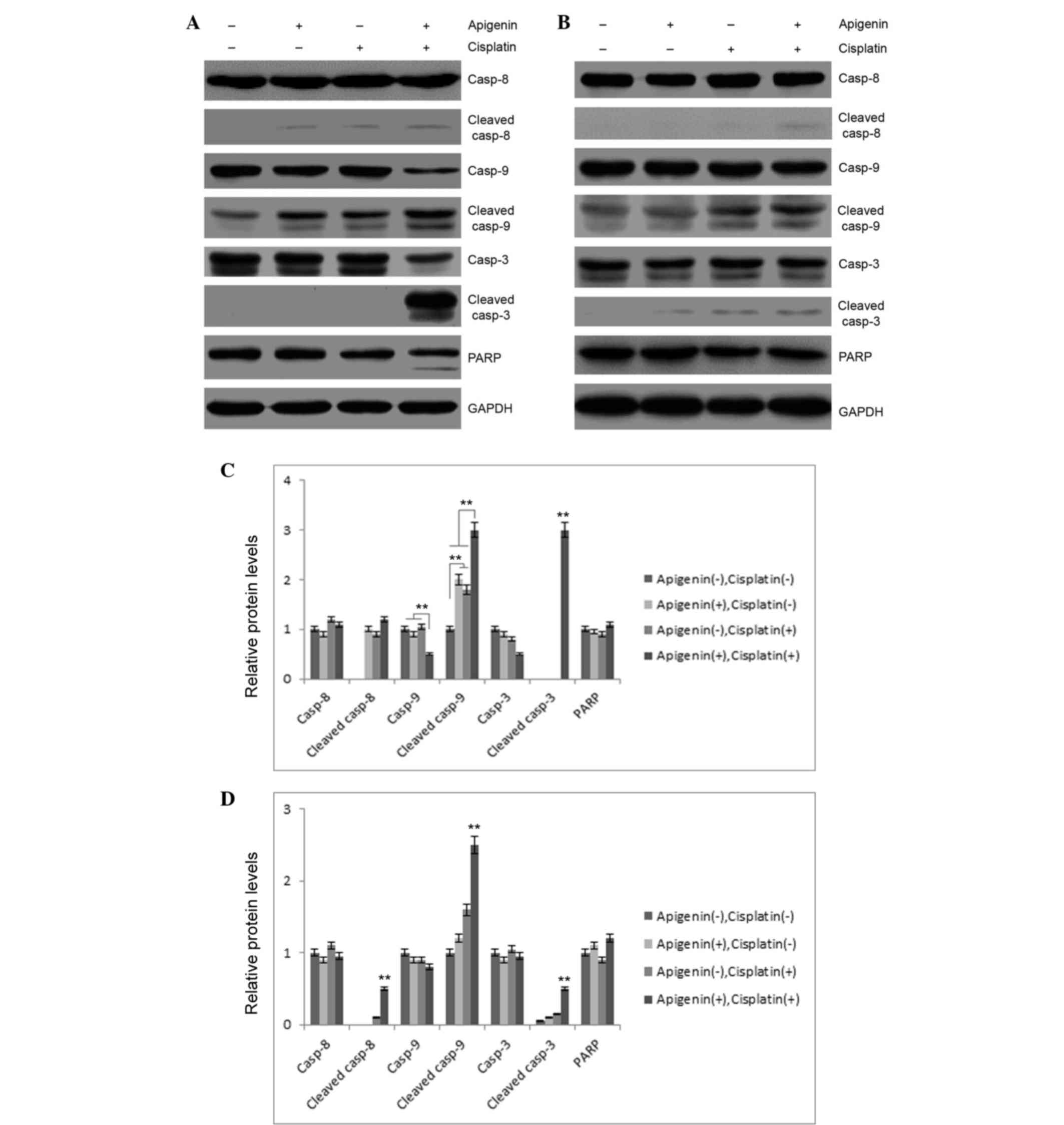
To investigate these results, the role of apigenin
and cisplatin treatment in caspase activation was examined. As
reported in Fig. 3A, apigenin (30 µM)
or cisplatin (10 µM) treatment in A549 cells resulted in partial
cleavage of caspase-9, while no marked cleavage of caspase-3 or −8
was detected in these samples. Combined application of apigenin and
cisplatin caused significant cleavage of caspase-3 and −9.
Concomitantly, PARP cleavage was also detected in apigenin and
cisplatin-treated cells (Fig. 3A),
indicating that combined application of apigenin and cisplatin
promoted activation of caspase-3 and −9. However, combinatorial
treatment did not cause significant cleavage of these caspases in
H1299 cells, which are p53-null (Fig.
3B). These results indicated that apigenin amplified the
apoptosis-inducing activity of cisplatin in A549 cells.
Apigenin promotes p53 phosphorylation
and accumulation
p53 may be functionally compromised by its
interaction with several proteins. Among those proteins, MDM2
serves as a pivotal negative regulator and counteracts p53
activation (31,32). p53 is maintained at constant levels in
quiescent cells as the protein undergoes constant degradation
mediated by MDM2 and the proteasome (33). Factors that promote N-terminal
phosphorylation of p53 protein cause disruption of p53-MDM2
interaction, resulting in p53 accumulation and transcriptional
regulation (34). In the present
study, immunoblotting was performed in A549 cells to determine
whether apigenin treatment caused p53 accumulation. In addition to
increased accumulation of p53 protein in apigenin-treated samples,
apigenin also dose-dependently induced p53 phosphorylation, as
detected by a phospho-Ser15 specific antibody (Fig. 4A). Compared with treatment using
cisplatin alone, combined treatment with apigenin and cisplatin
significantly elevated the levels of p53 protein in the sample
(Fig. 4B). Elevated p53 expression
could may result from increased gene expression or from
posttranslational modification, an event that can lead to p53
stabilization and accumulation (35).
However, the levels of p53 mRNA in the A549 cells that were treated
with apigenin and cisplatin for 6, 12 and 24 h were not
significantly altered, as determined by RT-PCR (data not shown);
this suggests that elevated expression of p53 protein was likely to
be associated with decreased degradation. Notably, increased p53
accumulation was associated with increased detection of Bax, a
proapoptotic protein, in the samples (Fig. 4B), suggesting that apigenin amplified
the cisplatin cytotoxic effect through induction of p53
accumulation and p53-regulated proapoptotic gene expression.
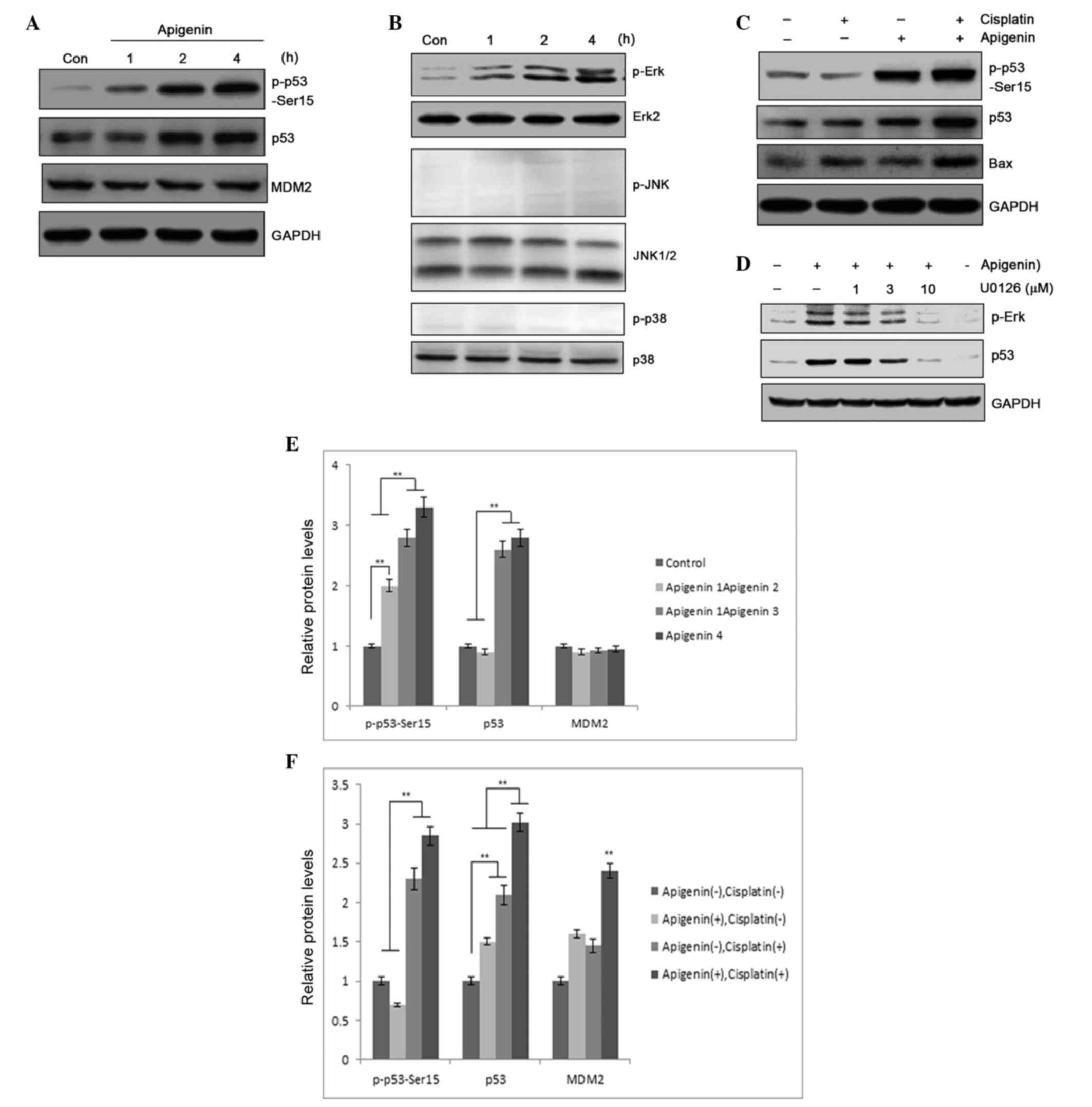 | Figure 4.(A) p53 expression and p53 Ser-15
phosphorylation was determined by western blotting, following
treatment of A549 cells with 30 µM apigenin for 1, 2 or 4 h. (B)
p53 and Bax expression, and p53 phosphorylation, as detected by
western blotting, following treatment of A549 cells with 30 µM
apigenin for 2 h, then 10 µM cisplatin for another 24 h. (C) MAPK
pathway molecule activation, determined by protein phosphorylation
levels, in A549 cells treated with 30 µM apigenin for 15, 30, 60 or
120 min, reported by western blotting. (D) Erk/MAPK activation
relative to p53 accumulation in A549 cells pretreated with U0126,
an Erk/MAPK inhibitor, then treated with 10 µM apigenin to induce
Erk/MAPK activation. (E) Semi-quantification of (A). (F)
Semi-quantification of (B). Con, control; MDM2, mouse double minute
2 homolog; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Erk,
extracellular signal-regulated kinase; JNK, c-Jun N-terminal
kinase; MAPK, mitogen-activated protein kinase.**P<0.01. |
Erk/mitogen-activated protein kinase
(MAPK) activation is responsible for induced p53
phosphorylation
MAPK activation has a critical role in p53
phosphorylation, which may promote p53 stabilization (36). An immunoblotting analysis was
performed in A549 cells to determine whether apigenin treatment
activated MAPK. Apigenin treatment was revealed to selectively
activate the Erk/MAPK pathway, while no activation of JNK or p38
MAPK was detected in apigenin-treated A549 cells (Fig. 4C). Erk/MAPK activation was determined
to be responsible for p53 phosphorylation, as pretreatment with
U0126, a specific inhibitor that blocks Erk/MAPK pathway activation
(37), dose-dependently blocked
apigenin-induced p53 phosphorylation (Fig. 4D). This suggests that Erk/MAPK pathway
activation is a contributing factor in apigenin-induced p53
accumulation. These results indicate that apigenin sensitizes tumor
cells to the cisplatin-associated cytotoxic effect via modification
of the p53 protein.
Apigenin is a dietary flavonoid that is hypothesized
to have a beneficial role in cancer chemotherapy (38). This compound has been reported to
induce cell cycle arrest, in addition to apoptosis when used at
varying concentrations (39). In the
current study, apigenin had the ability to amplify
cisplatin-induced apoptosis of cancer cells. Apigenin treatment
induced MAPK activation and subsequent p53 phosphorylation, leading
to its accumulation and increased effector gene activation. In
addition, increased caspase activation was also detected in samples
treated with both cisplatin and apigenin, suggesting that apigenin
sensitizes A549 cells to induced apoptosis through p53 accumulation
and altered effector gene regulation.
As a tumor suppressor, p53 has an important role in
genome stability and functions as a critical tumor suppressor that
is involved in preventing cancer (40). Multiple studies have verified the
important role p53-dependent apoptosis has in inhibiting
carcinogenesis (41). Cell lines of
lung cancer (A549), breast cancer (MCF-7), colorectal cancer (HCT
116) and cervical cancer (HeLa) that possess wild-type p53, and the
lung cancer line H1299, which does not generate functional p53,
were therefore examined, and the effects of combined treatment of
cisplatin with apigenin was determined in these cells. Apigenin
notably amplified the inhibitory effect on proliferation of
cisplatin in cells with wild-type p53, but not in the p53-null
H1299 cells. This was supported by the observed increased
activation of caspases-9 and −3, and the nuclear morphological
changes in A549 cells.
Posttranslational modification has a critical role
in p53 function (42,43). Consistent with this, increased
phosphorylation and accumulation of p53 was detected in
apigenin-treated samples. Apigenin significantly enhanced p53
phosphorylation. Increased p53 phosphorylation was revealed to be
modulated by MAPK, as apigenin treatment promoted MAPK activation;
this was blocked by application of U0126, a specific inhibitor of
the Erk/MAPK pathway. It is of note that the timing of MAPK (60
min) and p53 (2 h) activation potentially suggest a sequential
effect of these events, indicating that MAPK activation may have a
crucial role in p53 accumulation and the proapoptotic effect.
Apigenin has demonstrable activity in reducing
oxidative stress, inducing cell cycle inhibition and apoptosis, and
as an anti-inflammatory agent (44).
Foods high in apigenin include celery, parsley, tomatoes and red
wine (45,46). Apigenin is generally considered to be
safe when consumed in plant foods, with no toxic or mutagenic
effects previously reported (47). In
a population-based case-control study that included 1,141 patients
with ovarian cancer and 1,183 women of similar age to assess the
content of their diets, it was reported that flavonoid intake
lowered the risk of ovarian cancer (48). The current results demonstrate that
dietary flavonoids like apigenin sensitize tumor cells to
chemotherapeutic agents. Therefore, combined treatment of tumor
cells with conventional chemotherapeutic drugs and dietary
supplements provides a promising direction for cancer
chemotherapy.
Apigenin enhances the cytotoxic effect of cisplatin
during apoptosis in tumor cells, promotes p53 phosphorylation and
accumulation, and the Erk/MAPK pathway was revealed to be focal in
apigenin-induced p53 accumulation.
Acknowledgements
The present work was supported by National Natural
Science Foundation of China (grant nos. 81201946 and 81372394),
Ministry of Education Research Fund for Doctoral Programmes (grant
no. 2012202120013), the Science Foundation of the Medical
University Of Tianjin (grant no. 2011KY15), the National Research
Platform of Clinical Evaluation Technology for New Anticancer Drugs
(grant no. 2013ZX09303001). The authors thank Xiaoyan Li and Ziying
Yang for their technical assistance and Dr Erguang Li for English
editing.
References
|
1
|
Afzal M, Safer AM and Menon M: Green tea
polyphenols and their potential role in health and disease.
Inflammopharmacology. 23:151–161. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Altemimi A, Watson DG, Kinsel M and
Lightfoot DA: Simultaneous extraction, optimization, and analysis
of flavonoids and polyphenols from peach and pumpkin extracts using
a TLC-densitometric method. Chem Cent J. 9:392015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shukla S and Gupta S: Apigenin: A
promising molecule for cancer prevention. Pharm Res. 27:962–978.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kris-Etherton PM, Hecker KD, Bonanome A,
Coval SM, Binkoski AE, Hilpert KF, Griel AE and Etherton TD:
Bioactive compounds in foods: Their role in the prevention of
cardiovascular disease and cancer. Am J Med. 113:(Suppl 9B).
71S–88S. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang S, Zhang H, Yang X, Zhu Y and Zhang
M: Evaluation of antioxidative and antitumor activities of
extracted flavonoids from Pink Lady apples in human colon and
breast cancer cell lines. Food Funct. 6:3789–3798. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Trudel D, Labbé DP, Bairati I, Fradet V,
Bazinet L and Têtu B: Green tea for ovarian cancer prevention and
treatment: A systematic review of the in vitro, in vivo and
epidemiological studies. Gynecol Oncol. 126:491–498. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gupta SC, Kannappan R, Reuter S, Kim JH
and Aggarwal BB: Chemosensitization of tumors by resveratrol. Ann N
Y Acad Sci. 1215:150–160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nessa MU, Beale P, Chan C, Yu JQ and Huq
F: Synergism from combinations of cisplatin and oxaliplatin with
quercetin and thymoquinone in human ovarian tumour models.
Anticancer Res. 31:3789–3797. 2011.PubMed/NCBI
|
|
9
|
Patel D, Shukla S and Gupta S: Apigenin
and cancer chemoprevention: Progress, potential and promise
(review). Int J Oncol. 30:233–245. 2007.PubMed/NCBI
|
|
10
|
Torkin R, Lavoie JF, Kaplan DR and Yeger
H: Induction of caspase-dependent, p53-mediated apoptosis by
apigenin in human neuroblastoma. Mol Cancer Ther. 4:1–11. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ruela-de-Sousa RR, Fuhler GM, Blom N,
Ferreira CV, Aoyama H and Peppelenbosch MP: Cytotoxicity of
apigenin on leukemia cell lines: Implications for prevention and
therapy. Cell Death Dis. 1:e192010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Horinaka M, Yoshida T, Shiraishi T, Nakata
S, Wakada M and Sakai T: The dietary flavonoid apigenin sensitizes
malignant tumor cells to tumor necrosis factor-related
apoptosis-inducing ligand. Mol Cancer Ther. 5:945–951. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Strouch MJ, Milam BM, Melstrom LG, McGill
JJ, Salabat MR, Ujiki MB, Ding XZ and Bentrem DJ: The flavonoid
apigenin potentiates the growth inhibitory effects of gemcitabine
and abrogates gemcitabine resistance in human pancreatic cancer
cells. Pancreas. 38:409–415. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu Y, Xin Y, Diao Y, Lu C, Fu J, Luo L and
Yin Z: Synergistic effects of apigenin and paclitaxel on apoptosis
of cancer cells. PLoS One. 6:e291692011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Angelini A, Di Ilio C, Castellani ML,
Conti P and Cuccurullo F: Modulation of multidrug resistance
p-glycoprotein activity by flavonoids and honokiol in human
doxorubicin- resistant sarcoma cells (MES-SA/DX-5): Implications
for natural sedatives as chemosensitizing agents in cancer therapy.
J Biol Regul Homeost Agents. 24:197–205. 2010.PubMed/NCBI
|
|
16
|
Choi EJ and Kim GH: 5-Fluorouracil
combined with apigenin enhances anticancer activity through
induction of apoptosis in human breast cancer MDA-MB-453 cells.
Oncol Rep. 22:1533–1537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wong IL, Chan KF, Tsang KH, Lam CY, Zhao
Y, Chan TH and Chow LM: Modulation of multidrug resistance protein
1 (MRP1/ABCC1)-mediated multidrug resistance by bivalent apigenin
homodimers and their derivatives. J Med Chem. 52:5311–5322. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cai X and Liu X: Inhibition of Thr-55
phosphorylation restores p53 nuclear localization and sensitizes
cancer cells to DNA damage. Proc Natl Acad Sci USA.
105:16958–16963. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chabner BA and Roberts TG Jr: Timeline
Chemotherapy and the war on cancer. Nat Rev Cancer. 5:65–72. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Billecke C, Finniss S, Tahash L, Miller C,
Mikkelsen T, Farrell NP and Bögler O: Polynuclear platinum
anticancer drugs are more potent than cisplatin and induce cell
cycle arrest in glioma. Neuro Oncol. 8:215–226. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mendes F, Groessl M, Nazarov AA, Tsybin
YO, Sava G, Santos I, Dyson PJ and Casini A: Metal-based inhibition
of poly (ADP-ribose) polymerase-the guardian angel of DNA. J Med
Chem. 54:2196–2206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dhandapani KM, Mahesh VB and Brann DW:
Curcumin suppresses growth and chemoresistance of human
glioblastoma cells via AP-1 and NFκB transcription factors. J
Neurochem. 102:522–538. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Z, Musich PR and Zou Y: Differential
DNA damage responses in p53 proficient and deficient cells:
Cisplatin-induced nuclear import of XPA is independent of ATR
checkpoint in p53-deficient lung cancer cells. Int J Biochem Mol
Biol. 2:138–145. 2011.PubMed/NCBI
|
|
25
|
Losert D, Pratscher B, Soutschek J, Geick
A, Vornlocher HP, Müller M and Wacheck V: Bcl-2 downregulation
sensitizes nonsmall cell lung cancer cells to cisplatin, but not to
docetaxel. Anticancer Drugs. 18:755–761. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He F, Wang Q, Zheng XL, Yan JQ, Yang L,
Sun H, Hu LN, Lin Y and Wang X: Wogonin potentiates
cisplatin-induced cancer cell apoptosis through accumulation of
intracellular reactive oxygen species. Oncol Rep. 28:601–605.
2012.PubMed/NCBI
|
|
27
|
Chen X, Wang Z, Yang Z, Wang J, Xu Y, Tan
RX and Li E: Houttuynia cordata blocks HSV infection through
inhibition of NF-κB activation. Antiviral Res. 92:341–345. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang Z, Lee J, Ahn HJ, Chong CK, Dias RF
and Nam HW: Western Blot Detection of Human Anti-Chikungunya Virus
Antibody with Recombinant Envelope 2 Protein. Korean J Parasitol.
54:239–241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin DL and Chang C: p53 is a mediator for
radiation-repressed human TR2 orphan receptor expression in MCF-7
cells, a new pathway from tumor suppressor to member of the steroid
receptor superfamily. J Biol Chem. 271:14649–14652. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stindt MH, Muller PA, Ludwig RL,
Kehrloesser S, Dötsch V and Vousden KH: Functional interplay
between MDM2, p63/p73 and mutant p53. Oncogene. 34:4300–4310. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Heyne K, Förster J, Schüle R and Roemer K:
Transcriptional repressor NIR interacts with the p53-inhibiting
ubiquitin ligase MDM2. Nucleic Acids Res. 42:3565–3579. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ahn HJ, Kim KS, Shin KW, Lim KH, Kim JO,
Lee JY, Kim J, Park JH, Yang KM, Baek KH, et al: Ell3 stabilizes
p53 following CDDP treatment via its effects on ubiquitin-dependent
and -independent proteasomal degradation pathways in breast cancer
cells. Oncotarget. 6:44523–44537. 2015.PubMed/NCBI
|
|
34
|
Zhao Z, Wu L, Shi H and Wu C: p53
N-terminal binding and stabilisation by PIAS3 inhibits MDM2-induced
p53 ubiquitination and regulates cell growth. Mol Med Rep.
9:1903–1908. 2014.PubMed/NCBI
|
|
35
|
Wang L, Wang L, Zhang S, Qu G, Zhang D, Li
S and Liu S: Downregulation of ubiquitin E3 ligase TNF
receptor-associated factor 7 leads to stabilization of p53 in
breast cancer. Oncol Rep. 29:283–287. 2013.PubMed/NCBI
|
|
36
|
Kitamura T, Fukuyo Y, Inoue M, Horikoshi
NT, Shindoh M, Rogers BE, Usheva A and Horikoshi N: Mutant p53
disrupts the stress MAPK activation circuit induced by
ASK1-dependent stabilization of Daxx. Cancer Res. 69:7681–7688.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cui C, Wang P, Cui N, Song S, Liang H and
Ji A: Sulfated polysaccharide isolated from the sea cucumber
Stichopus japonicas promotes the SDF-1α/CXCR4 axis-induced NSC
migration via the PI3K/Akt/FOXO3a, ERK/MAPK, and NF-κB signaling
pathways. Neurosci Lett. 616:57–64. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim B, Jung N, Lee S, Sohng JK and Jung
HJ: Apigenin Inhibits Cancer Stem Cell-Like Phenotypes in Human
Glioblastoma Cells via Suppression of c-Met Signaling. Phytother
Res. 30:1833–1840. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Seo HS, Ku JM, Choi HS, Woo JK, Jang BH,
Go H, Shin YC and Ko SG: Apigenin induces caspase-dependent
apoptosis by inhibiting signal transducer and activator of
transcription 3 signaling in HER2-overexpressing SKBR3 breast
cancer cells. Mol Med Rep. 12:2977–2984. 2015.PubMed/NCBI
|
|
40
|
Cho Y, Gorina S, Jeffrey PD and Pavletich
NP: Crystal structure of a p53 tumor suppressor-DNA complex:
Understanding tumorigenic mutations. Science. 265:346–355. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vazquez A, Bond EE, Levine AJ and Bond GL:
The genetics of the p53 pathway, apoptosis and cancer therapy. Nat
Rev Drug Discov. 7:979–987. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vucic D, Dixit VM and Wertz IE:
Ubiquitylation in apoptosis: A post-translational modification at
the edge of life and death. Nat Rev Mol Cell Biol. 12:439–452.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Meek DW: Tumour suppression by p53: A role
for the DNA damage response? Nat Rev Cancer. 9:714–723.
2009.PubMed/NCBI
|
|
44
|
Guo H, Kong S, Chen W, Dai Z, Lin T, Su J,
Li S, Xie Q, Su Z, Xu Y and Lai X: Apigenin mediated protection of
OGD-evoked neuron-like injury in differentiated PC12 cells.
Neurochem Res. 39:2197–2210. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gazzani G, Daglia M and Papetti A: Food
components with anticaries activity. Curr Opin Biotechnol.
23:153–159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tang D, Chen K, Huang L and Li J:
Pharmacokinetic properties and drug interactions of apigenin, a
natural flavone. Expert Opin Drug Metab Toxicol. 2:1–8. 2016.
View Article : Google Scholar
|
|
47
|
Chen L and Zhao W: Apigenin protects
against bleomycin-induced lung fibrosis in rats. Exp Ther Med.
11:230–234. 2016.PubMed/NCBI
|
|
48
|
Gates MA, Vitonis AF, Tworoger SS, Rosner
B, TitusErnstoff L, Hankinson SE and Cramer DW: Flavonoid intake
and ovarian cancer risk in a population-based case-control study.
Int J Cancer. 124:1918–1925. 2009. View Article : Google Scholar : PubMed/NCBI
|