Introduction
Esophageal cancer (EC) is the sixth leading cause of
cancer mortality, with 17,990 new cases and 15,210 mortalities
reported in 2013 (1). The major
histological type in China is squamous cell carcinoma (SCC), which
accounts for >90% of all types of EC (2,3). Locally
advanced esophageal carcinoma is known to be refractory to a single
modality of treatment (4). Patients
with unresectable or medically inoperable disease are usually
treated with radiation therapy and concurrent chemotherapy
(4,5).
However, hypoxic environments contribute to malignant behavior,
including tumor progression, invasion and radiation resistance
(6), which are major barriers to the
success of radiation therapy. Therefore, improving the hypoxic
environment to enhance the radiation sensitivity of cancer cells
has become an urgent task.
Recently, a number of studies have focused on the
oncogenic phosphatidylinositol-3-kinase (PI3K)/AKT pathway, also
known as the protein kinase B pathway (7). The PI3K/AKT pathway phosphorylates and
activates AKT to phosphorylated (p)-AKT, which plays a critical
role in promoting a malignant phenotype and has prognostic
significance in a number of solid tumors (7,8). Previous
reports also suggested that p-AKT overexpression may correlate with
a poor prognosis (9,10). Hypoxic environments can activate a
specific set of tumor-promoting factors, including signal
transducer and activator of AKT and hypoxia-inducible factor
(HIF)-1 (11,12). HIF-1 can activate the vascular
endothelial growth factor (VEGF) gene in response to PI3K/AKT and
mammalian target of rapamycin (mTOR) signaling to regulate the
adaptation to hypoxic conditions (13). In addition, p-AKT enhances the
expression of the HIF-1α gene and its target genes such as VEGF
(14,15). HIF-1α is an oxygen-sensitive subunit,
and regulates >100 genes involved in cell survival, tumor
metabolism, proliferation, invasion and angiogenesis to resist
various treatments (14–17).
Triciribine (TCN), as an effective AKT inhibitor,
has been demonstrated to effectively inhibit p-AKT (18). It has been reported that TCN potently
and selectively inhibits the activation, dimerization and nuclear
translocation of AKT, resulting in an increase in the apoptosis of
prostate carcinoma cells (19).
Thus, we hypothesize that TCN inhibits AKT and
HIF-1α expression, and improves tumor microenvironment in
esophageal SCC (ESCC) cells. In other words, TCN can radiosensitize
human ESCC cells by decreasing AKT and HIF-1α expression in
hypoxia. To confirm the hypothesis described above, the present
study examined the effects of TCN and/or X-rays on human ESCC cells
in hypoxia both in vitro and in vivo.
Materials and methods
Reagents
The AKT inhibitor TCN (>99%) was purchased from
Selleck Chemicals (Houston, TX, USA), while RPMI-1640 medium and
fetal bovine serum (FBS) were obtained from Gibco (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Cell Counting Kit (CCK) 8,
streptomycin, penicillin and dimethyl sulfoxide were obtained from
Beyotime Institute of Biotechnology (Haimen, China). Antibodies
against AKT (9272S) and p-AKT (4060S) were purchased from Cell
Signaling Technology, lnc. (Danvers, MA, USA), while antibodies
against HIF-1α (sc-13515), VEGF (sc-117031) and β-actin (sc-47778)
were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA).
Mouse anti-phospho-histone H2AX, Ser139 (γ-H2.AX) antibody
(MAB504A) was obtained from EMD Millipore (Billerica, MA, USA).
Cell culture
The ESCC cell line ECA109 (supplied by Shanghai
Institute of Cell Biology, Shanghai, China) was cultured in
RPMI-1640 medium supplemented with 10% Gibco FBS (Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 mg/ml streptomycin.
ECA109 cells were maintained at 37°C in a humidified incubator at
5% CO2, 20% O2 or 1% O2. Hypoxic
conditions were created in an hypoxia chamber, where the cells were
incubated in a modular chamber flushed with a complex air of 1%
O2, 5% CO2 and 94% N2 at 37°C.
Treatment with irradiation
ECA109 cells were irradiated with 6 MV X-rays
(Elekta Instrument AB, Stockholm, Sweden) at a dose rate of 5.66
Gy/min (2, 4, 6 and 8 Gy) at room temperature. The tumors of nude
mice were irradiated with 4 MV X-rays (6 Gy) using an RS-2000
biological irradiator (Shanghai Bio-Chain Institute of Cell
Biology, Shanghai, China) at a dose rate of 4.48 Gy/min at room
temperature.
CCK8 assay
Cell viability was determined using CCK8 assay
(Beyotime Institute of Biotechnology, Haimen, China). ECA109 cells
(4,000 cells/well) were cultured in 96-well plates for 24 h
following TCN treatment with different concentrations (0, 0.5, 2, 4
and 8 µmol/l). Cell density was measured by CCK8 assay following
the manufacturer's protocol. Briefly, 10 µl of CCK8 (at 5 mg/ml)
was added to each well at a final concentration of 0.5 mg/ml, and
the cells were incubated for 4 h at 37°C. Then, the samples
absorbance at 490 nm was read with a microplate reader (model 630;
Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Clonogenic survival assay
ECA109 cells were plated in 6-well plates at a
specific density on the basis of the dose of X-rays received. Then,
cells treated with or without TCN with different doses of X-rays
were assessed by clonogenic assay. Briefly, the cells were treated
with or without TCN for 24 h, and then irradiated with 0, 2, 4, 6
and 8 Gy at room temperature. The cells were grown at 37°C for 12
days, fixed with methanol, stained with Giemsa, and then scored by
counting with an inverted microscope, using the standard definition
of a colony consisting of ≥50 cells. The surviving fraction (SF)
was defined as follows: SF = (mean number of colonies)/(number of
cells inoculated × plating efficiency).
Measurement of apoptosis by flow
cytometry
Treated ECA109 cells were plated in 6-well plates at
a specific density. The cells were exposed to X-rays (4 Gy) after
TCN treatment in normoxia or hypoxia for 24 h. After 48 h, the
cells were collected and labeled with Annexin V and propidium
iodide according to the manufacturer's protocol. Cell death was
analyzed by flow cytometry using light scatter characteristics (BD
Biosciences, Franklin Lakes, NJ, USA). The apoptosis rate was
determined by Annexin V-FITC Apoptosis Detection kit Nanjing KeyGen
Biotech Co., Ltd. (Nanjing, China). This experiment was repeated ≥3
times.
Western blot assay
Treated ECA109 cells were lysed in SDS Lysis Buffer
(Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) and then
centrifuged at 14,000 × g (15 min, 4°C). Protein
concentrations of the samples were calculated with a bicinchoninic
acid kit (Beyotime Institute of Biotechnology). Equal amounts of
protein were separated by 6 or 10% SDS-PAGE, transferred to
Protran® nitrocellulose membranes (Schleicher &
Schuell BioScience GmbH, Inc., Dassel, Germany), and then blocked
with TBS (pH 7.4) containing 0.05% Tween 20 and 5% nonfat milk. The
membranes were incubated overnight at 4°C by gentle agitation with
various primary antibodies: Anti-AKT antibody (1:500), anti-p-AKT
antibody (1:500), anti-HIF-1α antibody (1:500), anti-VEGF antibody
(1:250) and anti-β-actin antibody (1:250). The following day, the
membrane was incubated with alkaline phosphatase-conjugated goat
anti-mouse immunoglobulin (Ig) G or goat anti-rabbit IgG as
secondary antibody (1:2,000; BS13271; Bioworld Technology, Inc.,
St. Louis Park, MN, USA) for 1 h at room temperature. The blots
were visualized using the SuperSignal West Femto kit (Pierce;
Thermo Fisher Scientific, Inc.).
Immunofluorescence staining of
γ-H2AX
ECA109 cells were grown on glass coverslips, treated
with or without TCN, and then administered X-rays (1 Gy). The cells
were glass-fixed with acetone and permeabilized with 0.1% Triton
X-100 in PBS for 5 min at room temperature. Next, the cells were
incubated with an anti-γ-H2AX antibody (diluted 1:200) at 4°C
overnight and then incubated with fluorescein
isothiocyanate-conjugated secondary antibody (diluted 1:100;
ab2492; Abcam, Shanghai, China) for 1 h at room temperature. After
washing in PBS, cells were incubated in the dark with DAPI (at a
dilution of 1:35 in 4% paraformaldehyde) for 5 min. Then, the
slides were examined with at ×400 magnification with a confocal
laser scanning microscope (LSM 510; Zeiss GmbH, Jena, Germany). For
each treatment condition, the number of γ-H2AX foci were counted in
≥100 cells from randomly captured images.
In vivo experiment
A total of 24 four-week-old male BALB/c nude mice
(weight, 18–20 g) were acquired from Nanjing Medical University
Animal Center (Nanjing, China). The animals, which were allowed
free feeding and drinking, were housed under conventional
conditions with constant temperature and humidity, and were
maintained on a 12:12-h dark-light cycle. The mice were injected
subcutaneously with ECA109 cells (5×106 cells in 0.1 ml
PBS) at one site of the right armpit separately. When the tumor
mass became obvious (~150 mm3), the mice were randomly
divided into four subgroups: Control group, 25 mg/kg TCN group,
ionizing radiation (IR) (4 Gy) group and 25 mg/kg TCN plus IR
group. Each group contained 6 mice. For the control group, PBS
alone was injected every 2 days, and TCN was administered to the
mice 3 times/week for 4 weeks. On the fifth day, the mice were
administered a single dose of 4 Gy X-rays (4.48 Gy/min) with the
RS-2000 biological irradiator. The mice received TCN twice more
after IR. On day 25, the mice were sacrificed. Tumor volume was
calculated with the formula: Tumor volume (mm3) = length
diameter (mm) × width diameter (mm) 2/2. Mouse tumors
were analyzed by WB for p-AKT, AKT, HIF-1α and VEGF. All the
procedures were performed in accordance with the guidelines of the
laboratory animal ethics committee of Nanjing Medical University
(Nanjing, China).
Statistical analysis
All experiments were performed in triplicate. Each
datum represents the mean ± standard deviation or the mean ±
standard error of mean of different experiments under the same
conditions. Statistical analysis of the results was performed using
GraphPad Prism program version 5.0 (GraphPad Software, Inc., La
Jolla, CA, USA) and SPSS statistical software system for Windows
version 16.0 (SPSS, Inc., Chicago, IL, USA). Statistical
significance between each treated group and the control was
analyzed using the independent t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
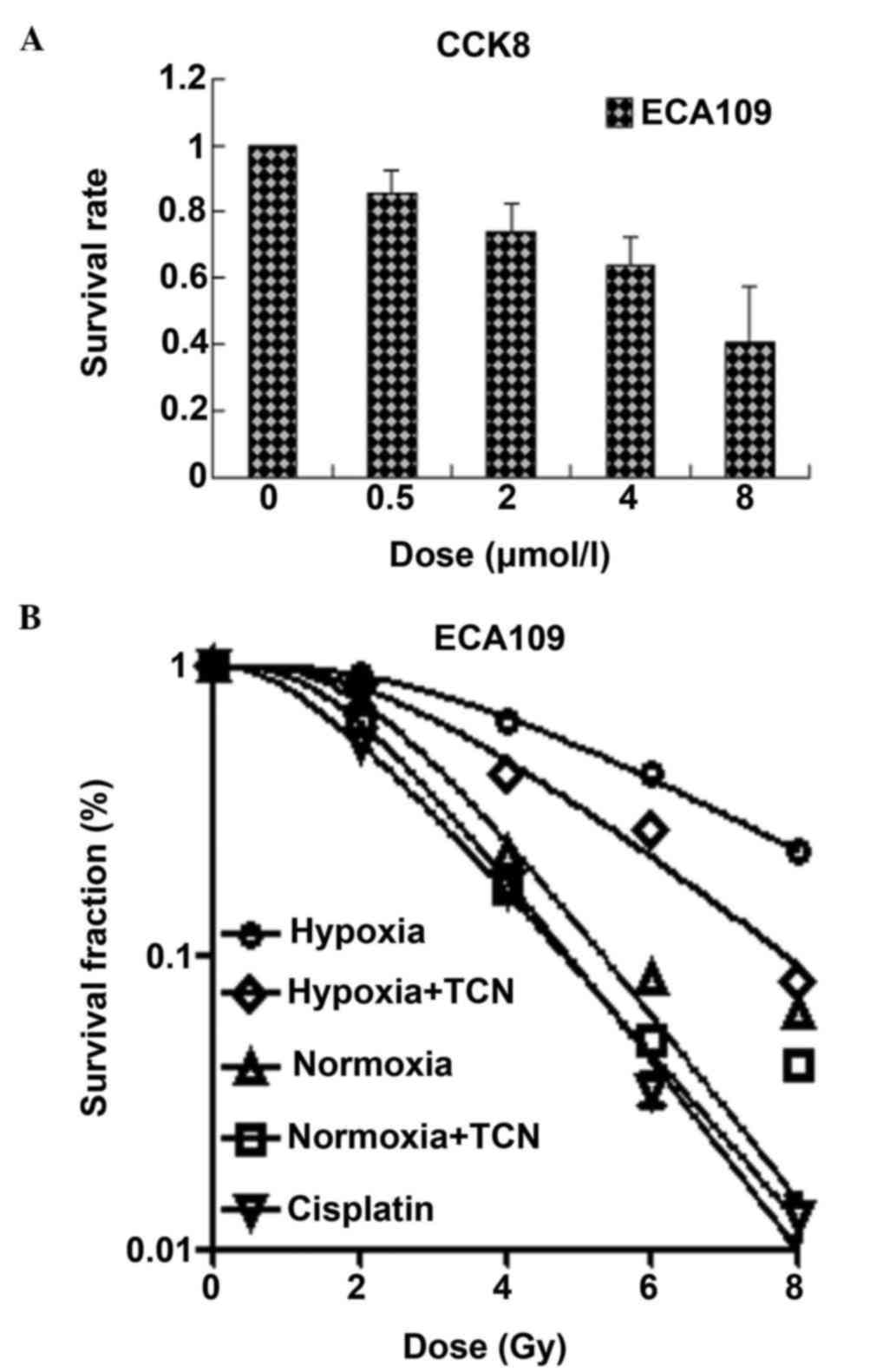
Half maximal inhibitory concentration
(IC50) for TCN in ESCC ECA109 cells
The viability of ECA109 cells was observed at
various concentrations of TCN (0, 0.5, 2, 4 and 8 µmol/l) for 24 h
according to CCK8 assay (Fig. 1A).
The IC50 value for the ECA109 cell line was extrapolated
at 24 h, and was calculated to be 6.155 µmol/l. Thus, a low
concentrations of TCN (2 µmol/l) was selected for subsequent
experiments with the ECA109 cell line.
TCN is an effective radiosensitizer of
ESCC cells in normoxia and hypoxia in vitro
ESCC ECA109 cells were treated for 24 h with TCN (4
µmol/l) to investigate the effects of TCN on radiotherapy
sensitization of ESCC cells in normoxia and hypoxia. Clonogenic
assays revealed the radiation dose-response survival curves for
ESCC ECA109 cells, and suggested radioresistance of ESCC cells
(Fig. 1B). Notably, the effect of TCN
was more obvious in hypoxia as compared with normoxia in ECA109
cells. When a low cytotoxic concentration (4 µmol/l) was selected
in vitro, TCN diminished clonogenic survival to a great
extent and radiosensitized ECA109 similarly under conditions of
hypoxia [sensitization enhancement ratio (SER)=1.36] and normoxia
(SER=1.50). By contrast, hypoxic cells in normoxia reduced their
ability to form colonies after irradiation, indicating
hypoxia-induced radioresistance (normoxia, SER=1.50; normoxia +
TCN, SER=1.65). Furthermore, this reduction was observed in
cisplatin-treated cells, which served as positive controls
(SER=1.89) (Table I). These data
revealed that TCN could significantly suppress clonogenic
proliferation in normoxia and hypoxia, which was particularly
evident in hypoxia, and reversed the radio-resistance induced by
normoxic or hypoxic conditions in ESCC cells.
 | Table I.Radiosensitization activity of TCN in
ECA109 cells. |
Table I.
Radiosensitization activity of TCN in
ECA109 cells.
| ECA109 | D0
(Gy) | Dq
(Gy) | SF2 | SERD0 |
|---|
| Hypoxia | 5.01 | 2.56 | 0.85 | 1.00 |
| Hypo + TCN | 3.68 | 2.21 | 0.65 | 1.36 |
| Normoxia | 3.34 | 2.15 | 0.63 | 1.50 |
| Nor + TCN | 3.03 | 1.14 | 0.61 | 1.65 |
| Cisplatin | 2.65 | 0.71 | 0.47 | 1.89 |
The early apoptosis of ESCC cells treated by IR with
or without TCN (4 µmol/l) was tested in normoxia and hypoxia. As
shown in Fig. 2A, treatment with TCN
for 24 h did not significantly induce apoptosis, and no distinct
increase was observed in either normoxia or hypoxia. Notably, the
proportion of apoptotic cells was considerably increased in the
irradiation-treated group compared with the control group
(normoxia, P=0.002; hypoxia, P=0.006). In addition, the number of
apoptotic cells in the combined treatment group was significantly
higher than the number of cells treated with irradiation alone
under both normoxic and hypoxic conditions (both P=0.003) (Fig. 2B).
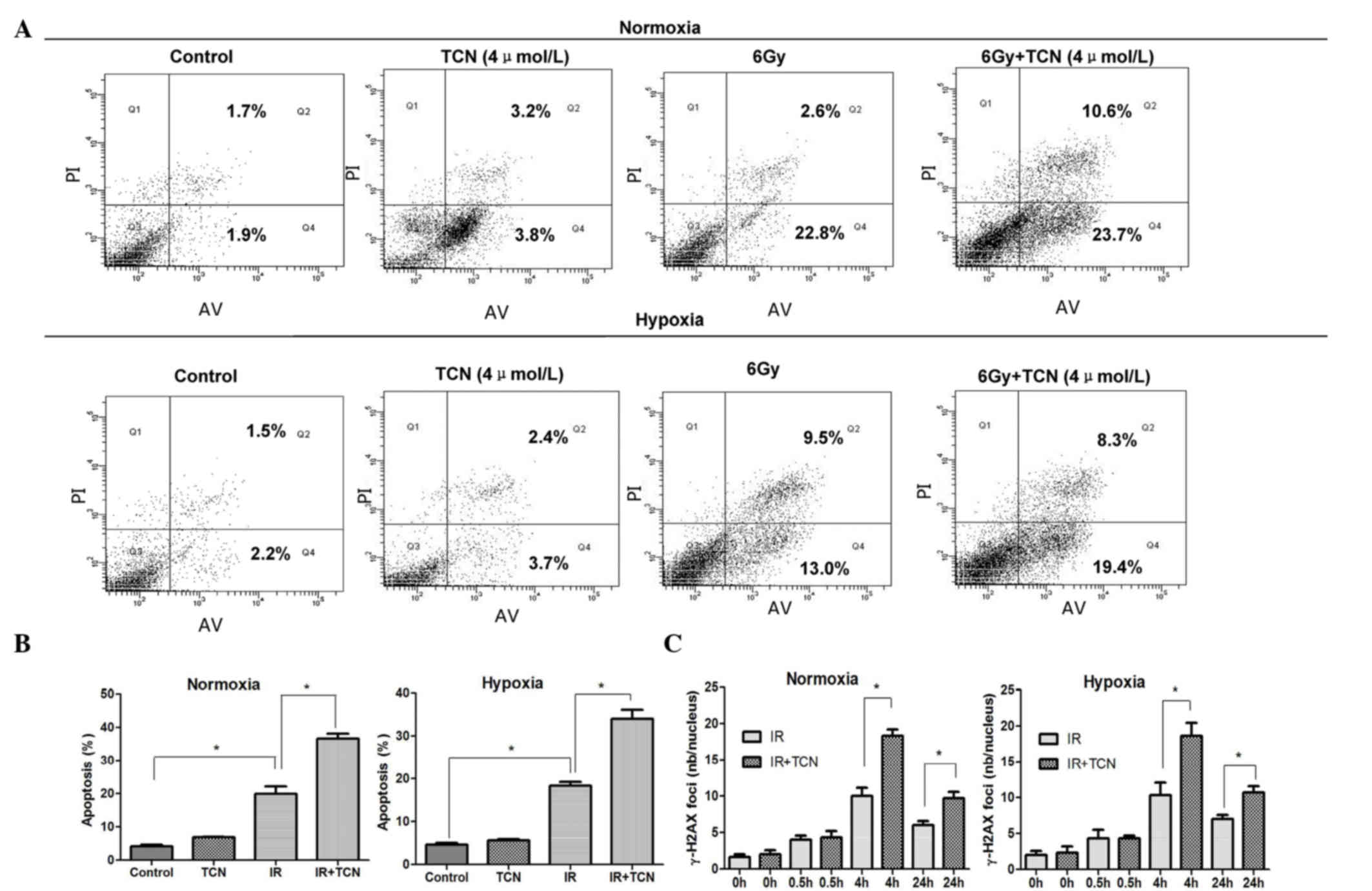 | Figure 2.(A) The effect of 6 Gy+TCN in cell
apoptosis is significant. *Normoxia, P=0.002; hypoxia, P=0.006. (B)
TCN (4 µmol/l) significantly enhanced the irradiation-induced
apoptosis of ECA109 cells under hypoxia and normoxia (*P=0.003).
(C) Determination of TCN-induced γ-H2AX foci in ECA109 cells at
0.5, 4 and 24 h after 1 Gy±4 µmol/l TCN treatment (mean ± standard
error of the mean, n=3). *P=0.0003 and P=0.0008 at 4 and 24 h,
respectively. TCN, triciribine; IR, ionizing radiation; γ-H2AX,
phospho-histone H2A.X, Ser139; nb, number. |
It was speculated that the effect of TCN on ESCC
cells to IR may be based on the impairment in the repair of DNA
double-strand breaks (DSBs). Therefore, the levels of DSBs in
ECA109 cells were detected at different times after exposure to IR
under either normoxia or hypoxia by immunofluoresence staining of
γ-H2AX foci. As shown in Fig. 2C, the
majority of γ-H2AX foci were cleared at 4 h after exposure to 1 Gy
of X-rays in ECA109 cells without TCN, while γ-H2AX foci persisted
in ECA109 cells that were pretreated with TCN (4 µmol/l). The
average of γ-H2AX foci per cell in cells subjected to combined
treatment with TCN and radiation was evident, compared with the
radiation-only group, at 0.5, 4 and 24 h under normoxia and hypoxia
(Fig. 2C) (P<0.0001, P=0.0003 and
P=0.0008, respectively). TCN was not as obvious in inducing DNA
damage in terms of γ-H2AX foci induction compared with the controls
(P=0.5493).
Upregulation of AKT and HIF-1α by
hypoxia can be attenuated by TCN in ESCC cells and xenograft in
vivo
As aforementioned, the inhibition of AKT
phosphorylation was considered to possibly facilitate
radiosensitivity in hypoxic environments, and this effect was
possibly mediated through the inhibition of the critical
transcription factor HIF-1α and its target gene VEGF. To
investigate whether AKT, p-AKT, HIF-1α and VEGF were involved in
combined treatment-induced cell death, western blotting was
utilized to examine the protein expression of ESCC ECA109 cells and
xenograft of ECA109 in vivo. Accumulation of HIF-1α, VEGF
and p-AKT was observed upon the first 6 h of hypoxic incubation,
and reached a maximum at 24 h (Fig.
3A). Thus, the 24 h time point was chosen for further
experiments. Of note, the hypoxia-stimulated levels of p-AKT,
HIF-1α and VEGF were significantly decreased by TCN (Fig. 3B).
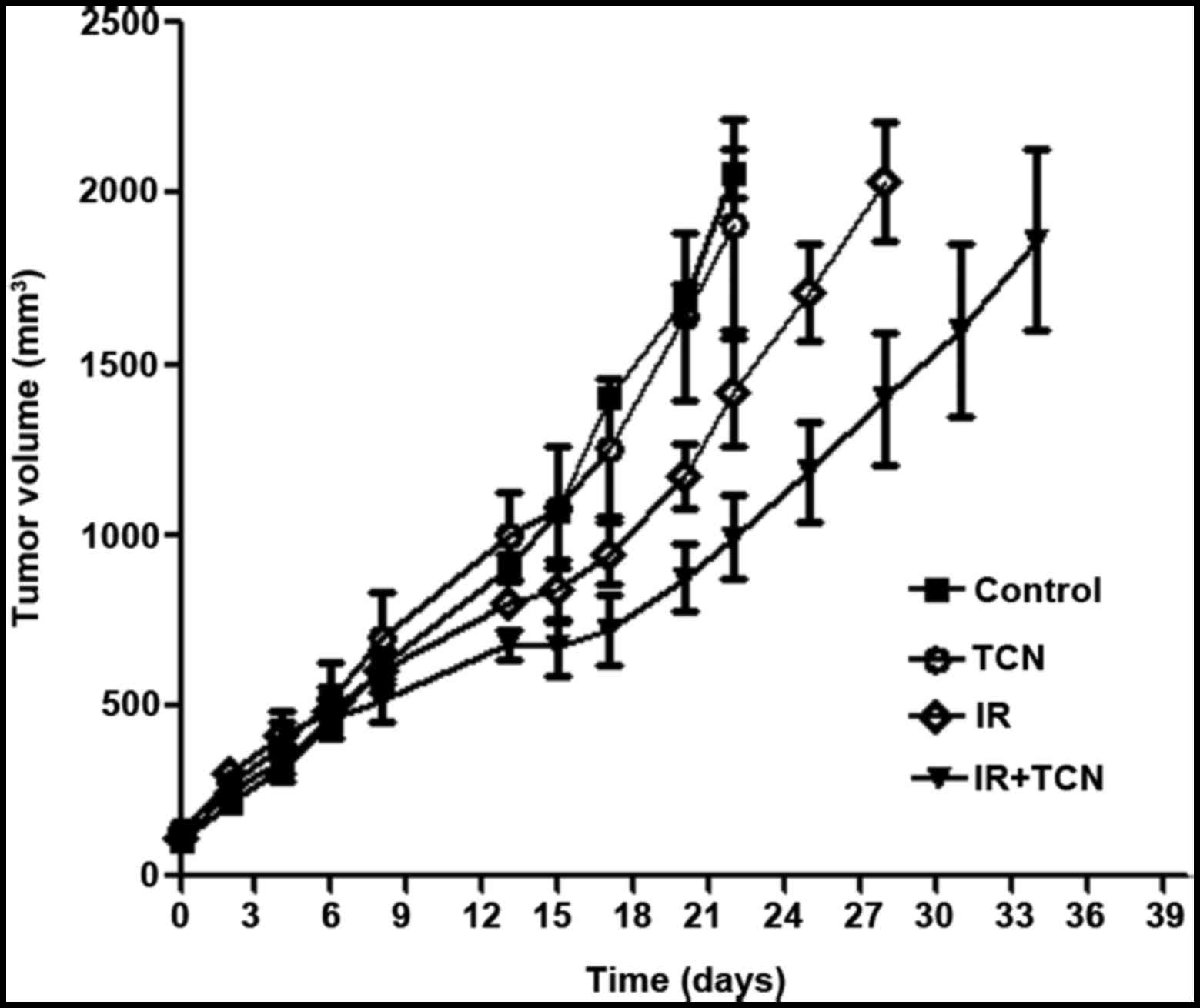
TCN promotes radiation sensitivity in
nude mice
To confirm whether TCN exerts a radiosensitization
effect on ESCC xenograft in vivo, ECA109 tumor-bearing mice
were treated with 6 Gy irradiation and received intraperitoneal
injection of TCN (25 mg/kg) every day for 1 week before
irradiation.
It was detected that TCN inhibits p-AKT, HIF-1α and
VEGF protein expression in hypoxic ESCC xenografts. By temporarily
blocking the tumor blood supply for 5 min before irradiation, an
hypoxic model was established. Following drugs and irradiation
administration, either irradiation or combined treatment
effectively delayed tumor growth and reduced tumor weight (P=0.025
for irradiation and P=0.002 for TCN + IR on the 22th day) (Fig. 4).
In addition, the doubling time of ECA109 tumors was
calculated. In the control group and the TCN alone group, the
doubling time was 7.3±0.6 and 7.9±0.7 days, respectively. For the
irradiation-treated group, the combination treatment significantly
extended the doubling time to 13.8±0.8 days (P=0.001), while in the
irradiation alone group, this value was 10.6±1.3 days (P=0.037). In
general, these results demonstrated that intraperitoneal injection
of TCN enhances the suppression effect of radiation on ESCC in
vivo.
Discussion
To enhance the radiotherapy efficacy against cancer,
several strategies have been utilized. Modulation of DNA damage
repair, cellular antioxidant machinery, pro-survival signaling,
tumor hypoxia state and cell cycle distribution are commonly used
targets for the development of radiation sensitizers (20–24).
A central role in the PI3K/AKT pathway is the
serine-threonine kinase AKT, since it enables PI3K to phosphorylate
membrane-bound phosphatidylinositol diphosphate to generate
phosphatidylinositol trisphosphate, and allows p-AKT on Thr308 and
Ser473 residues to interact with pyruvate dehydrogenase kinase 1
and mTOR complex 2 (9). Furthermore,
p-AKT not only inhibits apoptosis via B-cell lymphoma
(Bcl)-2-associated death promoter/Bcl-extra large and glycogen
synthase kinase 3 beta/myeloid cell leukemia 1, but also
downregulates the transcription factors forkhead box and p53,
upregulates nuclear factor-κB activity, and inhibits pro-caspase 9
(10). In addition, p-AKT mediates a
series of pro-survival signals for anti-apoptosis, proliferation,
cell growth and angiogenesis (8). In
the PI3K/AKT pathway, activated p-AKT mediates diverse pro-survival
signals, promotes the malignant phenotype of cancer cells through
multiple downstream pathways and correlates with a poor prognosis
(7–10). p-AKT also increases expression of the
HIF-1α gene and that of its target genes, including VEGF (12). HIF-1α is an oxygen-sensitive subunit
(14), which, under hypoxic
conditions, is an important factor for tumor cells to resist IR
(15). TCN, which was initially
described as a DNA synthesis inhibitor, has recently been shown to
function as an inhibitor of AKT (18). Previous studies demonstrated that TCN
inhibits AKT phosphorylation at Thr308 and Ser473 and AKT activity
in the human prostate cancer cell line PC-3 (18). In addition, TCN sensitized PC-3 cells
to tumor necrosis factor-related apoptosis-inducing ligand- and
anti-cluster of differentiation 95-induced apoptosis, whereas the
cells remained resistant to DNA damaging chemotherapeutics
(16). The observed sensitization
essentially depended on the phosphorylation status of AKT (16). Despite the lack of studies focused on
the radiosensitivity of TCN and p-AKT, we speculated an association
between TCN and p-AKT and radiosensitization of ESCC.
In our study, for the first time it was demonstrated
that TCN reduced colony formation and induced apoptosis or
impairment in the repair of DSBs combined with IR. In addition, TCN
downregulated p-AKT, HIF-1α and VEGF in ESCC cells, and suppressed
the growth of ESCC xenografts. These results may complement those
from previous studies on TCN, which suggested that TCN may be an
antitumor agent in different cancers, and may expand our
understanding of the mechanisms of TCN activity (18).
In conclusion, the present results provide evidence
that the combined application of TCN and IR may be an effective
treatment for ESCC. TCN at low concentrations substantially
radiosensitized normoxic and hypoxic ESCC cells by downregulating
AKT and HIF-1α, which contributes to tumor aggressiveness,
invasiveness and resistance to radiation therapy. Furthermore, our
data indicate that this treatment critically depends on a high
constitutive AKT phosphorylation level.
References
|
1
|
National Cancer Institute, . Cancer
Statistics: SEER Stat Fact Sheets: Esophageal Cancer. http://seer.cancer.gov/statfacts/html/esoph.htmlAccessed
January 1, 2014.
|
|
2
|
Gholipour C, Shalchi RA and Abbasi M: A
histopathological study of esophageal cancer on the western side of
the Caspian littoral from 1994 to 2003. Dis Esophagus. 21:322–327.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tran GD, Sun XD, Abnet CC, Fan JH, Dawsey
SM, Dong ZW, Mark SD, Qiao YL and Taylor PR: Prospective study of
risk factors for esophageal and gastric cancers in the Linxian
general population trial cohort in China. Int J Cancer.
113:456–463. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tepper J, Krasna MJ, Niedzwiecki D, Hollis
D, Reed CE, Goldberg R, Kiel K, Willett C, Sugarbaker D and Mayer
R: Phase III trial of trimodality therapy with cisplatin,
fluorouracil, radiotherapy, and surgery compared with surgery alone
for esophageal cancer: CALGB 9781. J Clin Oncol. 26:1086–1092.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu HC, Hung SK, Huang CJ, Chen CC, Chen
MJ, Chang CC, Tai CJ, Tzen CY, Lu LH and Chen YJ: Esophagectomy for
locally advanced esophageal cancer, followed by chemoradiotherapy
and adjuvant chemotherapy. World J Gastroenterol. 11:5367–5372.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Karar J and Maity A: Modulating the tumor
microenvironment to increase radiation responsiveness. Cancer Biol
Ther. 8:1994–2001. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Davies MA: Regulation, role, and targeting
of Akt in cancer. J Clin Oncol. 29:4715–4717. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Courtney KD, Corcoran RB and Engelman JA:
The PI3K pathway as drug target in human cancer. J Clin Oncol.
28:1075–1083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hasselblom S, Hansson U, Olsson M, Torén
L, Bergström A, Nilsson-Ehle H and Andersson PO: High
immunohistochemical expression of p-AKT predicts inferior survival
in patients with diffuse large B-cell lymphoma treated with
immunochemotherapy. Br J Haematol. 149:560–568. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hong JY, Hong ME, Choi MK, Kim YS, Chang
W, Maeng CH, Park S, Lee SJ, Do IG, Jo JS, et al: The impact of
activated p-AKT expression on clinical outcomes in diffuse large
B-cell lymphoma: A clinicopathological study of 262 cases. Ann
Oncol. 25:182–188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cummins EP and Taylor CT:
Hypoxia-responsive transcription factors. Pflugers Arch.
450:363–371. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jung JE, Lee HG, Cho IH, Chung DH, Yoon
SH, Yang YM, Lee JW, Choi S, Park JW, Ye SK and Chung MH: STAT3 is
a potential modulator of HIF-1-mediated VEGF expression in human
renal carcinoma cells. FASEB J. 19:1296–1298. 2005.PubMed/NCBI
|
|
13
|
Xu Q, Briggs J, Park S, Niu G, Kortylewski
M, Zhang S, Gritsko T, Turkson J, Kay H, Semenza GL, et al:
Targeting Stat3 blocks both HIF-1 and VEGF expression induced by
multiple oncogenic growth signaling pathways. Oncogene.
24:5552–5560. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liao D and Johnson RS: Hypoxia: A key
regulator of angiogenesis in cancer. Cancer Metastasis Rev.
26:281–290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang X, Zhu HC, Zhang C, Qin Q, Liu J, Xu
LP, Zhao LJ, Zhang Q, Cai J, Ma JX, et al: HIF-1α; 1772 C/T and
1790 G/A polymorphisms are significantly associated with higher
cancer risk: An updated meta-analysis from 34 case-control studies.
PLoS One. 8:e803962013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang X, Zhang C, Zhu HC, Qin Q, Zhao LJ,
Liu J, Xu LP, Zhang Q, Cai J, Ma JX, et al: HIF-1α P582S and A588T
polymorphisms and digestive system cancer risk-a meta-analysis.
Tumour Biol. 35:2825–2830. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo Q, Dai SB, Shen F, Yu D, Shen ST,
Zhang Q, Huang JX and Wu ZD: VEGF+405G/C (rs2010963) polymorphisms
and digestive system cancer risk: A meta-analysis. Tumour Biol.
35:4977–4982. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gürsel DB, ConnellAlbert YS, Tuskan RG,
Anastassiadis T, Walrath JC, Hawes JJ, Amlin-Van Schaick JC and
Reilly KM: Control of proliferation in astrocytoma cells by the
receptor tyrosine kinase/PI3K/AKT signaling axis and the use of
PI-103 and TCN as potential anti-astrocytoma therapies. Neuro
Oncol. 13:610–621. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dieterle A, Orth R, Daubrawa M, Grotemeier
A, Alers S, Ullrich S, Lammers R, Wesselborg S and Stork B: The Akt
inhibitor triciribine sensitizes prostate carcinoma cells to
TRAIL-induced apoptosis. Int J Cancer. 125:932–941. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kesari S, Advani SJ, Lawson JD, Kahle KT,
Ng K, Carter B and Chen CC: DNA damage response and repair:
Insights into strategies for radiation sensitization of gliomas.
Future Oncol. 7:1335–1346. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pajonk F, Vlashi E and McBride WH:
Radiation resistance of cancer stem cells: The 4 R's of
radiobiology revisited. Stem Cells. 28:639–648. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ahmed KM and Li JJ: NF-κB-mediated
adaptive resistance to ionizing radiation. Free Radic Biol Med.
44:1–13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang C, Yang X, Zhang Q, Guo Q, He J, Qin
Q, Zhu H, Liu J, Zhan L, Lu J, et al: STAT3 inhibitor NSC74859
radiosensitizes esophageal cancer via the downregulation of HIF-1α.
Tumour Biol. 35:9793–9799. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Q, Zhang C, He J, Guo Q, Hu D, Yang
X, Wang J, Kang Y, She R, Wang Z, et al: STAT3 inhibitor stattic
enhances radiosensitivity in esophageal squamous cell carcinoma.
Tumour Biol. 36:2135–2142. 2015. View Article : Google Scholar : PubMed/NCBI
|