Introduction
Gastrointestinal stromal tumors (GISTs) are the most
common mesenchymal tumors of the gastrointestinal tract, and
although the exact incidence of GISTs worldwide is not known, the
approximate annual incidence of GISTs in Japan is 1500–2000 cases.
GISTs are believed to originate from the interstitial cells of
Cajal (ICC) (1). The ICC are
electrical pacemakers that regulate the motility of the
gastrointestinal tract, and are located around the myenteric plexus
and the muscularis propria (1).
Approximately 85% of GISTs harbor activating mutations in the KIT
receptor tyrosine kinase or platelet-derived growth factor
receptor-α (PDGFRA) (2,3). Constitutive activation of these receptor
tyrosine kinases induces multiple signaling pathways that are
essential for the pathogenesis of GISTs (2,3). For
example, oncogenic mutations of KIT promote the activation of
various signaling cascades, including the Ras/mitogen-activated
protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)/Akt and
Janus kinase (JAK)/signal transducer and activator of transcription
(STAT) signaling pathways, which have been shown to be activated in
various tumors (4,5). Imatinib, a potent inhibitor of KIT, is
effective for the treatment of unresectable or metastatic GISTs.
However, patients with metastatic GISTs eventually develop
resistance to imatinib (6); thus, it
is necessary to further define the molecular pathogenesis of GISTs,
in order to develop alternative therapeutic strategies.
Adenosine monophosphate (AMP) deaminases (AMPDs)
catalyze the hydrolytic deamination of AMP to inosine
monophosphate, which is a critical step in nucleotide metabolism
(7). There are three members of the
AMPD family: AMPD1, AMPD2 and AMPD3 (8,9). AMPD1 is
exclusively expressed in skeletal muscle, whereas AMPD2 and AMPD3
are ubiquitously expressed. Deficiencies in all three AMPDs have
been reported in humans (7), and
mutations in AMPD1 were associated with muscle weakness or pain in
certain patients (10,11). AMPD2 deficiency is associated with
pontocerebellar hypoplasia, which is a rare inherited and
progressive neurodegenerative disorder (12). Conversely, individuals with a complete
AMPD3 deficiency did not exhibit any significant disorders
(13), although elevated levels of
adenosine triphosphate (ATP) were detected in the cells of
AMPD3-deficient mice (14).
Accumulating evidence has suggested that metabolic alterations are
associated with the progression of tumor formation (15–19). A
recent report suggested that AMPD2 and AMPD3 may be potential
targets for cancer treatment (20).
The present study aimed to determine the expression and role of
AMPD3 in GISTs.
Materials and methods
Patients
A total of 16 GIST samples and 6 normal
gastrointestinal tract tissues samples were obtained from 16
patients with GIST who underwent surgery at Nagoya University
Hospital (Nagoya, Japan) between September 2008 and April 2014.
Informed consent was obtained from all patients. Ethical approval
was obtained from the Nagoya University Graduate School of Medicine
(Nagoya, Japan). Patients were histologically diagnosed. The
clinical characteristics of the patients with GIST are shown in
Table I.
 | Table I.Clinical characteristics of patients
with GISTs. |
Table I.
Clinical characteristics of patients
with GISTs.
| Variable | Value |
|---|
| Gender |
|
| Male | 6 |
|
Female | 10 |
| Age (years) | 63 (46–81) |
| Anatomic site of
GIST |
|
|
Stomach | 12 |
| Small
intestine | 4 |
| c-KIT exon 11
mutation |
|
|
Deletion | 6 |
|
Insertion | 5 |
| Point
mutation | 5 |
| Malignancy |
|
|
Benign | 8 |
|
Malignant | 8 |
| Imatinib
treatment |
|
|
Yes/no | 4/9 |
|
Unknown | 3 |
Cell lines
The human GIST cell line, GIST-T1, was purchased
from Cosmo Bio, Co., Ltd. (Tokyo, Japan). GIST-T1 cells were
cultured in Dulbecco's modified Eagle's medium (DMEM; Wako Pure
Chemical Industries, Osaka, Japan) supplemented with 10% fetal
bovine serum (FBS; Biowest, Nuaille, France), and penicillin and
streptomycin (100 U/ml; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) at 37°C with 5% CO2 for at least 1 month. SKOV3
(ovarian cancer) and HCT116 (colorectal cancer) cell lines were
obtained from the American Type Culture Collection (Manassas VA
USA), and KP4 (pancreatic cancer), TE1 (esophagus cancer), KASUMI1
(acute myeloid leukemia), MKN28 (stomach cancer) and QG90 (lung
cancer) cell lines were obtained from RIKEN BioResource Center
(Tsukuba, Japan). The cells were maintained in DMEM supplemented
with 10% FBS and antibiotics.
Small interfering RNA (siRNA)
transfection
siRNAs were obtained from Hokkaido System Science,
Co., Ltd. (Sapporo, Japan). The sequences of the siRNAs targeting
AMPD3 were 5′-CGGGACUUCUAUAACGUGAGA-3′ (siAMPD3-1) and
5′-CCGGAUGGCAUUCCGAUAUGA-3′ (siAMPD3-2). The sequences of the
siRNAs used for KIT knockdown were 5′-GACGAGAUUAGGCUGUUAUGC-3′
(siKIT-1) and 5′-GCAUCACGGUGACUUCAAUUA-3′ (siKIT-2). The sequence
of the control siRNA that targeted luciferase was
5′-CUUACGCUGAGUACUUCGATT-3′. GIST-T1 cells were transfected with 50
nM siRNAs using Lipofectamine RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was purified from the GIST and normal
gastrointestinal tract tissue samples, the non-transfected cell
lines and the siRNA-transfected cells using the RNeasy Mini kit
(QIAGEN Benelux B. V., Venlo, The Netherlands). cDNA was
synthesized using PrimeScript™ Reverse Transcriptase (Takara Bio
Inc., Shiga, Japan). qPCR was performed on a
LightCycler® Nano Instrument using a FastStart Essential
DNA Green Master kit (Roche Diagnostics, Basel, Switzerland),
according to the manufacturer's protocol. Thermal cycling
conditions were 95°C for 10 sec and 60°C for 30 sec for 35 cycles.
The relative mRNA expression levels were normalized to GAPDH using
LightCyclerR® Nano Software 1.0 (Roche Life Science,
Tokyo, Japan). The sequences of primers used to amplify each gene
were as follows: GAPDH forward, 5′-AGGTGGAGGAGTGGGTGTCGCTGTT-3′ and
reverse, 5′-CCGGGAAACTGTGGCGTGATGG-3′; AMPD3 forward,
5′-ACATCCTGGCTCTCATCACC-3′ and reverse, 5′-CAGCAGATGCTTTTGGTTCA-3′;
AMPD2 forward, 5′-CGTAGTGCCCCGTATGAGTT-3′ and reverse,
5′-CGAGTCACTGTCCGTCTTCA-3′; and KIT forward,
5′-GAAGTCACCGTGATGCCAGC-3′ and reverse,
5′-CTCTGTCTGCATTGTTCTGTG-3′. RT-minus was used for negative control
and three independent experiments were performed.
Western blotting
Total protein was extracted from the non-transfected
and siRNA-transfected cell lines using laemmli sample buffer [20%
glycerol, 135 mM Tris-HCl (pH 6.8), 4% SDS, 10% 2-Mercaptoethanol
and 0.003% BPB]. The protein concentrations of the lysates were
measured using the RC-DC Protein assay (Bio-Rad Laboratories,
Hercules, CA). Proteins were separated by 0.01% SDS-PAGE and
transferred onto polyvinylidene difluoride membranes. Following
blocking with 0.5% skimmed milk in phosphate-buffered saline (PBS),
membranes were incubated with anti-KIT (catalog no., 3392; rabbit
polyclonal; 1,000 dilution; Cell Signaling Technology, Inc.,
Danvers, MA, USA) and anti-β-actin (catalog no., A1978; clone,
AC15; mouse monoclonal; 1:5,000 dilution; Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany) antibodies overnight at 4°C. After
washing with Tris-buffered saline plus Tween-20 [20 mM Tris (pH
7.5), 150 mM NaCl and 0.1% Tween 20], the membranes were incubated
with horseradish peroxidase-conjugated secondary antibodies,
followed by 3,3-diaminobenzidine.
Imatinib sensitivity assay
GIST-T1 cells were transfected with siRNAs, and 48 h
later, the cells were treated with different concentrations of
imatinib for 48 h. A cell proliferation assay was performed and the
ratio of viable cells to control cells (not treated with imatinib)
was calculated. Imatinib was obtained from Sigma-Aldrich (Merck
Millipore).
Cell proliferation assay
The siRNA-transfected cells were plated onto a
96-well plate at a density of 1,000 cells/well. The day of
transfection was set as day 0, and the number of viable cells on
days 1, 2, 3 and 4 were assessed using the Cell Counting kit-8
(CCK-8) assay (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan).
EdU incorporation assay
EdU incorporation assays were performed using the
Click-iT® Plus EdU Alexa Fluor® 594 Imaging
kit (Thermo Fisher Scientific, Inc.). The siRNA-transfected cells
were incubated with EdU for 24 h and fixed with 4%
paraformaldehyde. The cells were permeabilized using 0.5% Triton
X-100, and stained with a reaction cocktail (0.5% Triton X-100 in
PBS) and Hoechst stain, according to the manufacturer's protocol.
Images of the cells were captured using a fluorescence microscope,
and the percentage of EdU-positive cells was evaluated.
Invasion assay
The invasion assay was performed using 8-µm pore
filters inserted into 24-well Boyden Chambers (Corning
Incorporated, Corning, NY, USA). The filter was pre-coated with
Matrigel (BD Bioscience, San Jose, CA, USA). GIST-T1 cells
(1.5×105) were seeded into the upper chamber and allowed
to invade the lower surface of the filter. After 18 h, the cells
were fixed with 100% ethanol and stained with 0.5% crystal violet.
The number of cells in five randomly selected fields was counted,
and three independent experiments were performed.
Migration assay
Cell migration was determined using a wound healing
assay and a Boyden chamber assay. For the wound healing assay,
confluent monolayers of siRNA-transfected cells were scratched with
a 200 µl sterile pipette tip and imaged 24 h later. The distance
between the leading edges of the wound was measured in five
randomly selected fields, and three independent experiments were
performed. To assess cell migration using the Boyden chamber (8 µm
pore size and 6.5 mm membrane diameter), siRNA-transfected cells
were seeded onto the upper chamber and allowed to migrate to the
lower surface of the filter that had been pre-coated with
fibronectin. After 12 h, the cells were fixed in 100% ethanol and
0.5% crystal violet. The number of cells in five randomly selected
fields was counted, and three independent experiments were
performed.
Statistical analysis
Data are expressed as the mean ± standard error. An
unpaired t-test was performed to evaluate P-values. The correlation
of KIT expression with AMPD3 expression was analyzed by Pearson's
correlation analysis using Analyse-it 3.0 (Analyse-it Software,
Ltd., Leeds, UK). A difference was considered statistically
significant when P<0.05.
Results
AMPD3 is highly expressed in GISTs and
is positively correlated with KIT
To obtain further insight into the molecular basis
of GISTs, genes whose expression levels were upregulated in GISTs
were searched for using the Oncomine database (21). This search suggested that AMPD3 is
upregulated in GISTs. The present study examined the expression
levels of AMPD1, AMPD2 and AMPD3 in 16 GIST specimens and 6 normal
gastrointestinal tissues (2 gastric and 4 colon tissues) using
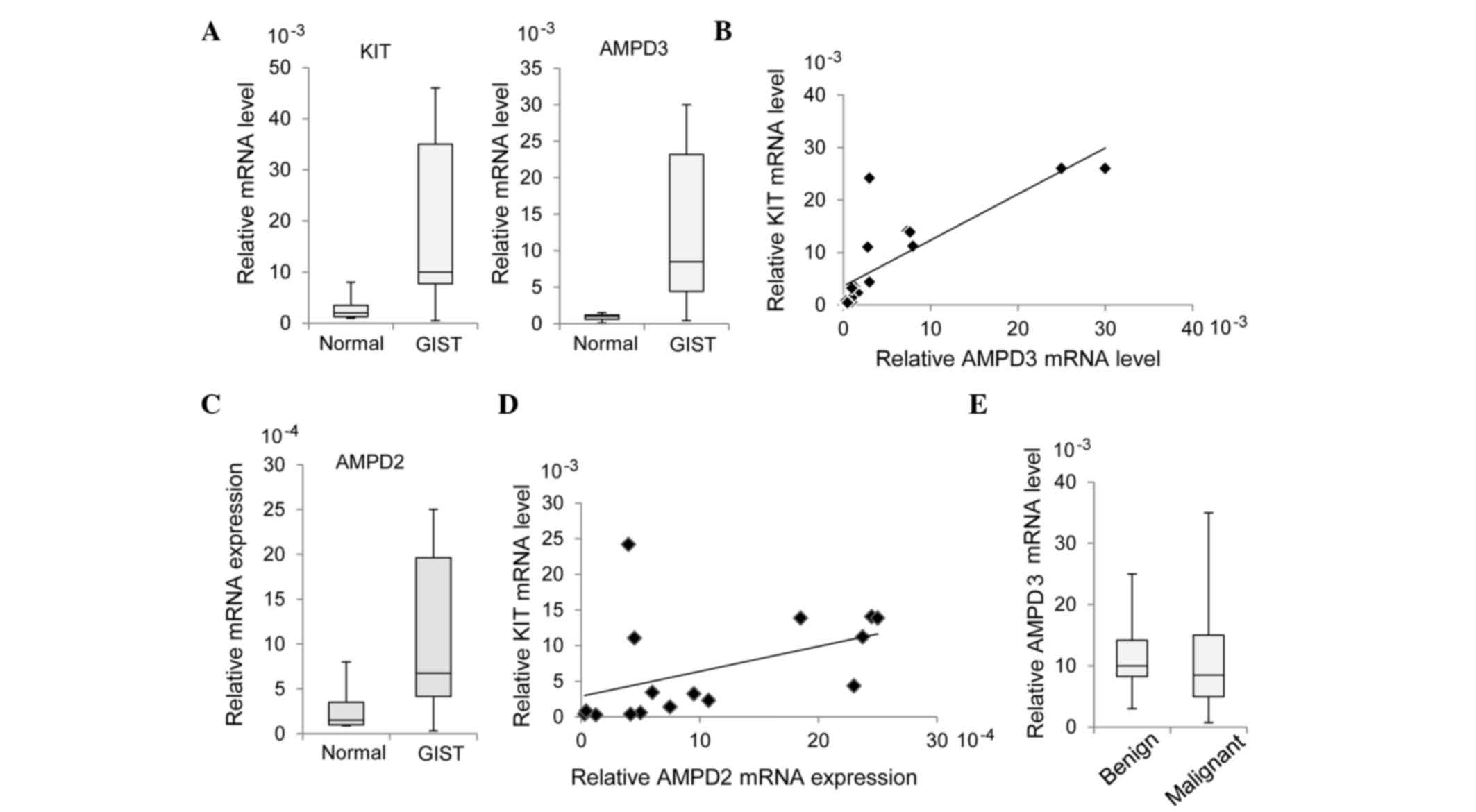
RT-qPCR. As is shown in Fig. 1A, the
mRNA expression levels of AMPD3 were significantly increased in the
GIST specimens, as compared with the normal tissue specimens
(P=0.0002). In addition, there was a significant correlation
between KIT and AMPD3 expression levels in the GIST samples
(r=0.687; P=0.0047; Fig. 1B).
The expression levels of AMPD1 and AMPD2 in the GIST samples were
~10-times lower than that of AMPD3. Similar to AMPD3, increased
expression of AMPD2 in GIST specimens was observed by RT-qPCR
(P=0.0045; Fig. 1C), whereas AMPD1
expression was not increased in GIST specimens (data not shown).
AMPD2 expression was also significantly correlated with KIT
expression (r=0.449; P=0.0806; Fig. 1D), although to a lesser extent than
AMPD3 and KIT. The present study assessed whether AMPD3 expression
was correlated with the malignancy of GISTs; however, there was no
significant difference in AMPD3 expression between benign and
malignant GISTs (P=0.455; Fig.
1E).
AMPD3 and KIT regulate the expression
of each other's protein
The present study examined whether AMPD3 expression
was regulated by KIT in GIST-T1 cells, which were originally
established using GISTs from a Japanese woman (22). The GIST-T1 cell line contains
heterozygous deletion mutations at 57 bases in exon 11 of the KIT
gene (22). Western blot analysis
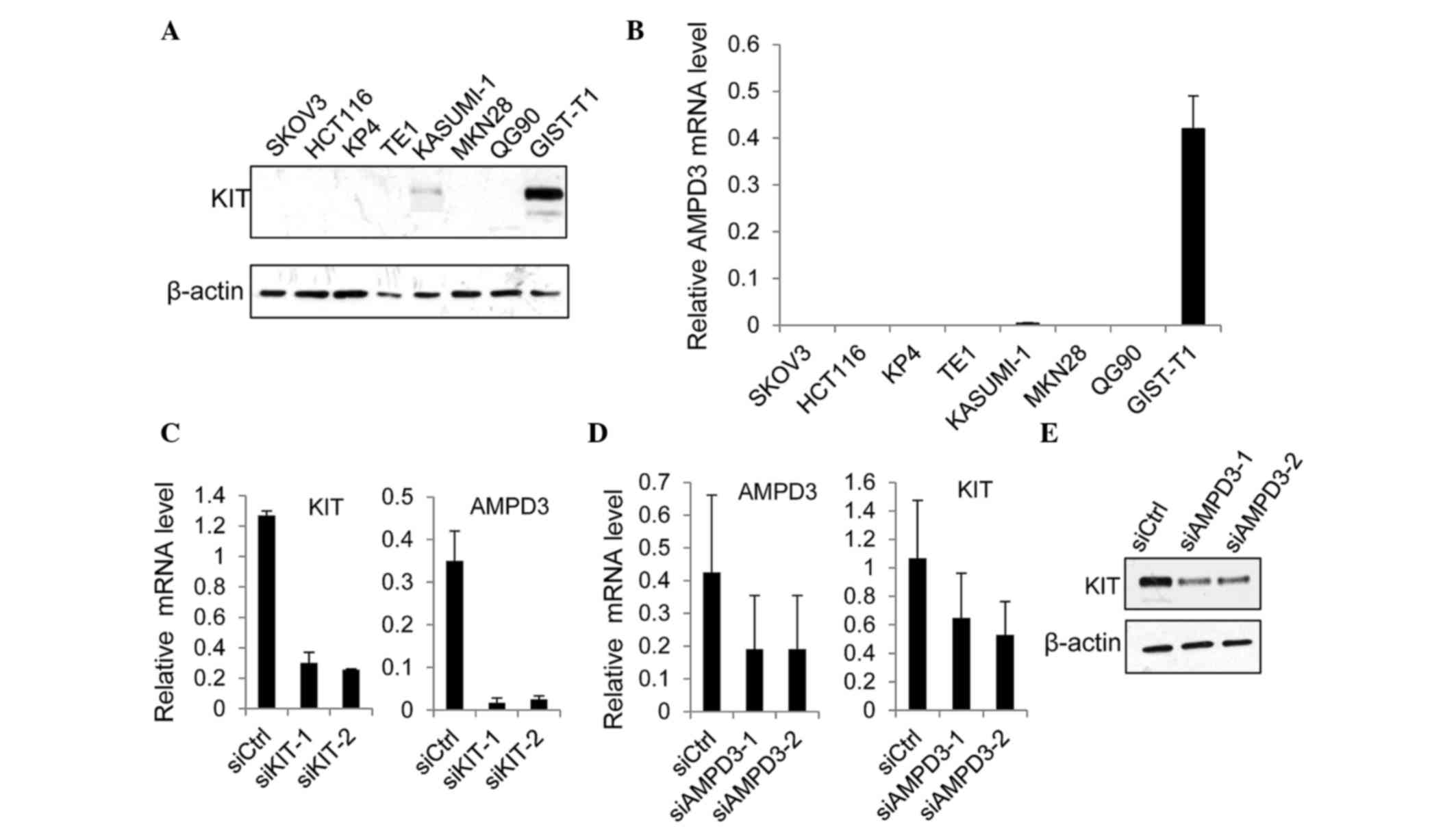
demonstrated that the protein expression levels of KIT were
markedly increased in GIST-T1 cells, as compared with the other
cancer cell lines examined (Fig. 2A).
Notably, the level of AMPD3 mRNA was significantly increased in
GIST-T1 cells, as compared with the other cancer cell lines
(Fig. 2B). The expression of AMPD2
was also upregulated in GIST-T1 cells, as compared with other cell
lines, although its expression was ~10-times lower than AMPD3
expression (data not shown).
Subsequently, the effect of KIT depletion on AMPD3
expression was assessed. The expression of KIT was suppressed using
two different siRNAs, and the level of AMPD3 mRNA was determined
using RT-qPCR. Both KIT-specific siRNAs markedly reduced the KIT
mRNA level (Fig. 2C). In addition,
the level of AMPD3 mRNA was significantly reduced by KIT knockdown
(Fig. 2C). Furthermore, whether AMPD3
knockdown using two AMPD3-specific siRNAs was able to affect the
level of KIT mRNA in GIST-T1 cells was evaluated using RT-qPCR. As
is shown in Fig. 2D and E, AMPD3
knockdown reduced KIT expression at the mRNA and protein level.
Notably, depletion of AMPD3 did not affect the level of AMPD2 mRNA
(data not shown). These results indicate that KIT and AMPD3
regulate the expression of each other's transcript.
Depletion of AMPD3 reduces the
proliferation, migration and invasion of GIST cells
To determine the role of AMPD3 in the malignant
characteristics of GIST-T1 cells, the effect of AMPD3 knockdown on
cell proliferation was assessed using CCK-8 assays. GIST-T1 cells
were transfected with siRNAs, and the number of cells was evaluated
at various time points. As is shown in Fig. 3A, the proliferation of AMPD3-knockdown
cells was reduced compared with the control-siRNA-transfected
cells. To examine whether the suppression of cell proliferation was
mediated by the inhibition of cell cycle progression or by the
induction of apoptosis, EdU incorporation and TUNEL assays (data
not shown) were performed. EdU is a thymidine analog and is
incorporated into newly synthesized DNA during the S phase. As is
shown in Fig. 3B, depletion of AMPD3
significantly reduced the ratio of EdU-positive cells (siAMPD3-1,
P=0.01; siAMPD3-2, P=0.02), whereas apoptosis was not induced by
AMPD3 knockdown (data not shown). These results indicate that AMPD3
is associated with the progression of the GIST cell cycle.
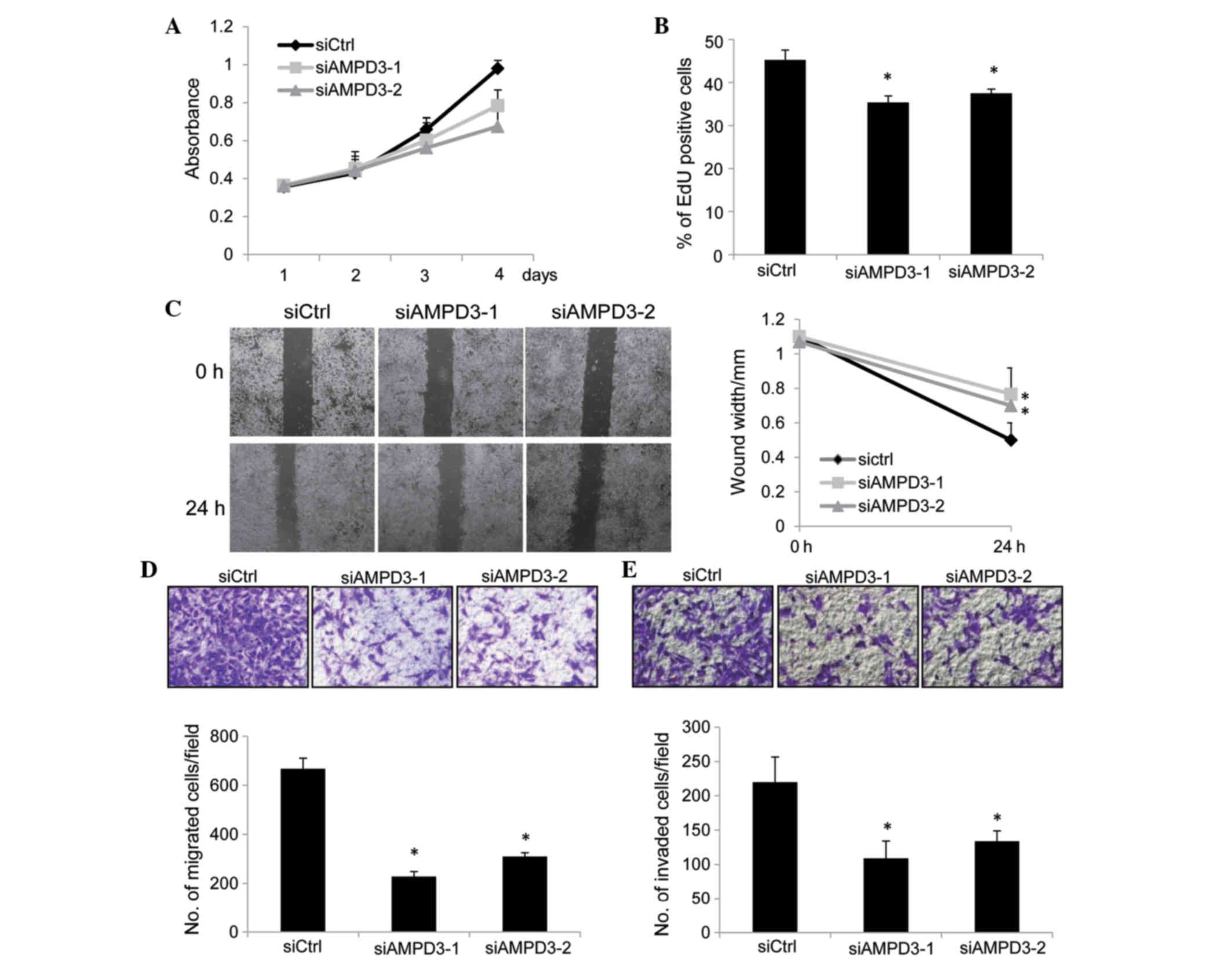 | Figure 3.AMPD3 depletion suppressed the
proliferation, migration and invasion of gastrointestinal stromal
tumor (GIST) cells. (A) GIST-T1 cells were transfected with small
interfering RNAs (siRNAs), and the number of viable cells was
evaluated using the Cell Counting kit-8 assay. (B) GIST-T1 cells
were transfected with siRNAs and, after 48 h, cell proliferation
was assessed using the EdU incorporation assay. The graph shows the
percentage of EdU-positive cells. Three independent experiments
were performed, and the data are presented as the mean ± standard
deviation (SD) (*P<0.05). (C) Confluent monolayers of
siRNA-transfected cells were scratched, and the distances between
the leading edges were measured at 0 and 24 h. Representative
images of the migrated cells are shown. The graph shows the average
distance of the wound edge at the indicated time point
(*P<0.05). (D) siRNA-transfected GIST-T1 cells were subjected to
a migration assay. Representative images of the migrated cells are
shown (0.5% crystal violet stain), and the graph indicates the
average number of migrated cells per field. Three independent
experiments were performed, and the data are shown as the mean ± SD
(*P<0.05). (E) siRNA-transfected GIST-T1 cells were subjected to
a cell invasion assay. Representative images of the invaded cells
are shown (0.5% crystal violet stain), and the graph indicates the
average number of invaded cells per field. Three independent
experiments were performed and data are shown as the mean ± SD
(*P<0.05). Original magnification, ×40. AMPD, adenosine
monophosphate deaminase 3; siCtrl, control-siRNA; siAMPD3, siRNA
targeting AMPD3. |
The migration of AMPD3-knockdown cells was evaluated
by performing a scratch assay. Confluent monolayers of
siRNA-transfected GIST-T1 cells were scratched, and the migration
of the cells into the scratch was observed after 24 h. The
migration of AMPD3-knockdown cells was delayed compared with the
control-siRNA-transfected cells (siAMPD3-1, P=0.008; siAMPD3-2,
P=0.002; Fig. 3C). A modified Boyden
chamber assay was performed to further confirm this result. The
siRNA-transfected cells were placed on the upper surface of the
filter and allowed to migrate to the bottom surface, which was
coated with fibronectin. Cells that migrated to the bottom surface
were counted to evaluate cell migration. Notably, the migration of
AMPD3-depleted cells was suppressed compared with the
control-siRNA-transfected cells (siAMPD3-1, P=0.008; siAMPD3-2,
P=0.002; Fig. 3D). Finally, the
invasion of AMPD3-depleted GIST-T1 cells was examined using a
Matrigel-coated Boyden chamber. As is shown in Fig. 3E, AMPD3 suppression significantly
delayed the invasion of GIST-T1 cells (siAMPD3-1, P=0.0004;
siAMDP3-2, P=0.0007).
AMPD3 depletion increases GIST-T1 cell
sensitivity to imatinib
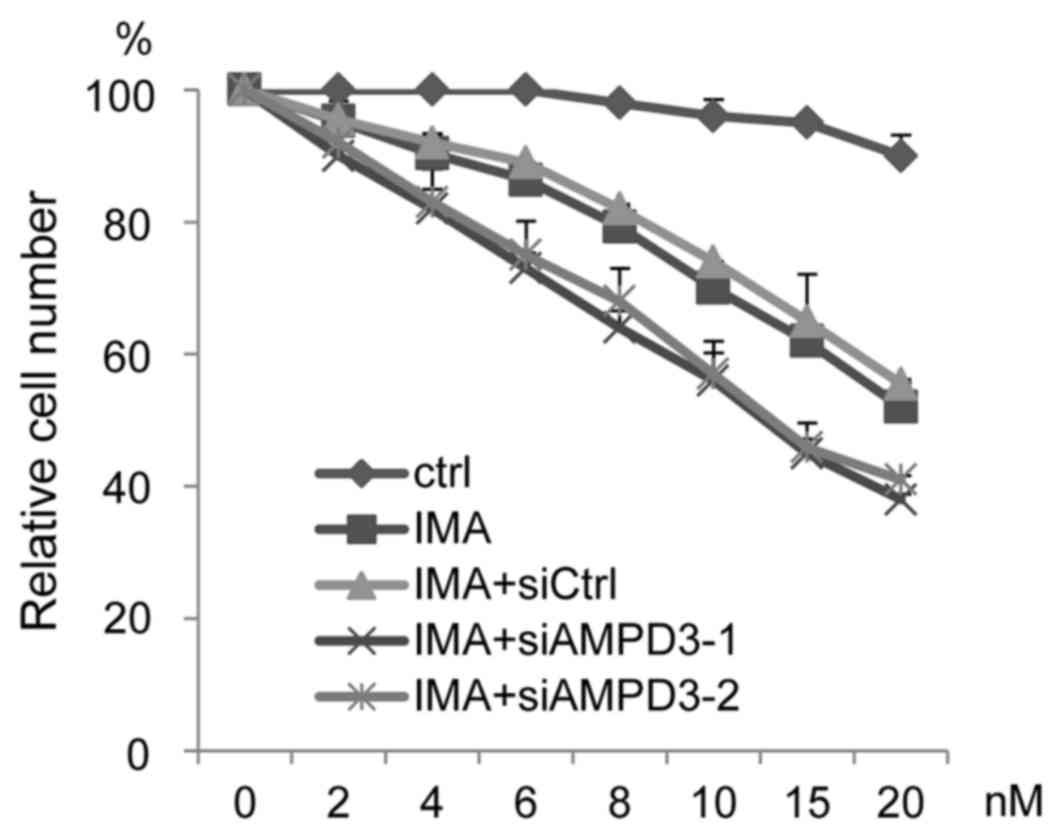
The ability of AMPD3 depletion to affect the
sensitivity of GIST-T1 cells to imatinib was evaluated. The
siRNA-transfected GIST-T1 cells were treated with various
concentrations of imatinib, and cell growth was assessed. As shown
in Fig. 4, AMPD3-siRNA-transfected
cells were more sensitive to imatinib than the
control-siRNA-transfected cells.
Discussion
Previous studies have demonstrated that activating
mutations in the receptor tyrosine kinases KIT and PDGFRA are
critical for the pathogenesis of GISTs (23–25).
Recent studies have shown the important function of ETS variant 1
(ETV1), an ETS family transcriptional factor, in the progression of
GISTs, and suggested that targeting both KIT and ETV1 may be
effective for the treatment of this type of tumor (26,27). KIT
is known to activate multiple pathways, including the RAS/MAPK and
PI3K/AKT signaling pathways (28,29); thus,
activated KIT may promote the expression of various
cancer-associated genes. The present study demonstrated that AMPD3
was significantly upregulated in GIST tissue samples, as compared
with normal tissue samples, and that AMPD3 expression was
correlated with KIT expression. In addition, KIT depletion was
shown to suppress the expression of AMPD3 in GIST-T1 cells at the
mRNA level. These results suggested that KIT regulates the
expression of AMPD3 in GISTs. Notably, it was also demonstrated
that AMPD3 knockdown suppressed KIT expression in GIST-T1 cells.
Therefore, KIT and AMPD3 may form a positive feedback loop to
promote their expression and the progression of GISTs.
AMPD3 is ubiquitously expressed, and mice with a
total AMPD3 deficiency showed increased levels of ATP in their
cells (14), indicating that AMPD3
contributes to the energy balance in cells. In the present study,
the depletion of AMPD3 in GIST-T1 cells suppressed the cell
proliferation, migration and invasion. These results indicated that
AMPD3 is associated with the malignant characteristics of GIST-T1
cells. AMPD3 depletion reduced KIT expression; thus, the
suppression of malignant characteristics by AMPD3 may be partly
mediated by KIT suppression. AMPD3 may also promote cancer
progression by contributing to the energy-dependent activation of
cancer-associated pathways. For example, AMP-activated protein
kinase (AMPK) is known to suppress the progression of various
tumors (30–32). Therefore, increases in the levels of
AMP as a result of AMPD3 knockdown may promote the activation of
AMPK, which can subsequently inhibit anabolic pathways that are
essential for cancer cell growth and survival. AMPD3 depletion may
promote the activation of protein kinases, such as AMPK, and
suppress cancer cell migration and invasion.
In summary, we have shown that the expression levels
of KIT and AMPD3 were correlated in GIST-T1 cells, and that the two
proteins likely form a positive feedback loop. In addition, it was
demonstrated that AMPD3 depletion suppressed the migration and
invasion of GIST-T1 cells. Although imatinib is effective in GIST
treatment, the drug cannot completely eradicate the tumor. The
results of the present study suggested that the combined inhibition
of KIT and AMPD3 may be effective for the treatment of GIST.
Acknowledgements
The authors would like to thank the members of the
Division of Cancer Biology, Nagoya University Graduate School of
Medicine for their helpful discussions and technical assistance.
This research was funded by a grant from the Ministry of Education,
Culture, Sports, Science and Technology of Japan (grant no.
24790689), and supported by the Naito Foundation.
References
|
1
|
Corless CL, Fletcher JA and Heinrich MC:
Biology of gastrointestinal stromal tumors. J Clin Oncol.
22:3813–3825. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rubin BP, Singer S, Tsao C, Duensing A,
Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, et al: KIT
activation is a ubiquitous feature of gastrointestinal stromal
tumors. Cancer Res. 61:8118–8121. 2001.PubMed/NCBI
|
|
3
|
Corless CL and Heinrich MC: Molecular
pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol.
3:557–586. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Corless CL, Barnett CM and Heinrich MC:
Gastrointestinal stromal tumours: Origin and molecular oncology.
Nat Rev Cancer. 11:865–878. 2011.PubMed/NCBI
|
|
5
|
Duensing A, Medeiros F, McConarty B,
Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD and
Fletcher JA: Mechanisms of oncogenic KIT signal transduction in
primary gastrointestinal stromal tumors (GISTs). Oncogene.
23:3999–4006. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heinrich MC, Corless CL, Demetri GD,
Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den
Abbeele AD, Druker BJ, et al: Kinase mutations and imatinib
response in patients with metastatic gastrointestinal stromal
tumor. J Clin Oncol. 21:4342–4349. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morisaki T, Sabina RL and Holmes EW:
Adenylate deaminase. A multigene family in humans and rats. J Biol
Chem. 265:11482–11486. 1990.PubMed/NCBI
|
|
8
|
Mahnke DK and Sabina RL: Calcium activates
erythrocyte AMP deaminase [isoform E (AMPD3)] through a
protein-protein interaction between calmodulin and the N-terminal
domain of the AMPD3 polypeptide. Biochemistry. 44:5551–5559. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mahnke-Zizelman DK and Sabina RL: Cloning
of human AMP deaminase isoform E cDNAs. Evidence for a third AMPD
gene exhibiting alternatively spliced 5′-exons. J Biol Chem.
267:20866–20877. 1992.PubMed/NCBI
|
|
10
|
Coley W, Rayavarapu S, Pandey GS, Sabina
RL, Van der Meulen JH, Ampong B, Wortmann RL, Rawat R and Nagaraju
K: The molecular basis of skeletal muscle weakness in a mouse model
of inflammatory myopathy. Arthritis Rheum. 64:3750–3759. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
van Adel BA and Tarnopolsky MA: Metabolic
myopathies: Update 2009. J Clin Neuromuscul Dis. 10:97–121. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Akizu N, Cantagrel V, Schroth J, Cai N,
Vaux K, McCloskey D, Naviaux RK, Van Vleet J, Fenstermaker AG,
Silhavy JL, et al: AMPD2 regulates GTP synthesis and is mutated in
a potentially treatable neurodegenerative brainstem disorder. Cell.
154:505–517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamada Y, Goto H, Wakamatsu N and
Ogasawara N: A rare case of complete human erythrocyte AMP
deaminase deficiency due to two novel missense mutations in AMPD3.
Hum Mutat. 17:782001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cheng J, Morisaki H, Toyama K, Ikawa M,
Okabe M and Morisaki T: AMPD3-deficient mice exhibit increased
erythrocyte ATP levels but anemia not improved due to PK
deficiency. Genes Cells. 17:913–922. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Desman G, Waintraub C and Zippin JH:
Investigation of cAMP microdomains as a path to novel cancer
diagnostics. Biochim Biophys Acta. 1842:2636–2645. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fu QF, Liu Y, Fan Y, Hua SN, Qu HY, Dong
SW, Li RL, Zhao MY, Zhen Y, Yu XL, et al: Alpha-enolase promotes
cell glycolysis, growth, migration, and invasion in non-small cell
lung cancer through FAK-mediated PI3K/AKT pathway. J Hematol Oncol.
8:222015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pantano F, Santoni M, Procopio G, Rizzo M,
Iacovelli R, Porta C, Conti A, Lugini A, Milella M, Galli L, et al:
The changes of lipid metabolism in advanced renal cell carcinoma
patients treated with everolimus: A new pharmacodynamic marker?
PLoS One. 10:e01204272015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen H, Wang JP, Santen RJ and Yue W:
Adenosine monophosphate activated protein kinase (AMPK), a mediator
of estradiol-induced apoptosis in long-term estrogen deprived
breast cancer cells. Apoptosis. 20:821–830. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shieh JM, Chen YC, Lin YC, Lin JN, Chen
WC, Chen YY, Ho CT and Way TD: Demethoxycurcumin inhibits energy
metabolic and oncogenic signaling pathways through AMPK activation
in triple-negative breast cancer cells. J Agric Food Chem.
61:6366–6375. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang J, Wang Y, Shang D, Yu F, Liu W,
Zhang Y, Feng C, Wang Q, Xu Y, Liu Y, et al: Characterizing and
optimizing human anticancer drug targets based on topological
properties in the context of biological pathways. J Biomed Inform.
54:132–140. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rhodes DR, Yu J, Shanker K, Deshpande N,
Varambally R, Ghosh D, Barrette T, Pandey A and Chinnaiyan AM:
ONCOMINE: A cancer microarray database and integrated data-mining
platform. Neoplasia. 6:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Taguchi T, Sonobe H, Toyonaga S, Yamasaki
I, Shuin T, Takano A, Araki K, Akimaru K and Yuri K: Conventional
and molecular cytogenetic characterization of a new human cell
line, GIST-T1, established from gastrointestinal stromal tumor. Lab
Invest. 82:663–665. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Buleje J, Acosta Ó, Guevara-Fujita M,
Enriquez Y, Taxa L, Machicado E, Lizaraso-Caparó F and Fujita R:
Mutational profile of KIT and PDGFRA genes in gastrointestinal
stromal tumors in Peruvian samples. Rev Esp Enferm Dig. 107:72–78.
2015.PubMed/NCBI
|
|
24
|
Comandone A and Boglione A: The importance
of mutational status in prognosis and therapy of GIST. Recenti Prog
Med. 106:17–22. 2015.(In Italian). PubMed/NCBI
|
|
25
|
Joensuu H, Rutkowski P, Nishida T, Steigen
SE, Brabec P, Plank L, Nilsson B, Braconi C, Bordoni A, Magnusson
MK, et al: KIT and PDGFRA mutations and the risk of GI stromal
tumor recurrence. J Clin Oncol. 33:634–642. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ran L, Sirota I, Cao Z, Murphy D, Chen Y,
Shukla S, Xie Y, Kaufmann MC, Gao D, Zhu S, et al: Combined
inhibition of MAP kinase and KIT signaling synergistically
destabilizes ETV1 and suppresses GIST tumor growth. Cancer Discov.
5:304–315. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Y, Gu ML, Zhou XX, Ma H, Yao HP and
Ji F: Altered expression of ETV1 and its contribution to
tumorigenic phenotypes in gastrointestinal stromal tumors. Oncol
Rep. 32:927–934. 2014.PubMed/NCBI
|
|
28
|
Liang J, Wu YL, Chen BJ, Zhang W, Tanaka Y
and Sugiyama H: The C-kit receptor-mediated signal transduction and
tumor-related diseases. Int J Biol Sci. 9:435–443. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu K: Stem cell factor (SCF)-kit mediated
phosphatidylinositol 3 (PI3) kinase signaling during mammalian
oocyte growth and early follicular development. Front Biosci.
11:126–135. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim A, Im M and Ma JY: Ethanol extract of
Remotiflori radix induces endoplasmic reticulum stress-mediated
cell death through AMPK/mTOR signaling in human prostate cancer
cells. Sci Rep. 5:83942015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schuster S, Penke M, Gorski T, Gebhardt R,
Weiss TS, Kiess W and Garten A: FK866-induced NAMPT inhibition
activates AMPK and downregulates mTOR signaling in hepatocarcinoma
cells. Biochem Biophys Res Commun. 458:334–340. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Popovics P, Frigo DE, Schally AV and Rick
FG: Targeting the 5′-AMP-activated protein kinase and related
metabolic pathways for the treatment of prostate cancer. Expert
Opin Ther Targets. 19:617–632. 2015. View Article : Google Scholar : PubMed/NCBI
|