Introduction
Pancreatic cancer is the tenth most common form of
cancer, and is the fourth leading cause of cancer-associated
mortality (1). Pancreatic cancer is
one of the deadliest forms of cancer, with a 5-year survival rate
of only 4.4%.
A number of risk factors have been identified for
pancreatic cancer, including gender, ethnicity and history of
chronic pancreatitis (2). The
greatest environmental risk factor for pancreatic cancer is
cigarette smoking (2). The relative
risk for cigarette smoking is between 2 and 3 fold, but can be
higher in heavy smokers, suggesting a dose-response association
(2). It is not clear how cigarette
smoke contributes to pancreatic cancer development. Components of
cigarette smoke likely reach the pancreas through the general
circulation, but also may come from the GI tract through reflux.
The chemical component(s) of cigarette smoke that are responsible
for increasing pancreatic cancer risk are not known, although some
evidence supports a role for aromatic amines or N-nitroso compounds
(2). The molecular mechanisms of the
enhancement of pancreatic cancer by cigarette smoke are not
understood.
One approach to understanding the molecular
mechanisms of pancreatic cancer development is to use animal
models. Several chemical carcinogenesis models exist (3). A transgenic model has been developed,
which uses an oncogenic Kras (KRasG12D) inserted into
the endogenous Kras locus (4). The
gene has a Lox-STOP-Lox (LSL) construct inserted upstream. These
mice are interbred with mice containing the Cre recombinase
downstream from a pancreatic specific promoter, either PDX-1 or
P48. The PDX-1-Cre;LSL-KRasG12D mice develop pancreatic
intraepithelial neoplasias (PanINs), which progress over time
(4). In addition, when these mice are
crossed to mice containing p53 mutations or Ink4a/Arf deficiency,
the rapid development of pancreatic adenocarcinomas is observed
(5,6).
The present study examined the hypothesis that
exposure to cigarette smoke or overexpression of the oncogenic Kras
oncogene would affect cell proliferation in the pancreas in mice.
PDX-1-Cre; LSL-KRasG12D and wild-type mice were exposed
to cigarette smoke for 2 weeks. It has been observed previously
that a 5-day exposure to cigarette smoke is sufficient to increase
cell proliferation in the lung (7,8). Mice were
treated with bromodeoxyuridine (BrdU) during the exposure period,
and the rate of cell proliferation and gene expression associated
with cell proliferation were examined.
Materials and methods
Experimental design
PDX-1-Cre and LSL-KRASG12D mice were
obtained from the National Cancer Institute Mouse Repository
(Frederick, MD, USA). The two strains were bred to obtain 16
PDX-1-Cre; LSL-KRASG12D mice. PDX-1-Cre-positive mice
were used as controls. Male 9–10 week old mice were used. Half of
the mice in each group were exposed to cigarette smoke for a period
of 2 weeks. In total, 7–8 mice were in each of the 4 groups, with a
total of 30 mice. An inhalation exposure to smoke was performed in
a whole-body Hinners type stainless steel/glass chamber, as
previously described (9). Cigarette
smoke was generated from 3R4F University of Kentucky (Lexington,
KY, USA) research cigarettes. The concentration of smoke
particulates in the exposure chamber atmosphere averaged 46±3 mg
TPM/m3. The mice received smoke exposure for a total of
6 h each day, 5 days per week, Monday to Friday, for 2 weeks (10
days total exposure). Mice were euthanized 3 days after the last
exposure (the next Monday morning). Mice were administered BrdU in
the drinking water (0.5 mg/ml), starting at the same time as smoke
exposure, and continuing until they were euthanized. Subsequently,
the pancreas was split into two pieces, with one half fixed in
buffered neutral formalin (and subsequently processed to paraffin
blocks) and the other half flash frozen in liquid nitrogen, and
then stored at −80°C.
Analysis of cell proliferation
Subsequent to fixation and processing, 5-µm sections
were prepared from the paraffin blocks. The sections of
paraffin-embedded tissue samples were deparaffinized, thoroughly
washed in water, and then placed in 3% H2O2
in methanol for 10 min. The sections were then stained
immunohistochemically by the avidin-biotin-peroxidase complex
method using Vectastain ABC reagent (Vector Laboratories,
Burlingame, CA, USA), with mouse anti-BrdU monoclonal antibodies
(cat. no. 555627; BD Biosciences, Franklin Lakes, NJ, USA), at 100
µl/slide using a 1:40 dilution of BrdU. The reaction product was
then visualized using diaminobenzidine (Vector Laboratories
Peroxidase Substrate kit) and the slides were counterstained with
hematoxylin. Having brown nuclei identified cells that had
incorporated BrdU. Labeling indexes were determined for the
following 3 regions in the pancreas: ductal, acinar and islet
cells. In total, 500 cells per slide were counted and labeling
indexes were determined.
Western blot analysis
Levels of tumor necrosis factor (TNF) a, p53 and
cyclin D1 proteins in pancreas homogenates were determined by
western analyses. Antibodies were obtained from Santa Cruz
Biotechnology (Dallas, TX, USA). The frozen pancreases were
homogenized in extraction buffer (Pierce Biotechnology, Inc.,
Rockford, IL, USA) containing protease inhibitors (Sigma-Aldrich;
Merck Millipore, Darmstadt, Germany). Lysed tissue was centrifuged
at 8,000 × g for 30 min at 4°C. Protein levels of the
supernatants were determined by bicinchoninic acid assay (Pierce
Biotechnology, Inc.) and stored at −80°C. Protein samples (30 µg
per treatment) were separated using 10% SDS-PAGE and subsequently
were transferred onto nitrocellulose membranes. Membranes were
blocked with 5% non-fat milk buffer and incubated overnight at 4°C
with primary antibodies. Subsequent to washing, membranes were
incubated with secondary antibodies conjugated with horseradish
peroxidase and visualized using enhanced chemiluminescence
detection reagents (Thermo Fisher Scientific Inc., Waltham, MA,
USA). Bands were quantified using ImageJ software (National
Institutes of Health, Bethesda, MD, USA) and normalized to β-actin
expression.
Statistical analysis
Results were first analyzed by two-way analysis of
variance (ANOVA), using Sigmaplot for Windows (version 13.0; Systat
Software, Inc., San Jose, CA). If significant interactions were
identified, differences between means were determined using the
Holm-Sidak post-hoc test. The results are reported as the mean ±
standard error of the mean. P≤0.05 was considered to indicate a
statistically significant difference. The results of the ANOVAs are
shown in Table I.
 | Table I.Results of two-way analysis of
variance for the study endpoints. |
Table I.
Results of two-way analysis of
variance for the study endpoints.
|
| P-values |
|---|
|
|
|
|---|
| Study endpoint | Main effect for smoke
exposurea | Main effect for Kras
overexpressionb | Smoke/Kras
interaction |
|---|
| Final body
weight |
0.033 | 0.41 | 0.27 |
| Ductal cell LI |
0.016 | <0.001 | 0.12 |
| Acinar cell LI |
0.013 | <0.001 | 0.28 |
| Islet cell LI | 0.72 | <0.001 |
0.066 |
| TNFα | 0.58 | 0.10 | 0.84 |
| P53 | 0.59 |
0.090 | 0.75 |
| Cyclin
D1 | 0.39 | 0.38 | 0.89 |
Results
The present study examined the effects of mutant
Kras overexpression and smoke exposure on pancreatic cell
proliferation and associated gene expression. Mice were exposed to
sidestream tobacco smoke for 2 weeks. Mutant Kras overexpression
did not affect body weights, but short-term cigarette smoke
exposure significantly reduced body weights (P=0.033; data not
shown). Mice were administered BrdU in the drinking water, and then
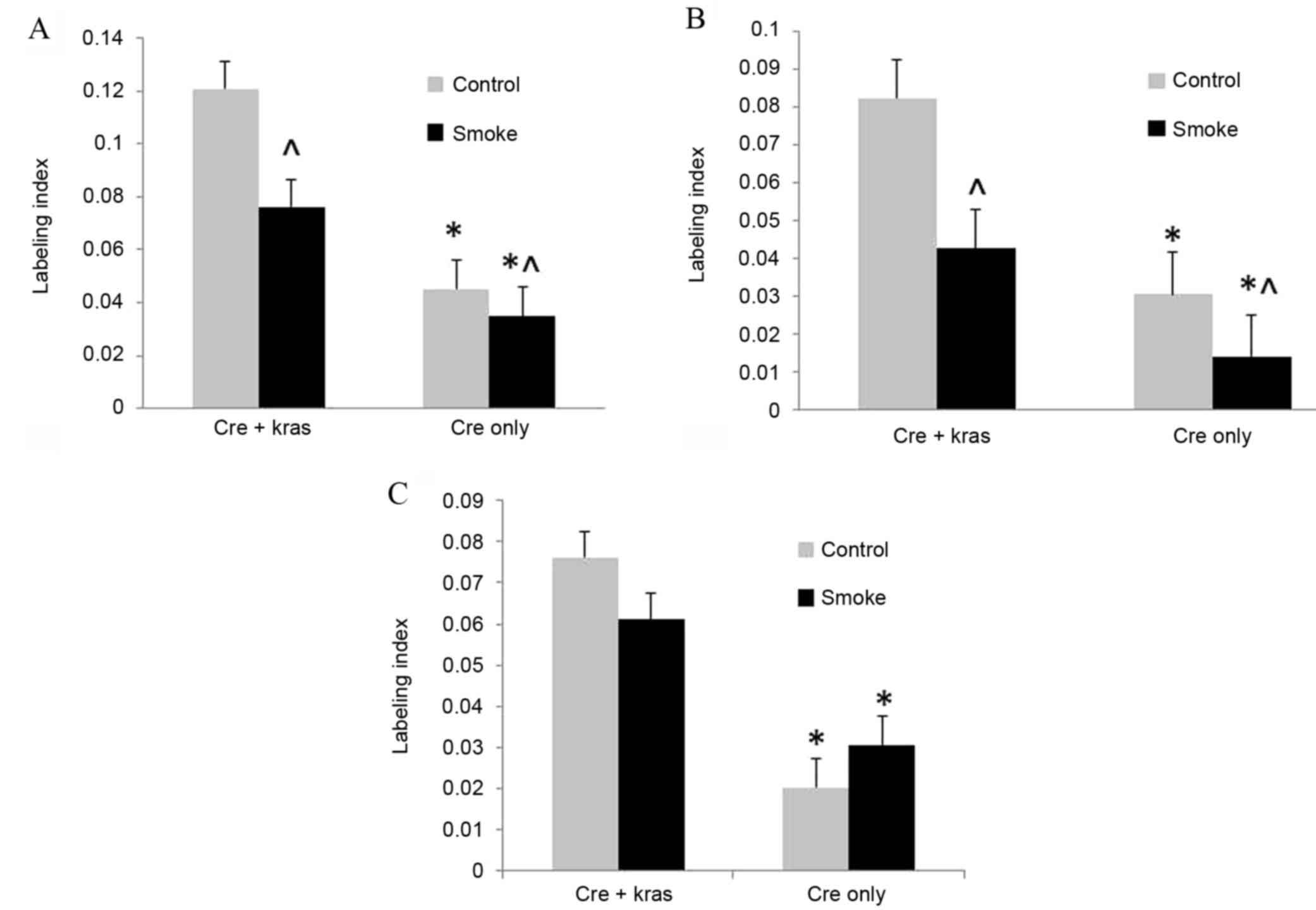
labeling indexes of BrdU-stained nuclei were used to quantify cell
proliferation. Cell proliferation in ductal cells, from which the
majority of human pancreatic cancers are derived, was increased
(P<0.001) in mice overexpressing mutant Kras; however, cigarette
smoke exposure decreased (P=0.016) cell proliferation (Fig. 1). In acinar cells, cell proliferation
was similarly increased (P<0.001) by mutant Kras overexpression
and decreased (P=0.013) by cigarette smoke exposure. In islet
cells, cell proliferation was increased (P<0.001) by mutant Kras
overexpression, but not affected by cigarette smoke exposure.
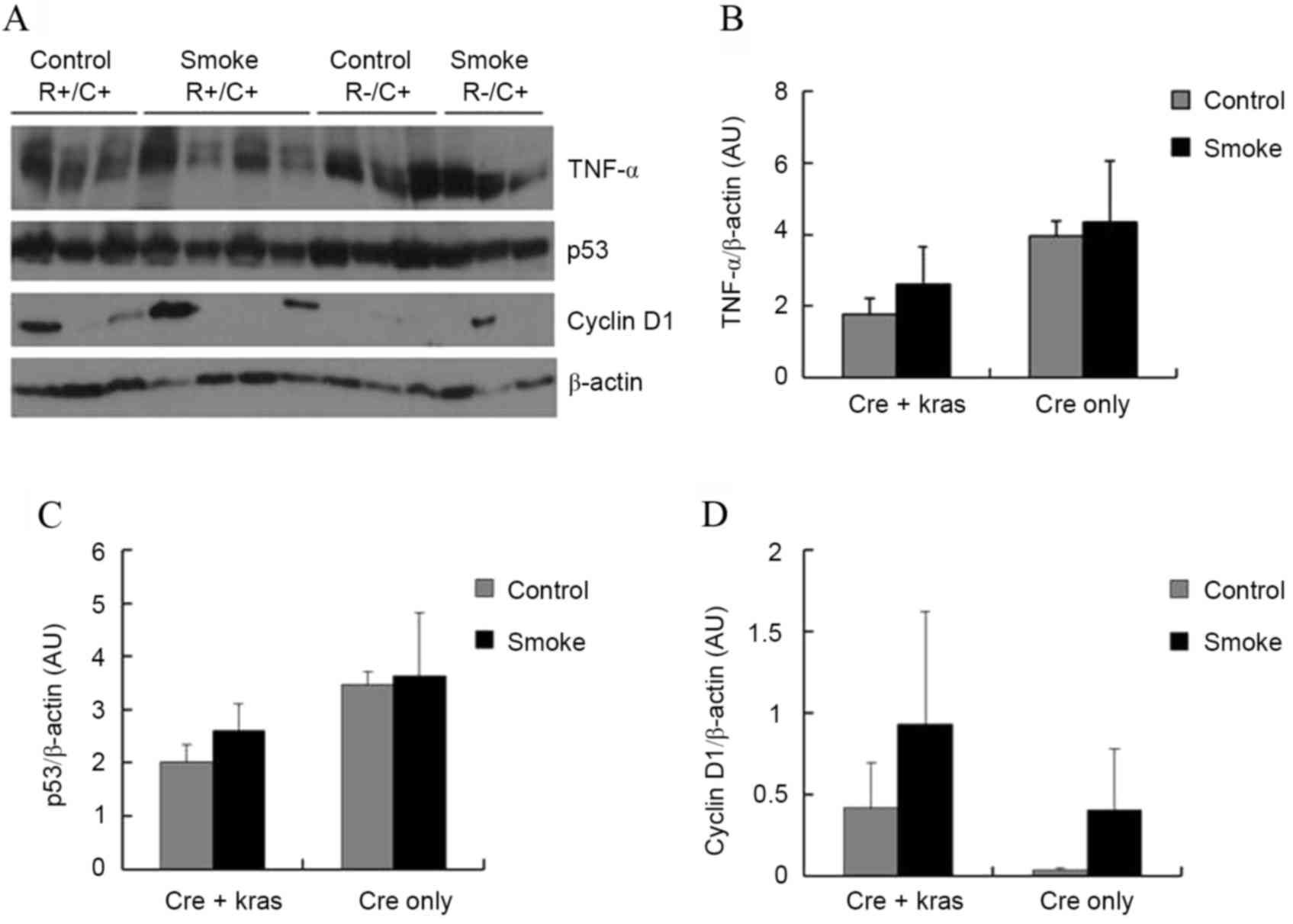
Subsequently, the levels of several proteins in the
pancreas that could be affecting cell proliferation and
tumorigenesis were examined. TNFα is a pro-inflammatory cytokine
that may have promoting or inhibitory effects in pancreatic
carcinogenesis (10). p53 is a tumor
suppressor gene that promotes apoptosis but also inhibits cell
proliferation (11). Cyclin
D1 regulates cyclin-dependent kinases (CDKs), which
increase cell proliferation (12).
Neither cigarette smoke exposure nor Kras overexpression, however,
significantly affected the protein levels of TNFα, p53, or cyclin
D1 in the pancreas (Fig.
2). Kras overexpression slightly, but not significantly,
decreased the protein levels of TNFα (P=0.10) and p53 (P=0.09).
Discussion
In the present study, it was observed that Kras
overexpression increased but cigarette smoke exposure decreased
cell proliferation in pancreatic ductal cells, the cell type from
which most human pancreatic cancers are derived. It was observed
that cigarette smoke induced decreases in cell proliferation in
pancreatic ductal cells and acinar cells but had no effect in islet
cells. A previous study has quantified cell proliferation in
response to cigarette smoke; Wisniewska et al (13) observed inconsistent effects of
cigarette smoke. Wisniewska et al (13), however, did not differentiate between
the different types of pancreatic cells. Cigarette smoke was found
not to promote 7,12-dimethylbenzanthracene-induced pancreatic
carcinogenesis in mice (14). Cell
proliferation was increased in the lung after short-term exposure
to cigarette smoke in several studies (7,8,15,16). In
other studies examining cell proliferation in the pancreas, Xue
et al (17) found that diets
high in fat and phosphorus and low in calcium and vitamin D
increased cell proliferation in pancreatic ductal and acinar cells.
Ledda-Columbano et al (18)
found that the administration of thyroid hormone increased cell
proliferation in pancreatic acinar cells.
The present study observed that the expression of a
mutant Kras oncogene in the pancreas increased cell proliferation
in ductal, acinar and islet cells. The expression of this oncogene
has previously been shown to induce PanINs and pancreatic tumors
(4). The expression of the mutant
Kras produces metabolic changes that are supportive of an increase
in cell proliferation (19).
Neither KrasG12D nor smoke exposure
significantly affected the levels of three proteins that could
affect cell proliferation. One factor could have been that whole
pancreas was used in the analysis, which could have prevented the
observation of changes in individual cell types.
KrasG12D's inhibitory effect on p53 protein levels is
consistent with the effect of p53 on increasing apoptosis but
inhibiting cell proliferation (11).
TNFα has been found to increase pancreatic cell proliferation
(20,21); therefore, it is not clear how Kras
inhibition of TNFα contributes to the increase in cell
proliferation observed in the mutant Kras mice.
In summary, the present mouse model does not appear
to be a good model for cigarette smoke-induced human pancreatic
carcinogenesis. Cigarette smoking is the number one environmental
risk factor for human pancreatic cancer. Therefore, the inhibition
of pancreatic ductal and acinar cell proliferation by smoke
exposure in the present study is not associated with smoke effects
on humans. It would be expected that decreased cell proliferation
would be protective against pancreatic carcinogenesis.
Acknowledgments
The authors thank Ms. Ruth Holland and Mr. Chris
Holland (both University of Kentucky) for technical assistance.
This study was supported by the Institute for Science and Health
(grant no. 09-1830-01RFA07) and the Kentucky Agricultural
Experiment Station.
Glossary
Abbreviations
Abbreviations:
|
ANOVA
|
analysis of variance
|
|
BrdU
|
bromodeoxyuridine
|
|
LSL
|
Lox-STOP-Lox
|
|
PanINs
|
pancreatic intraepithelial
neoplasias
|
|
TNF
|
tumor necrosis factor
|
References
|
1
|
American Cancer Society, . Cancer Facts
and Figures. American Cancer Society; Atlanta, GA: 2015
|
|
2
|
Li D and Jiao L: EpidemiologyPancreatic
Cancer. Von Hoff DD, Evans DB and Hruban RH: Jones and Bartlett
Publishers; Sudbury, MA: pp. 103–112. 2005
|
|
3
|
Grippo PJ and Sandgren EP: Modeling
pancreatic cancer in animals to address specific hypothesesMethods
in Moleculear Medicine. 103. (Pancreatic Cancer: Methods and
Protocols). Su G: Humana Press; Totowa, NJ: pp. 217–243. 2005
|
|
4
|
Hingorani SR, Petricoin EF, Maitra A,
Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD,
Hitt BA, et al: Preinvasive and invasive ductal pancreatic cancer
and its early detection in the mouse. Cancer Cell. 4:437–450. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hingorani SR, Wang L, Multani AS, Combs C,
Deramaudt TB, Hruban RH, Rustgi AK, Chang S and Tuveson DA:
Trp53R172H and KrasG12D cooperate to promote chromosomal
instability and widely metastatic pancreatic ductal adenocarcinoma
in mice. Cancer Cell. 7:469–483. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aguirre AJ, Bardeesy N, Sinha M, Lopez L,
Tuveson DA, Horner J, Redston MS and DePinho RA: Activated Kras and
Ink4a/Arf deficiency cooperate to produce metastatic pancreatic
ductal adenocarcinoma. Genes Dev. 17:3112–3126. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Witschi H, Oreffo VI and Pinkerton KE:
Six-month exposure of strain A/J mice to cigarette sidestream
smoke: Cell kinetics and lung tumor data. Fundam Appl Toxicol.
26:32–40. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li J, Tharappel JC, Han SG, Cantor AH, Lee
EY, Gairola CG and Glauert HP: Effect of dietary selenium and
cigarette smoke on pulmonary cell proliferation in mice. Toxicol
Sci. 111:247–253. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gairola CG: Animal models of 2nd hand
smokingMolecular Mechanisms for Tobacco-Induced Diseases. Wang XL
and Scott D: Nova Science Publishers; New York, NY: pp. 121–132.
2006
|
|
10
|
Chopra M, Lang I, Salzmann S, Pachel C,
Kraus S, Bäuerlein CA, Brede C, Garrote AL, Mattenheimer K, Ritz M,
et al: Tumor necrosis factor induces tumor promoting and
anti-tumoral effects on pancreatic cancer via TNFR1. PLoS One.
8:e757372013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kruiswijk F, Labuschagne CF and Vousden
KH: p53 in survival, death and metabolic health: A lifeguard with a
licence to kill. Nat Rev Mol Cell Biol. 16:393–405. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim JK and Diehl JA: Nuclear cyclin D1: An
oncogenic driver in human cancer. J Cell Physiol. 220:292–296.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wisniewska E, Dylik A, Kulza M, Florek E,
Piekoszewski W, Seńczuk-Przybyłowska M and Marszałek A: Exposure to
ethanol and tobacco smoke in relation to level of PCNA antigen
expression in pancreatic and hepatic rat cells. Pharmacol Rep.
65:914–926. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bersch VP, Osvaldt AB, Edelweiss MI, Rde C
Schumacher, Wendt LR, Abreu LP, Blom CB, Abreu GP, Costa L,
Piccinini P and Rohde L: Effect of nicotine and cigarette smoke on
an experimental model of intraepithelial lesions and pancreatic
adenocarcinoma induced by 7,12-dimethylbenzanthracene in mice.
Pancreas. 38:65–70. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
March TH, Kolar LM, Barr EB, Finch GL,
Ménache MG and Nikula KJ: Enhanced pulmonary epithelial replication
and axial airway mucosubstance changes in F344 rats exposed
short-term to mainstream cigarette smoke. Toxicol Appl Pharmacol.
161:171–179. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhong CY, Zhou YM, Douglas GC, Witschi H
and Pinkerton KE: MAPK/AP-1 signal pathway in tobacco smoke-induced
cell proliferation and squamous metaplasia in the lungs of rats.
Carcinogenesis. 26:2187–2195. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xue L, Lipkin M, Newmark H and Wang J:
Influence of dietary calcium and vitamin D on diet-induced
epithelial cell hyperproliferation in mice. J Natl Cancer Inst.
91:176–181. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ledda-Columbano GM, Perra A, Pibiri M,
Molotzu F and Columbano A: Induction of pancreatic acinar cell
proliferation by thyroid hormone. J Endocrinol. 185:393–399. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bryant KL, Mancias JD, Kimmelman AC and
Der CJ: KRAS: Feeding pancreatic cancer proliferation. Trends
Biochem Sci. 39:91–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song SY, Gannon M, Washington MK, Scoggins
CR, Meszoely IM, Goldenring JR, Marino CR, Sandgren EP, Coffey RJ
Jr, Wright CV and Leach SD: Expansion of Pdx1-expressing pancreatic
epithelium and islet neogenesis in transgenic mice overexpressing
transforming growth factor alpha. Gastroenterology. 117:1416–1426.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Egberts JH, Cloosters V, Noack A,
Schniewind B, Thon L, Klose S, Kettler B, von Forstner C, Kneitz C,
Tepel J, et al: Anti-tumor necrosis factor therapy inhibits
pancreatic tumor growth and metastasis. Cancer Res. 68:1443–1450.
2008. View Article : Google Scholar : PubMed/NCBI
|