Introduction
Mammary serine protease inhibitor (maspin), encoded
by the serpin family B member 5 gene, belongs to the serine
protease inhibitor superfamily of proteins (1). Several studies have revealed that maspin
is an effective inhibitor of cancer cell invasion, metastasis and
angiogenesis (2). Maspin was
originally identified in normal mammary epithelial cells, and is
reduced or absent in carcinoma (3).
The induction of maspin gene expression in carcinoma cell lines
leads to an inhibition of cell invasion and metastasis in
vitro and in vivo (4).
Therefore, maspin possesses potential as a target for the prognosis
and diagnosis of and therapeutic intervention against cancer.
The regulation of maspin expression in cancer cells
is tissue-specific. Maspin was originally identified as a tumor
suppressor due to its high expression level in normal breast and
prostate, and low or absent expression levels in malignancies
(5,6).
Paradoxically, the expression of maspin is maintained during
carcinogenesis in a number of tissue types, including ovarian, lung
and pancreatic tissues (7–9). Additionally, the overexpression of
maspin has also been detected in inflammatory bowel disease
(10), and a high incidence of
aberrant maspin expression is associated with intestinal metaplasia
and carcinoma of the gall bladder (11).
The growth and invasion of transformed cells in
tumors are often accompanied by inflammation, as immune cells and
macrophages are recruited to the tumor site and release
pro-inflammatory cytokines, including interleukin-6 (IL-6),
transforming growth factor β1 (TGF-β1) and tumor necrosis factor α
(TNF-α) (12). A number of
inflammatory cytokines exert tumor suppressive properties against
cells at the early stages of tumorigenesis, but become tumor
inducers at the later stages of cancer progression (13). Depending upon the stage of
carcinogenesis, the effects of these cytokines vary according to
the signaling pathways activated. TGF-β1 stimulates signaling
responses via mothers against decapentaplegic homolog (Smad) and
non-Smad signaling pathways (14,15). Upon
activation by TGF-β1, Smad2 and Smad3 become phosphorylated and
form complexes with Smad4, which in turn regulate the transcription
of target genes. In the latter pathway, TGF-β1 signaling can occur
via several adapter proteins, such as p38 mitogen-activated protein
kinase (p38 MAPK), Erk MAP kinases, phosphoinositide-3-kinase
(PI3Kinase)/Akt, and c-Jun N-terminal kinases (JNK) (15).
Previous research has suggested that TGF-β1 serves a
role in several processes of carcinogenesis, including invasion,
migration, mesenchymal transition and extracellular matrix
synthesis, in a number of cancer cell types (16). Therefore, changes in maspin expression
may be associated with the inflammatory responses mediated by
TGF-β1, leading to the progression from hyperplasia to neoplasia.
Overexpression of TGF-β1 within the tumor microenvironment may
increase the metastatic potential of various types of tumor
(17).
Maspin promoters contain Smad- and p53-binding
elements, which are required for the upregulation of the maspin
gene by TGF-β1 in normal mammary epithelial cells (18). In the present study, the effects of
TGF-β1 and the pro-inflammatory cytokines TNF-α and IL-6 on maspin
expression in human cervical HeLa and oral squamous carcinoma HSC4
cell lines were investigated.
Materials and methods
Cell cultures
The human cervical carcinoma HeLa and oral squamous
carcinoma HSC4 cell lines were provided by the Institute of
Biotechnology and Genetic Engineering, Chulalongkorn University
(Bangkok, Thailand) and Associate Professor R. Surarit of Mahidol
University (Bangkok, Thailand), respectively. The cell cultures
were maintained in Dulbecco's modified Eagle's medium (DMEM;
Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% heat-inactivated fetal bovine serum (FBS;
Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) and 1X
antibiotic-antimycotic solution (Invitrogen; Thermo Fisher
Scientific, Inc.) at 37°C in an atmosphere of 5%
CO2.
Reverse transcription-quantitative
polymerase chain reaction (qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). A total of 1 µg RNA
was treated with DNase (Invitrogen; Thermo Fisher Scientific, Inc.)
and then the half amount of DNase-treated RNA was converted into
complementary DNA (cDNA) using oligo-(dT)18 and a
RevertAid™ First Strand cDNA Synthesis kit (Fermentas;
Thermo Fisher Scientific, Inc.). The reverse transcription reaction
was carried out at 42°C for 60 min. qPCR was then performed in an
ABI 7500 Real-time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Each reaction mixture contained 5 µl cDNA
(diluted 1:5), 10 µl 2X Maxima™ SYBR-Green/ROX qPCR
Master Mix (Fermentas; Thermo Fisher Scientific, Inc.) and 0.3 µM
primer pairs for maspin or GAPDH (internal control). The primer
pairs were as follows: Maspin forward,
5′-CGTAGAAAACTAATCAAGCGGCTCTAG-3′; maspin reverse,
5′-CCAATTCCTTTGCATAGGGTCTC-3′; GAPDH forward,
5′-CGTTGGGTGAAGGTCGGAGTCAAG-3′; and GAPDH reverse,
5′-GGCAACAATATCCACTTTACCAGA-3′. The thermocycling conditions were
as follows: 95°C for 10 min; and 45 cycles of 95°C for 15 sec and
60°C for 60 sec. Relative maspin expression levels were normalized
to GAPDH and calculated using the 2-ΔΔCq method
(19). All experiments were performed
3 times with 3 replicates per experiment.
Treatment of cell lines with cytokines
and inhibitors
The cytokines TGF-β1, TNF-α and IL-6 (Roche
Diagnostics; Indianapolis, IN, USA) were added at 0.1, 1, or 10
ng/ml to cancer cells in serum-free DMEM and incubated at 37°C in
an atmosphere of 5% CO2 for 48 h or as indicated. Smad3
inhibitor (SIS3; Merck KGaA, Billerica, MA, USA; 1, 5 or 10 µM),
100 µM pyrrolidine dithiocarbamate (PDTC; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany), 1 µM wortmannin (Sigma-Aldrich; Merck
KGaA), 10 µM SP600125 (Sigma-Aldrich; Merck KGaA), 10 µM U0126
(Sigma-Aldrich; Merck KGaA) or 10 µM SB202190 (Merck KGaA) were
incubated with confluent HeLa cells for 1 h at 37°C in a 5%
CO2 atmosphere. A total of 10 ng/ml TGF-β1 was then
added and the cells were incubated for an additional 48 h.
Western blotting
Subsequent to the aforementioned treatments, cells
were washed twice with cold PBS, lysed with a Mammalian Protein
Extraction buffer (GE Healthcare Life Sciences, Piscataway, NJ,
USA) containing a protease and phosphatase inhibitor cocktail
(Roche Diagnostics), and centrifuged at 14,000 × g for 10 min at
4°C. A total of 10 µg of protein, measured using a Bio-Rad™ Protein
Assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA), were
separated by 10% SDS-PAGE, then transferred onto a nitrocellulose
membrane. The blot was incubated with 5% non-fat dried milk in Tris
buffer saline (TBS) with 0.01% Tween-20. Membranes were then
treated with 0.5 µg/ml mouse monoclonal anti-human maspin (cat. no.
554292; BD Biosciences, San Jose, CA, USA), rabbit monoclonal
anti-Smad2 (cat. no. #3122; Cell Signaling Technology, Inc.,
Danvers, MA, USA; dilution 1:1,000), rabbit monoclonal
anti-phospho-Smad2 (cat. no. #8828; Cell Signaling Technology,
Inc.; dilution, 1:2,000), mouse polyclonal anti-p38
mitogen-activated protein kinase (cat. no. #9212; Cell Signaling
Technology, Inc.; dilution, 1:2,000), or mouse polyclonal
anti-phospho-p38 mitogen-activated protein kinase (p38 MAPK; cat.
no. #9211; Cell Signaling Technology, Inc.; dilution, 1:2,000)
primary antibodies. Subsequently, the membranes were incubated with
horseradish peroxidase-conjugated goat anti-mouse (cat. no.
1706516; dilution, 1:7,500; Bio-Rad Laboratories, Inc.) or goat
anti-rabbit immunoglobulin G (cat. no. 1706515; dilution, 1:7500;
Bio-Rad Laboratories, Inc.) secondary antibodies, and
immunoreactive protein bands were visualized using Western
Lightning® Plus-ECL substrate (PerkinElmer, Inc.,
Waltham, MA, USA). For normalization of protein loading, the blots
were then stripped and re-probed with primary rabbit polyclonal
anti-human actin antibodies (cat. no. #A2066; Sigma-Aldrich; Merck
KGaA; dilution, 1:1,000) as described previously. All experiments
were performed 3 times with 3 replicates per experiment.
Transfection and luciferase reporter
assay
A full-length maspin promoter in a luciferase
reporter plasmid (pLightSwitch; Active Motif, Carlsbad, CA, USA)
was used to generate two 5′ truncated maspin promoters (−600 maspin
promoter and −300 maspin promoter) in a pLightSwitch luciferase
reporter plasmid by a PCR-based method (20). Point mutations in two p53-binding
sites, p53 I and p53 II, in the maspin promoter region were
constructed using the QuickChange II Site-Directed Mutagenesis kit
(Stratagene, La Jolla, CA, USA) and a pLightSwitch plasmid
containing −300 maspin promoter as the template. A pair of primers,
5′-GCCCCTTCCTGCCCtatatacaacGAGGCCTTTTGGAAGC-3′ and
5′-GCTTCCAAAAGGCCTCgttgtatataGGGCAGGAAGGGGC-3′ (mutated nucleotides
illustrated in lowercase letters), were used to introduce sequence
substitution at the p53 I binding site. The primers for mutation of
p53 II binding site were
5′-GCCGAGAGGATTGCCGTAatataGTCTGTACGTATGCATG-3′ and
5′-CATGCATACGTACAGACtatatTACGGCAATCCTCTCGGC-3′. The correct changes
to the mutated sequences were confirmed by DNA sequencing at First
BASE Laboratories (Selangor, Malaysia). Each construct was
transfected into the HeLa cells using TurboFect™ in
vitro Transfection Reagent (Fermentas; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. After 4
h transfection, the medium was replaced with serum free-DMEM and
the cells were incubated for a further 2 h. The cells were then
incubated with 10 ng/ml TGF-β1, or without TGF-β1 as control, at
37°C in a 5% CO2 atmosphere for 48 h. Luciferase
activity was determined by adding LightSwitch Luciferase Assay
Reagent™ (Active Motif) according to manufacturer's
protocol, and measuring the luminescence in a Synergy™
HT Multi-Detection microplate reader (BioTek Instruments, Inc.,
Winooski, VT, USA). The results were recorded as the fold of the
induction of the reporter plasmid in the presence of TGF-β1,
subsequent to normalization with the transfection controls without
TGF-β1 treatment.
In vitro invasion assay
The in vitro invasion assay was performed
using polycarbonate 8 µM-pore Multiscreen MIC 96-well pre-coated
with 5 µg/ml Matrigel (BD Biosciences). A total of 5×104 HeLa
cells/well were pre-treated for 24 h with cytokines in serum-free
DMEM and then transferred to the upper wells of the filter plate.
DMEM culture medium containing 10% FBS was added to the lower
chambers. After 48 h incubation at 37°C in a 5% CO2
incubator, the number of invaded cells in the bottom wells were
measured using a CyQUANT® Cell Proliferation Assay kit
(Molecular Probes; Thermo Fisher Scientific, Inc.); fluorescence
(excitation at 450 nm and emission at 530 nm) was measured using a
Biotech K-40 spectrofluorometer (BioTek Instruments). Cells in
triplicate wells without Transwell inserts served as controls for
cell proliferation and/or death during the incubation period. Cell
invasion was calculated from the fluorescence values as follows:
Cell invasion=Relative fluorescence value of invaded cells/total
cells plated in upper chamber without Transwell insert.
Statistical analysis
All experiments were performed at least 3 times with
3–5 replicates per experiment. The differences in the mean values
among the groups were determined by one-way analysis of variance,
and differences between individual by a Dunnett post-hoc test using
SPSS v.18.0 software (SPSS Inc., Chicago, IL, USA). Data are
presented as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of pro-inflammatory cytokines
on maspin expression in HeLa and HSC4 cell lines
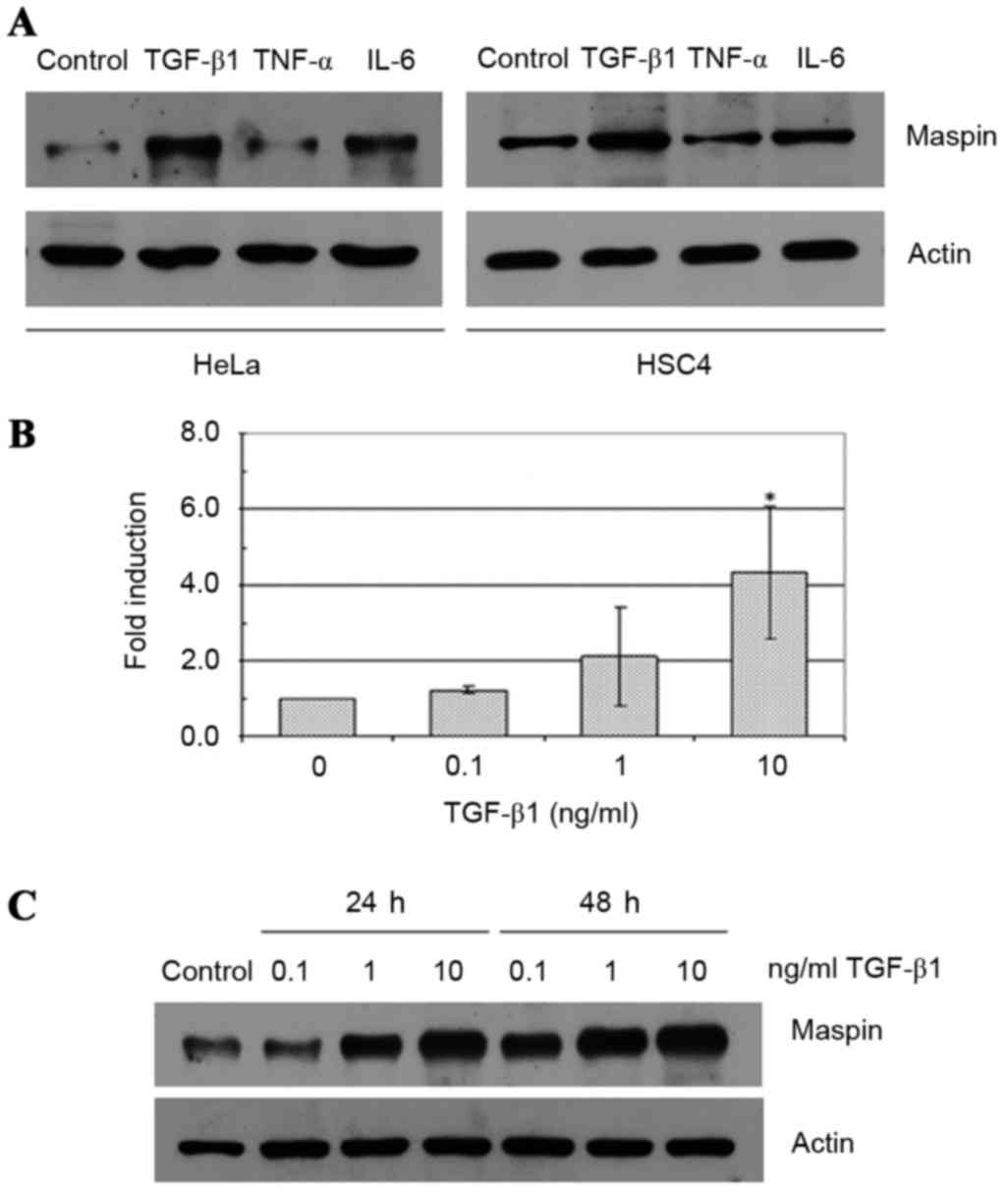
The effects of TGF-β1, TNF-α and IL-6 on maspin gene
expression were evaluated in two human cancer cell lines: HeLa
cervical carcinoma cells and HSC4 oral squamous cell carcinoma
cells. TGF-β1 treatment at a concentration of 10 ng/ml induced an
increase in the expression levels of maspin in HeLa and HSC4 cells
(Fig. 1A). By contrast, 10 ng/ml IL-6
exhibited no effect, and 10 ng/ml TNF-α demonstrated a slight
inhibitory effect on maspin levels in the two cell lines. In the
HeLa cells, 0.1–10 ng/ml TGF-β1 treatment increased maspin gene
expression levels at 24 and 48 h. in a dose-dependent fashion
compared with control cells, and this effect was demonstrated at
the mRNA (Fig. 1B) and protein levels
(Fig. 1C). Similar effects were
observed with HSC4 cells (data not shown).
Convergence of Smad and non-Smad
signaling pathways in TGF-β1-induced maspin gene expression in HeLa
cells
As expected, TGF-β1-treated HeLa cells produced
phospho-Smad2, and the presence of SIS3, a specific inhibitor of
Smad3 (21), inhibited the induction
of maspin gene expression in a dose-dependent manner (Fig. 2A). It is notable that the inhibition
of TGF-β1 signaling via the Smad pathway by SIS3 did not interfere
with the non-Smad pathway, as demonstrated by the presence of
phospho-p38 MAPK (Fig. 2A). In order
to determine whether the non-Smad signaling pathway was also
activated by TGF-β1, a series of kinase inhibitors, including PDTC
(a nuclear factor kB inhibitor), wortmannin (a PI3K inhibitor),
SP600125 (a JNK inhibitor), U0126 (a MEK1/2 inhibitor) and SB202190
(a p38 MAPK inhibitor), were employed. U0126 and SB202190 were
effective in inhibiting the TGF-β1-induced maspin gene expression,
whereas PDTC, wortmannin or SP600125 were not (Fig. 2B). Inhibition by U0126 of MEK1/2, an
upstream activator of p38 MAPK, resulted in an absence of p38 MAPK
phosphorylation, whereas SB202190 exhibited no effect on p38 MAPK
phosphorylation, as the inhibitor only binds to the ATP pocket of
p38 and blocks its intrinsic ATPase activity (22). Additionally, TGF-β1-dependent Smad2
phosphorylation was not affected by the presence of these kinase
inhibitors (Fig. 2B). Taken together,
these data suggest that Smad and non-Smad signaling pathways are
involved in TGF-β1-induced maspin gene expression in HeLa cells.
Notably, the blockage of TGF-β1-induced maspin expression was
achieved by inhibiting one pathway without affecting the activation
of the other.
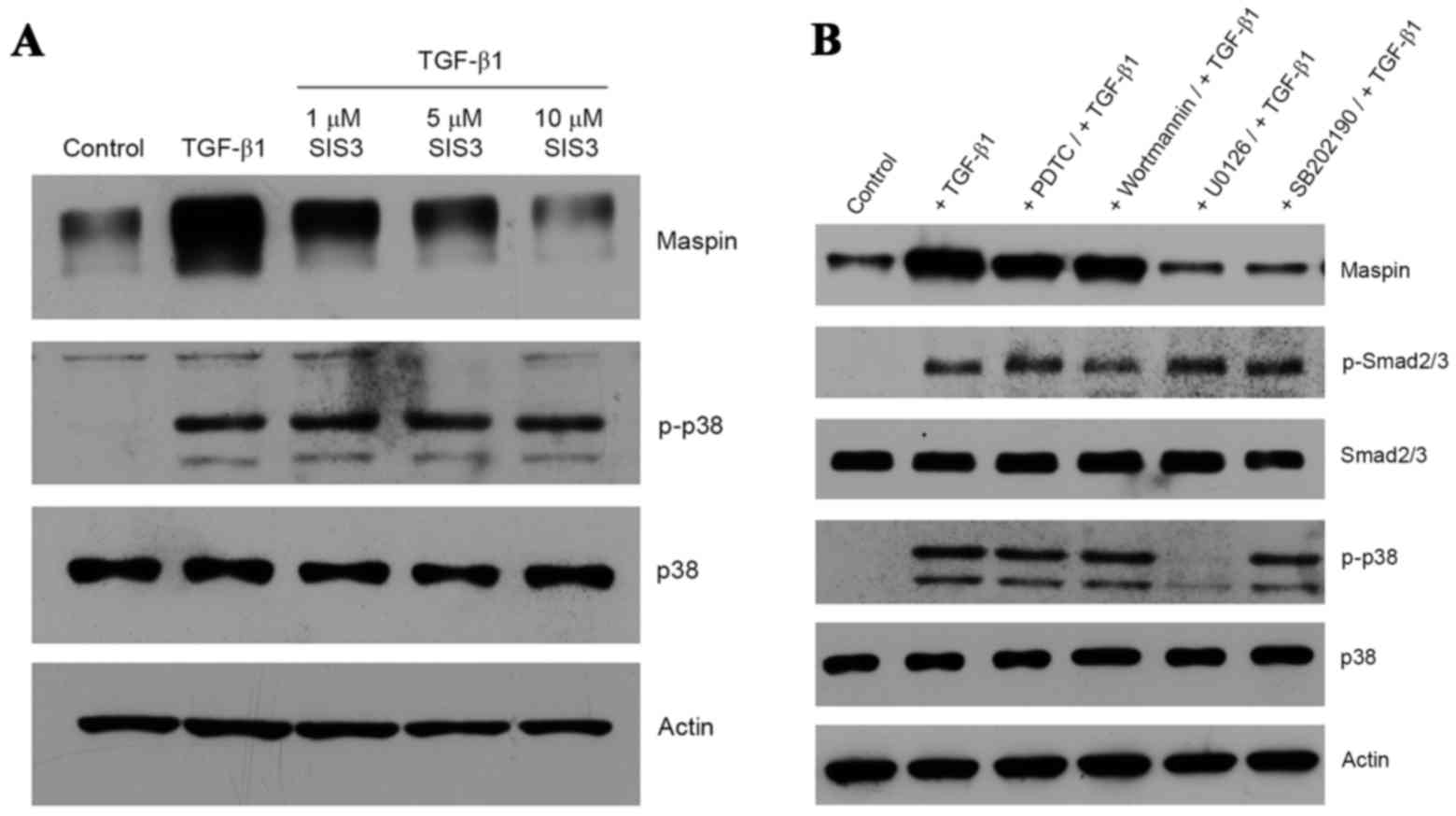 | Figure 2.Induction of maspin expression through
Smad and non-Smad signaling pathways. (A) Cultures of HeLa cells
were pre-treated with 1, 5 or 10 µM SIS3 prior to the addition of
10 ng/ml TGF-β1 and a 48-h incubation. Maspin, p38 and p-p38
proteins were detected by western blotting. (B) HeLa cells were
incubated with 100 µM PDTC, 1 µM wortmannin, 10 µM SP600125, 10 µM
U0126 or 10 µM SB202190 for 1 h at 37°C prior to the addition of 10
ng/ml TGF-β1, and then further incubated for 48 h. Maspin, p-Smad2,
Smad2, p-p38 and p38 were detected by western blotting. Actin was
used for normalization of gel loading. Smad, mothers against
decapentaplegic homolog; SIS3, Smad3 inhibitor; TGF-β1,
transforming growth factor β1; p-p38, phospho-p38; p-Smad2,
phospho-Smad2; PDTC, pyrrolidine dithiocarbamate. |
TGF-β1-induced maspin gene expression
in HeLa cells requires p53-binding sites in the maspin
promoter
The maspin promoter region contains two p53-binding
sites, p53 I and p53 II, which are located within the first
upstream 300 nucleotides (nt) (Fig.
3A). Using luciferase reporter plasmid constructs, which
contain point mutations in maspin promoter p53-binding sites that
have been previously demonstrated to abolish p53 protein binding
(18), transfection of HeLa cells and
subsequent addition of TGF-β1 revealed that mutations in either the
p53 I or p53 II binding sites significantly diminished the ability
of TGF-β1 to induce luciferase activity compared with control cells
transfected with a construct containing the full-length maspin
promoter sequence (nt −872 to +193) (Fig.
3B). Therefore, the two p53-binding sites in the maspin
promoter are necessary and probably sufficient in HeLa cells for
TGF-β1-induced maspin gene expression, as the luciferase expression
plasmid containing 5′ truncated maspin promoter element (from nt
−600 or −300 to −872) expressed luciferase activity comparable with
the cells containing the full length maspin promoter sequence
(Fig. 3B).
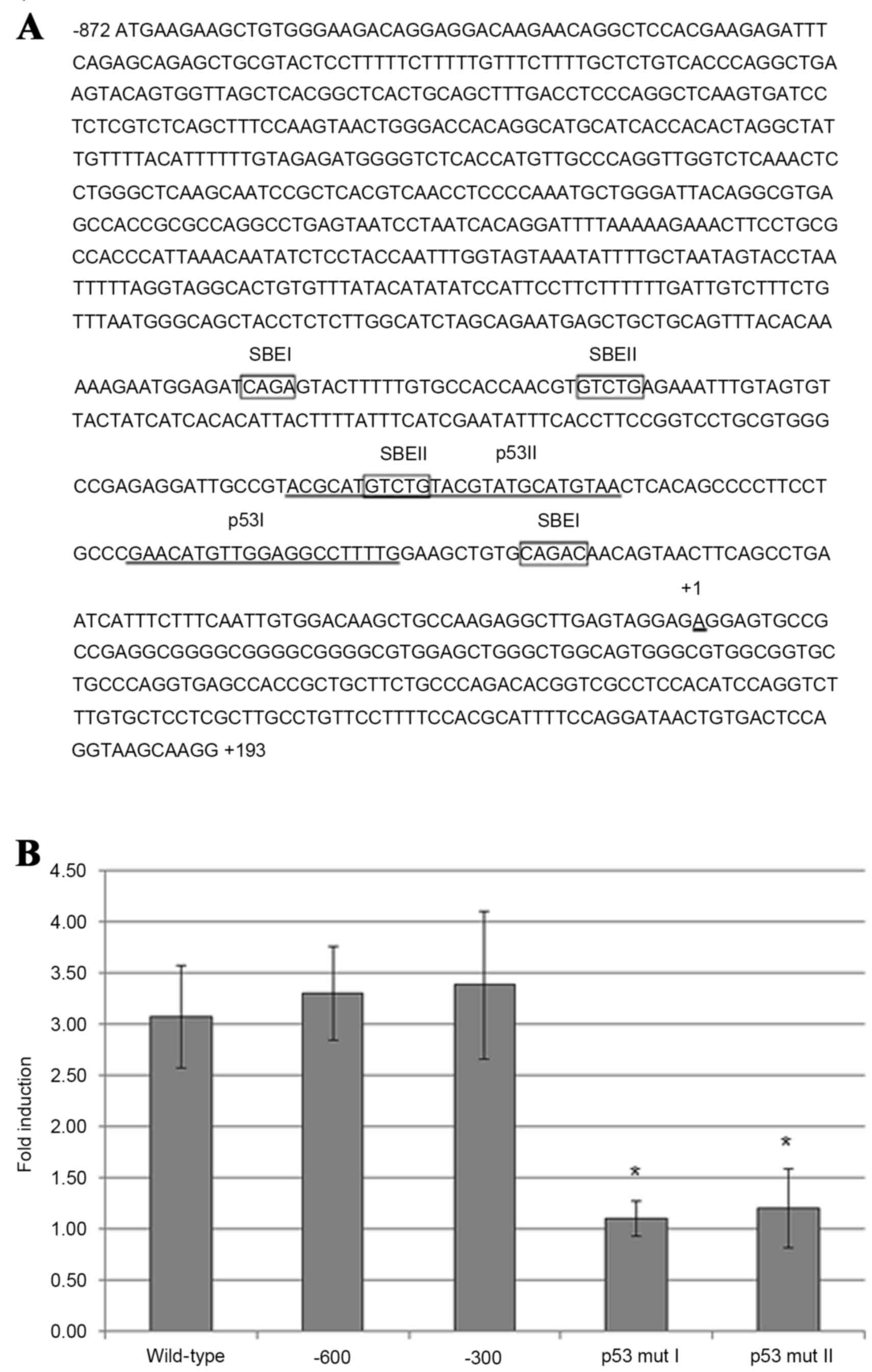 | Figure 3.Requirement of p53-binding sites in
maspin promoter for maspin induction by TGF-β1 in carcinoma cells.
(A) Maspin promoter sequence ranging from nucleotides −872 to +193
(wild-type), with putative Smad-binding elements (SBEI and SBEII;
boxed), and the two binding sites for p53 (p53 I and p53 II;
underlined). (B) HeLa cells were transfected with a luciferase
reporter plasmid containing wild-type maspin promoter, two 5′
truncated (−600 and −300) maspin promoters, or −300 maspin promoter
with point mutations in p53-binding sites, p53 mut I and p53 mut
II. After 4 h transfection, the medium was replaced with serum-free
Dulbecco's modified Eagle's medium and, after a further 2 h, 10
ng/ml TGF-β1 was added. After 48 h, the cells were harvested and
measured by a luciferase reporter assay. Results are presented as
the mean fold induction compared with the control without TGF-β1
treatment; errors bars represent standard deviation. *P<0.05 vs.
−300 promoter-transfected cells. TGF-β1, transforming growth factor
β1; p53, tumor protein p53. |
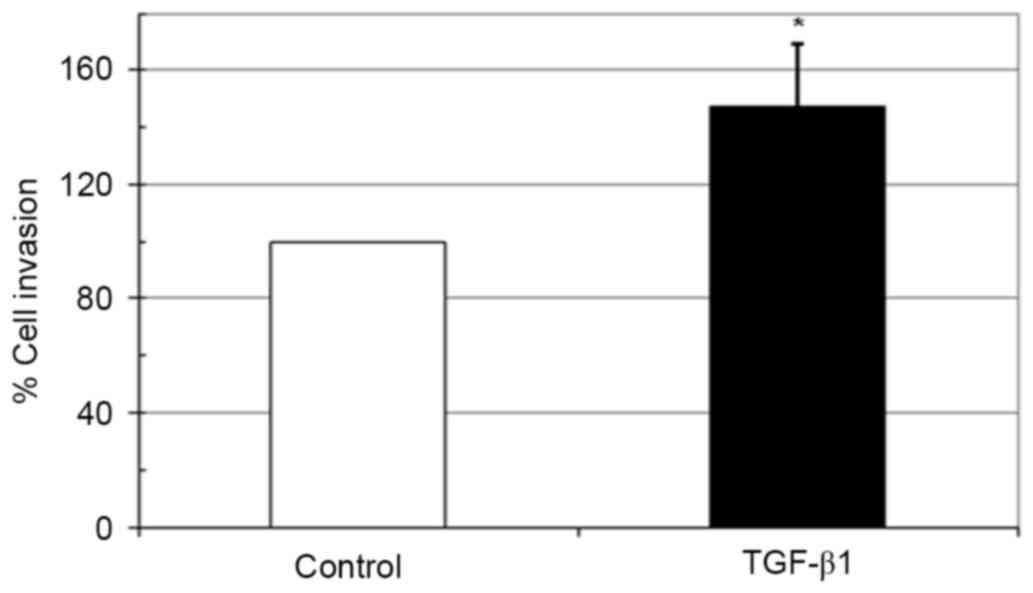
Effect of TGF-β1 on in vitro invasion
of HeLa cells
The biological activities of maspin involve the
inhibition of carcinoma cell migration and invasion (23). As incubation with TGF-β1 upregulated
the expression of maspin in HeLa cells, whether this inhibits the
invasive ability of HeLa cells was examined using a Matrigel
invasion assay. Notably, 10 ng/ml TGF-β1 significantly increased
the invasiveness of HeLa cells compared with the untreated control
(Fig. 4).
Discussion
The present study demonstrated that TGF-β1, but not
TNF-α or IL-6, induced maspin gene expression at the mRNA and
protein levels in cervical carcinoma HeLa and oral squamous
carcinoma HSC4 cell lines. In the HeLa cells, TGF-β1 was able to
induce maspin expression through the Smad-dependent and
-independent pathways.
Wang et al (18) suggested that the TGF-β1 induction of
maspin gene transcription, which occurs within 1 h following the
addition of TGF-β1, acts exclusively through the Smad signaling
pathway and depends on p53 and Smad2/3 binding to the maspin
promoter in normal mammary epithelial cells. However, in their
study, p53 binding to maspin promoter was detectable for 8 h in the
absence of bound Smad2/3, suggesting the possibility of a
Smad-independent TGF-β1-induced signaling pathway of p53-dependent
activation of maspin expression. Notably, one observation of the
present study in HeLa cells suggested the requirement of both Smad2
and p53 promoter binding for the induction of maspin gene
expression by TGF-β1 via the non-Smad pathway. This hypothesis is
concomitant with a crosstalk between the TGF-β1 signaling pathway
and p53, which has been demonstrated previously (24). Phosphorylated wild-type p53 physically
interacts with the phospho-Smad2/3-Smad4 complex upon TGF-β1
induction and in turn binds to promoters of tumor suppressive genes
(25).
The non-Smad pathway acts via MEK1/2 and p38 MAPK.
MEK1/2 is an upstream signaling molecule of p38 MAPK activation
(26). In the present study, the
inhibition of MEK1/2 and p38 MAPK in HeLa cells prevented the
stimulatory effect of TGF-β1 on maspin expression without affecting
Smad phosphorylation. The phosphorylation of p53 at Ser389 by p38
MAPK increases its DNA-binding capability (27). Cordenonsi et al (28) revealed that, in H1299 human lung
cancer cells, MEK1/2 inhibition by U0126 blocks the phosphorylation
of p53, which is concomitant with the loss of TGF-β1-induced p21
expression. The inhibition of the Smad-dependent TGF-β1 signaling
pathway by SIS3 in the present study did not interfere with the
non-Smad pathway, as demonstrated by the presence of phospho-p38
MAPK. This is consistent with previous data suggesting that
TGF-β1-mediated activation of p38 MAPK signaling is independent of
TGF-β1 type I receptor-mediated Smad activation (29). Therefore, it is possible that the
non-Smad TGF-β1 pathway of maspin gene expression induction is
mediated by MEK1/2 and p38 MAPK phosphorylation of p53, which
promotes p53 binding to maspin promoter.
Hypermethylation of the maspin promoter contributes
to maspin silencing in cancer cells (30). Previously, TGF-β1 was not observed to
have an effect on maspin expression in a number of cancer cell
lines, including the mammary carcinoma cell line MDA-MB-231 which
has mutant p53 and maspin promoter hypermethylation (18). In maspin-expressing transformed cells,
including MCF10A, the maspin promoter is hypomethylated (31). The present study also identified
hypomethylated maspin promoters in HeLa cells (data not shown).
TGF-β1 may induce the migration and invasion of
several types of carcinoma cells via the activation of the PI3K and
Akt pathway (32). In the present
study, a similar event, in which TGF-β1 stimulated the migration
and invasion of HeLa cells, was observed. Although maspin has been
demonstrated to inhibit cancer cell motility and invasiveness
(33), in HeLa cells the
TGF-β1-induced increase in maspin levels was apparently not
sufficient to attenuate the cancer cell migration and invasion
properties mediated by TGF-β1. Despite the tumor suppressive
activities of maspin in numerous types of cancer cells, a
paradoxical increase of maspin expression has been identified in
several malignant cell types compared with their normal cells of
origin, including those from lung (34), bladder (35), and ovarian tissues (36). An upregulation of maspin expression is
also associated with the advanced stages of several types of
tumors, including tumors of the cervix (37), endometrium (38), and pancreas (9). In addition, the subcellular localization
of maspin may serve a critical role in its biological function. For
example, the nuclear localization in cancer cells is essential for
the tumor suppressor activity of maspin (39). In addition, an increased cytoplasmic
localization of maspin was revealed in an invasive SKOV3 cell line,
but any tumor suppressive activity may have been inactivated, as
the invasion capabilities were not affected by a blocking antibody
(7). Therefore, TGF-β1-induced maspin
expression may not be able to inhibit tumor invasion and may serve
a role in cancer progression.
In conclusion, the present study demonstrated the
increased expression of maspin induced by TGF-β1 in human cervical
carcinoma HeLa and oral squamous carcinoma HSC4 cell lines. The
results indicated that the upregulation of maspin expression was
due to Smad and non-Smad MEK/MAPK TGF-β1 signaling pathways, acting
independently but converging to promote p53 binding to the maspin
promoter. However, the underlying molecular mechanisms of this
phenomenon require additional study, and may provide supporting
evidence for an association between the inflammatory response and
cancer progression, which may lead to the development of novel
cancer prevention and treatment strategies.
Acknowledgements
The present study was supported by grants from the
National Research Council of Thailand (grant no. 48499), the
Faculty of Medicine Research Fund of Chiang Mai University (grant
no. 077/2558), the Office of the Higher Education Commission and
Mahidol University under the Thailand National Research
Universities Initiative and the Faculty of Dentistry of Mahidol
University. The authors would like to thank Professor Prapon
Wilairat (Faculty of Science, Mahidol University) for critical
reading of the manuscript and useful comments.
References
|
1
|
Silverman GA, Bird PI, Carrell RW, Church
FC, Coughlin PB, Gettins PG, Irving JA, Lomas DA, Luke CJ, Moyer
RW, et al: The serpins are an expanding superfamily of structurally
similar but functionally diverse proteins. Evolution, mechanism of
inhibition, novel functions, and a revised nomenclature. J Biol
Chem. 276:33293–33296. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bodenstine TM, Seftor RE, Khalkhali-Ellis
Z, Seftor EA, Pemberton PA and Hendrix MJ: Maspin: Molecular
mechanisms and therapeutic implications. Cancer Metastasis Rev.
31:529–551. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zou Z, Anisowicz A, Hendrix MJ, Thor A,
Neveu M, Sheng S, Rafidi K, Seftor E and Sager R: Maspin, a serpin
with tumor-suppressing activity in human mammary epithelial cells.
Science. 263:526–529. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Beltran A, Parikh S, Liu Y, Cuevas BD,
Johnson GL, Futscher BW and Blancafort P: Re-activation of a
dormant tumor suppressor gene maspin by designed transcription
factors. Oncogene. 26:2791–2798. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maass N, Nagasaki K, Ziebart M, Mundhenke
C and Jonat W: Expression and regulation of tumor suppressor gene
maspin in breast cancer. Clin Breast Cancer. 3:281–287. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zou Z, Zhang W, Young D, Gleave MG, Rennie
P, Connell T, Connelly R, Moul J, Srivastava S and Sesterhenn I:
Maspin expression profile in human prostate cancer (CaP) and in
vitro induction of Maspin expression by androgen ablation. Clin
Cancer Res. 8:1172–1177. 2002.PubMed/NCBI
|
|
7
|
Bauerschlag DO, Habermann M, Weimer J,
Meinhold-Heerlein I, Hilpert F, Weigel M, Bauer M, Mundhenke C,
Jonat W, Maass N and Schem C: Heterogeneous expression of serine
protease inhibitor maspin in ovarian cancer. Anticancer Res.
30:2739–2744. 2010.PubMed/NCBI
|
|
8
|
Frey A, Soubani AO, Adam AK, Sheng S, Pass
HI and Lonardo F: Nuclear, compared with combined nuclear and
cytoplasmic expression of maspin, is linked in lung adenocarcinoma
to reduced VEGF-A levels and in Stage I, improved survival.
Histopathology. 54:590–597. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu H, Shi J, Anandan V, Wang HL, Diehl D,
Blansfield J, Gerhard G and Lin F: Reevaluation and identification
of the best immunohistochemical panel (pVHL, Maspin, S100P, IMP-3)
for ductal adenocarcinoma of the pancreas. Arch Pathol Lab Med.
136:601–609. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cao D, Wilentz RE, Abbruzzese JL, Ho L and
Maitra A: Aberrant expression of maspin in idiopathic inflammatory
bowel disease is associated with disease activity and neoplastic
transformation. Int J Gastrointest Cancer. 36:39–46. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim J, Jang KT, Kim KH, Park JW, Chang BJ,
Lee KH, Lee JK, Heo JS, Choi SH, Choi DW, et al: Aberrant maspin
expression is involved in early carcinogenesis of gallbladder
cancer. Tumour Biol. 31:471–476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dinarello CA: The paradox of
pro-inflammatory cytokines in cancer. Cancer Metastasis Rev.
25:307–313. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gohji K, Nomi M, Hara I, Arakawa S and
Kamidono S: Influence of cytokines and growth factors on matrix
metalloproteinase-2 production and invasion of human renal cancer.
Urol Res. 26:33–37. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Heldin CH and Moustakas A: Role of Smads
in TGFb signaling. Cell Tissue Res. 347:21–36. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mu Y, Gudey SK and Landström M: Non-Smad
signaling pathways. Cell Tissue Res. 347:11–20. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Arteaga CL, Dugger TC and Hurd SD: The
multifunctional role of transforming growth factor (TGF)-beta s on
mammary epithelial cell biology. Breast Cancer Res Treat. 38:49–56.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Drabsch Y and ten Dijke P: TGF-b signaling
in breast cancer cell invasion and bone metastasis. J Mammary Gland
Biol Neoplasia. 16:97–108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang SE, Narasanna A, Whitell CW, Wu FY,
Friedman DB and Arteaga CL: Convergence of p53 and transforming
growth factor beta (TGFbeta) signaling on activating expression of
the tumor suppressor gene maspin in mammary epithelial cells. J
Biol Chem. 282:5661–5669. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jung V, Pestka SB and Pestka S: Cloning of
polymerase chain reaction-generated DNA containing terminal
restriction endonuclease recognition sites. Methods Enzymol.
218:357–362. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jinnin M, Ihn H and Tamaki K:
Characterization of SIS3, a novel specific inhibitor of Smad3, and
its effect on transforming growth factor-beta1-induced
extracellular matrix expression. Mol Pharmacol. 69:597–607. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kumar S, Jiang MS, Adams JL and Lee JC:
Pyridinylimidazole compound SB 203580 inhibits the activity but not
the activation of p38 mitogen-activated protein kinase. Biochem
Biophys Res Commun. 263:825–831. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bailey CM, Khalkhali-Ellis Z, Seftor EA
and Hendrix MJ: Biological functions of maspin. J Cell Physiol.
209:617–624. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Elston R and Inman GJ: Crosstalk between
p53 and TGF-beta Signalling. J Signal Transduct. 2012:2940972012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cordenonsi M, Dupont S, Maretto S, Insinga
A, Imbriano C and Piccolo S: Links between tumor suppressors: p53
is required for TGF-beta gene responses by cooperating with Smads.
Cell. 113:301–314. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Poddar R and Paul S: Novel crosstalk
between ERK MAPK and p38 MAPK leads to homocysteine-NMDA
receptor-mediated neuronal cell death. J Neurochem. 124:558–570.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang C, Ma WY, Maxiner A, Sun Y and Dong
Z: p38 kinase mediates UV-induced phosphorylation of p53 protein at
serine 389. J Biol Chem. 274:12229–12235. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cordenonsi M, Montagner M, Adorno M,
Zacchigna L, Martello G, Mamidi A, Soligo S, Dupont S and Piccolo
S: Integration of TGF-beta and Ras/MAPK signaling through p53
phosphorylation. Science. 315:840–843. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu L, Hebert MC and Zhang YE: TGF-beta
receptor-activated p38 MAP kinase mediates Smad-independent
TGF-beta responses. EMBO J. 21:3749–3759. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oshiro MM, Watts GS, Wozniak RJ, Junk DJ,
Munoz-Rodriguez JL, Domann FE and Futscher BW: Mutant p53 and
aberrant cytosine methylation cooperate to silence gene expression.
Oncogene. 22:3624–3634. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Horswill MA, Narayan M, Warejcka DJ,
Cirillo LA and Twining SS: Epigenetic silencing of maspin
expression occurs early in the conversion of keratocytes to
fibroblasts. Exp Eye Res. 86:586–600. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vo BT, Morton D Jr, Komaragiri S, Millena
AC, Leath C and Khan SA: TGF-b effects on prostate cancer cell
migration and invasion are mediated by PGE2 through activation of
PI3K/AKT/mTOR pathway. Endocrinology. 154:1768–1779. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ngamkitidechakul C, Warejcka DJ, Burke JM,
O'Brien WJ and Twining SS: Sufficiency of the reactive site loop of
maspin for induction of cell-matrix adhesion and inhibition of cell
invasion. Conversion of ovalbumin to a maspin-like molecule. J Biol
Chem. 278:31796–31806. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yatabe Y, Mitsudomi T and Takahashi T:
Maspin expression in normal lung and non-small-cell lung cancers:
Cellular property-associated expression under the control of
promoter DNA methylation. Oncogene. 23:4041–4049. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Friedrich MG, Toma MI, Petri S, Cheng JC,
Hammerer P, Erbersdobler A and Huland H: Expression of Maspin in
non-muscle invasive bladder carcinoma: Correlation with tumor
angiogenesis and prognosis. Eur Urol. 45:737–743. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rose SL, Fitzgerald MP, White NO, Hitchler
MJ, Futscher BW, De Geest K and Domann FE: Epigenetic regulation of
maspin expression in human ovarian carcinoma cells. Gynecol Oncol.
102:319–324. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nosaka K, Horie Y, Shiomi T, Itamochi H,
Oishi T, Shimada M, Sato S, Sakabe T, Harada T and Umekita Y:
Cytoplasmic maspin expression correlates with poor prognosis of
patients with adenocarcinoma of the uterine cervix. Yonago Acta
Med. 58:151–156. 2015.PubMed/NCBI
|
|
38
|
Torres A, Torres K, Paszkowski T, Radej S,
Staśkiewicz GJ, Ceccaroni M, Pesci A and Maciejewski R: Highly
increased maspin expression corresponds with up-regulation of
miR-21 in endometrial cancer: A preliminary report. Int J Gynecol
Cancer. 21:8–14. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Goulet B, Kennette W, Ablack A, Postenka
CO, Hague MN, Mymryk JS, Tuck AB, Giguère V, Chambers AF and Lewis
JD: Nuclear localization of maspin is essential for its inhibition
of tumor growth and metastasis. Lab Invest. 91:1181–1187. 2011.
View Article : Google Scholar : PubMed/NCBI
|