Introduction
Breast cancer is a common malignancy in women and is
a significant cause of mortality. In spite of treatment, >4,000
individuals succumbed to breast cancer in the US in 2016 (1).
The testis-specific RNA polymerase II elongation
factor (Ell3) is known to increase the oncogenicity of breast
cancer cell lines by regulating the expression of cell cycle
regulators through the ERK signaling pathway and via the induction
of drug resistance through unknown mechanisms (2). The C-terminal domain of Ell3 exhibits
strong similarities to that of the eleven-nineteen lysine-rich
leukemia gene, which acts as a negative regulator of p53 and
regulates cell proliferation and survival (3–5).
Furthermore, Ell3 occupies enhancers in embryonic stem cells and
Ell3 binding to inactive or poised enhancers is essential for stem
cell specification (6).
Lipocalin-2 (LCN2), also known as neutrophil
gelatinase-associated lipocalin, is a member of the lipocalin
protein family and is upregulated in various types of epithelial
cancer, including breast, lung, thyroid, esophageal and pancreatic
duct adenocarcinomas (7–9). LCN2 has been reported to promote drug
resistance and tumor growth, and enhance tumor cell invasion
through its physical association with matrix metalloproteinase-9
(10). The functions of LCN2 during
cancer progression are yet to be fully elucidated. In human breast
cancer, LCN2 expression has been associated with markers of poor
prognosis, including estrogen receptor (ER)-negative status, poor
histological grading and lymph node metastasis, and LCN2 been shown
to be an independent prognostic marker for decreased survival
(11,12). LCN2 has been demonstrated to suppress
apoptosis in thyroid, lung, breast and pancreatic duct
adenocarcinomas (7,13).
The Wnt signaling pathway, named after its most
upstream ligands, the Wnts, is involved in various differentiation
events during embryonic development. Wnt signaling is also
associated with tumor formation (14). In the canonical Wnt signaling cascade,
adenomatous polyposis coli, axin and glycogen synthase kinase (GSK)
constitute the so-called ‘destruction complex’, which controls the
stability of β-catenin (15). In
cells that receive the Wnt signal, GSK is presumed not to
phosphorylate β-catenin. As a consequence, β-catenin accumulates
and forms nuclear complexes with transcription factors (15). In breast cancer, Wnt stimulates tumor
cell motility; conversely, Wnt pathway blockade reduces motility
(16). Furthermore, Wnt signaling has
been demonstrated to promote stem cell activity in mammosphere
assays of mammary gland cells (17).
In lung cancer, Wnt signaling has an essential role
in maintaining highly resistant cancer stem cells, and in
regulating cell cycle, metastasis and apoptosis. Thus, Wnt
antagonists decrease metastasis and induce apoptosis (18). Wnt signaling has also been reported to
be associated with drug resistance (19,20).
Dysregulation of the Wnt/β-catenin pathway was shown to be involved
in pancreatic cancer chemoresistance (19). Drug resistance of colon cancer cells
to 5-fluorouracil (5-FU) and irinotecan treatment has been linked
to Wnt signaling in a previous study (20). These features of Wnt signaling are
associated with apoptosis (21,22). Wnt
signaling regulates the early and late stages of apoptosis during
development and upon cellular injury of neurons, endothelial cells,
vascular smooth muscle cells and cardiomyocytes (20). In human melanoma, inhibition of Wnt-2
signaling, by either a novel monoclonal antibody against human
Wnt-2 ligand or Wnt-2 small interfering RNA (siRNA), downregulated
β-catenin and survivin, and induced apoptosis (22).
Survivin, which is a member of the family of
apoptosis protein inhibitors, functions as a key regulator of
mitosis and programmed cell death (23). Survivin has been identified as a
direct transcriptional target of Wnt/β-catenin (24), which involves the recognition of
discrete T-cell factor-4-binding elements in the survivin promoter.
Functionally, forced expression of non-destructible β-catenin
readily increases survivin levels and supports survivin-mediated
cytoprotection (25).
Resistance to chemotherapy is a major problem facing
current cancer treatment. The mechanisms of resistance to cytotoxic
chemotherapeutics share various features, including alterations in
the drug target, activation of pro-survival pathways and
ineffective induction of cell death (26).
Our group have previously demonstrated that ectopic
expression of Ell3 in breast cancer cell lines induced 5-FU drug
resistance (2). To understand the
underlying mechanism resistance of Ell3 over-expressing MCF-7
breast cancer cell line (Ell3 OE) to 5-FU, the present study
compared gene expression profiles of Ell3 OE cells with wild type
(control) cells after treatment with 5-FU. A number of genes and
signals related to drug resistance were activated in Ell3 OE cells
by treatment with 5-FU. The possible role of Ell3 as an upstream
regulator of these genes and signaling pathways is discussed
herein.
Materials and methods
Cell culture and reagents
MCF-7 cell lines were purchased from American Type
Culture Collection (Manassas, VA, USA). MCF-7 cells were cultured
in Dulbecco's modfiied Eagle medium supplemented with 10% fetal
bovine serum and 1% penicillin/streptomycin (all Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Ell3 OE, which are
Ell3-overexpressing MCF-7 cell lines, were generated by chromosomal
integration of an Ell3 expression plasmid, which was constructed by
cloning PCR-amplified Ell3 cDNA into a modified pcDNA3.1 vector
(Invitrogen; Thermo Fisher Scientific, Inc.) in which the CMV
promoter was replaced by an EF1a promoter. Three independent MCF-7
cell lines were established for Ell3-OE and MCF-7, respectively,
and all experiments were repeated in each cell line to confirm the
results. Nonspecific control siRNAs and siRNAs targeting Ell3 were
purchased from Dharmacon (Lafayette, CO, USA). MCF-7 cells were
transfected with either siRNA or plasmids using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions.
Microarray analysis
Biotinylated cRNAs were prepared from 500 ng total
RNA according to the standard Affymetrix protocol (Affymetrix,
Santa Clara, CA, USA). Following fragmentation, 12 µg aRNA was
hybridized for 16 h at 45°C using a GeneChip Human Genome Array.
GeneChips were washed and stained in the Affymetrix Fluidics
Station 450 then were scanned using the Affymetrix GeneChip Scanner
3000 7G. Data were analyzed via Robust Multi-array Analysis using
Affymetrix default analysis settings and global scaling as the
normalization method. The trimmed mean target intensity of each
array was arbitrarily set to 100. Normalized and log-transformed
intensity values were subsequently analyzed using GeneSpring GX
12.6 (Agilent Technologies, Santa Clara, CA, USA). Fold-change
filters included the requirement that the genes be expressed at
levels at least 150% of control levels for upregulated genes, and
<66% of control levels for downregulated genes. Hierarchical
clustering data were clustered groups that behaved similarly across
experiments using GeneSpring GX 12.6 (Agilent Technologies). The
clustering algorithm used was Euclidean distance at average
linkage.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the MCF-7 cell line
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
and 2 µg total RNA was reverse-transcribed into cDNA using the
SuperScript II First-Strand Synthesis System (Invitrogen; Thermo
Fisher Scientific, Inc.), which contained Primer, Script reverse
transcriptase, RNase inhibitor, deoxynucleotide mixture and
reaction buffer, according to the manufacturer's instructions. qPCR
was performed in triplicate using a Quantitect SYBR Green PCR kit
(Qiagen, Germantown, MD, USA) and CFX96 Real-time System (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) under the following cycling
conditions: 95°C for 30 sec for 35 cycles, 55°C for 30 sec for 40
cycles and 70°C for 30 sec for 30 cycles, respectively. Qiagen 2X
SYBR mix (10 µl), primers (10 pmol; forward and reverse each 1 µl),
cDNA (1 µg) and deionized water (20 µl) were used. For
quantification, target gene expression was normalized to the
expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The
following primer sequences were used: GAPDH forward,
5′-GGGTGTGAACCATGAGAA-3′ and reverse, 5′-GTCTTCTGGGTGGCAGTGAT-3′;
and LCN2 forward, 5′-CCTGGAGACATTGGGGACTTCA-3′ and reverse,
5′-GCCACTGCCTTCATAGTCAAACAC-3′. The data was normalized using the
2−ΔΔCq method (27).
Immunoblot assay
For protein analysis, cells were washed twice with
cold phosphate-buffered saline (PBS) and lysed with tissue lysis
buffer (20 mM Tris-base, pH 7.4, 137 mM NaCl, 2 mM EDTA, 1% Triton
X-100, 25 mM β-glycerophosphate, 2 mM sodium pyrophosphate, 10%
glycerol, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl
fluoride and 1 mM benzamidine). Lysates were centrifuged at 16,600
× g at 4°C for 10 min to remove cellular debris. Whole-cell
extracts were prepared and 50 mg of protein was separated by 10%
SDS-PAGE and transferred to polyvinylidene difluoride membranes
(Bio-Rad Laboratories, Inc.). The membranes were blocked with
blocking solution (5% skim milk in TBST; 50 mM Tris-base, pH 7.4,
0.15 M NaCl and 0.1% Tween-20) for 1 h prior to subsequent
incubation with anti-survivin (catalog no. sc-17779; 1:1,000
dilution; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and
anti-β-actin (catalog no. sc-47778; 1:1,000 dilution; Santa Cruz
Biotechnology, Inc.) primary antibodies overnight at 4°C. Following
this, the membranes were washed three times for 10 min in TBST and
incubated with an anti-mouse IgG secondary antibody (catalog no.
sc-516102; 1:5,000 dilution; Santa Cruz Biotechnology, Inc.) for 1
h at 37°C. Protein quantification was performed with ELISA
DuoSet® Human Carbonic Anhydrase IX (R&D Systems,
Inc., Minneapolis, MN, USA). The signals were analyzed following
treatment with TMB substrate and visualized by the ChemiDoc™ Touch
Imaging system (Bio-Rad Laboratories, Inc.).
Water-soluble tetrazolium salt-1 cell
proliferation assay
MCF-7 cells (4×104 cells per well) were
seeded into a 24-well cell culture plate.
Epigallocatechin-3-gallate (EGCG; 20 µM) or IWP-2 (5 µM), which are
LCN2 and Wnt signaling pathway inhibitors, respectively, were added
to the cultures at appropriate concentrations for 48 h. Cell
proliferation assays were performed using an EZ-CyTox Enhanced Cell
Viability Assay kit (Daeil Lab Services Co., Seoul, Korea).
Absorbance at 450 nm was measured using a microplate reader.
Immunocytochemical staining
MCF-7 cells were cultured on cover slips. Following
washing twice with PBS, cells were fixed with 4% paraformaldehyde
for 15 min. Cover slips were washed three times with PBS and the
cells were permeabilized with 0.1% Tween-20 in PBS for 20 min
followed by blocking for 30 min using blocking buffer (5% bovine
serum albumin in PBS). Following overnight incubation with the
primary antibodies, the cover slips were washed three times with
PBS and treated with Alexa Flour 594 donkey anti-rabbit IgG
(A21207; Invitrogen; Thermo Fisher Scientific, Inc.) for 1 h in the
dark. Cover slips were then washed three times in PBS and mounted
with VECTASHIELD Mounting Medium with DAPI (H-1200; Vector
Laboratories, Burlingame, CA, USA). Images were captured using a
Leica confocal laser scanning microscopy system (Leica Microsystems
GmbH, Wetzlar, Germany).
Statistical analysis
Graphical data are presented as the mean ± standard
deviation. Each experiment was performed at least three times and
subjected to statistical analysis. Statistical significance between
two groups was determined using the Student's t-test. P<0.05 was
considered to indicate a statistically significant difference. All
statistical analyses were performed using GraphPad Prism 6.0
software (GraphPad Software, Inc., La Jolla, CA, USA).
Results
5-FU treatment of Ell3 OE cells
induces upregulation of LCN2 gene expression and Wnt signaling
cDNA microarray analysis was performed to compare
the gene expression profiles of Ell3 OE and control MCF-7 cells
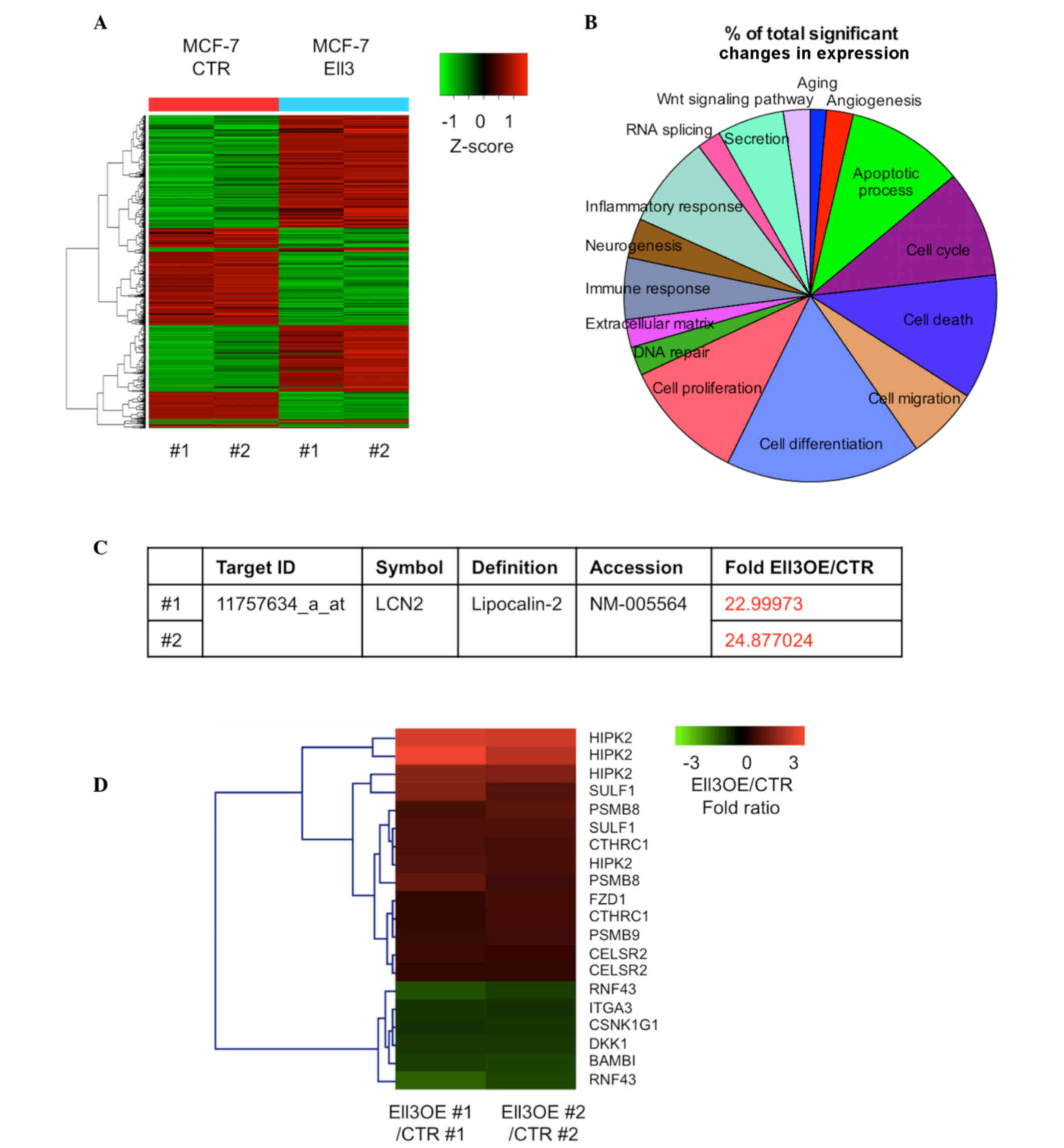
after 5-FU treatment. Fig. 1A
indicates the extensive alterations of total gene expression that
were detected in the present study. Among 20,811 genes in Ell3 OE
cells treated with 5-FU, expression levels of 694 genes (~3.33%)
were significantly altered by >2-fold (450 genes were
upregulated and 244 genes were downregulated). Genes with altered
expression were classified according to functional categories
(Fig. 1B). The cell differentiation
category had the most genes with altered expression (0.65%),
followed by the cell proliferation category (0.41%). The cell death
and apoptotic process categories represented 0.36 and 0.35%,
respectively. It was noted that expression of LCN2, which is able
to alter the sensitivity of certain types of cancers to
chemotherapeutic drugs (28), was
upregulated by >20-fold in Ell3 OE compared with control cells
(Fig. 1C). The Kyoto Encyclopedia of
Genes and Genomes (KEGG) is a knowledge base for systematic
analysis of gene functions in terms of the networks of genes and
molecules. The KEGG pathway database consists of graphical diagrams
of biochemical pathways including the majority of the known
metabolic pathways and some of the known regulatory pathways
(29). The KEGG pathway program was
utilized for the genes with >2-fold altered expression on the
microarray analysis. Notably, 66.6% of the Wnt signaling
pathway-related genes were activated in Ell3 OE cells (Fig. 1D) and several genes known to inhibit
Wnt signaling were downregulated. These results suggested that
activation of LCN2 and Wnt signaling pathway genes was the primary
cause of 5-FU drug resistance in Ell3 OE cells.
LCN2 reinforces 5-FU drug resistance
in Ell3 OE cells
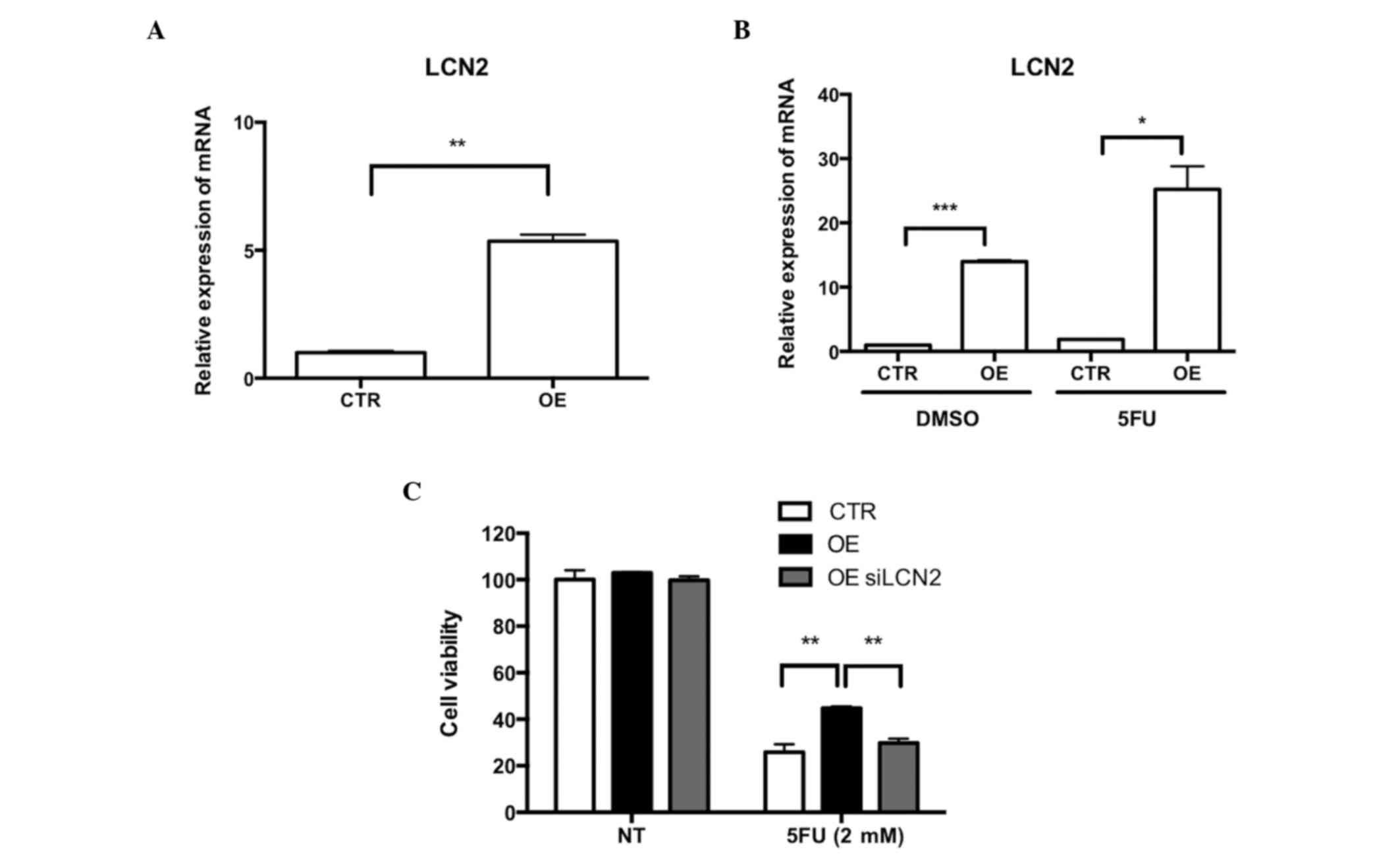
To demonstrate the role of LCN2 and Wnt signaling in
the resistance of Ell3 OE cells to 5-FU, the activation of LCN2
expression in Ell3 OE cells was confirmed by RT-qPCR analysis.
Notably, LCN2 expression was activated in Ell3 OE cells in the
absence of 5-FU and the expression level was further increased
after 5-FU treatment (Fig. 2A and B).
These results suggested that overexpression of LCN2 may be the
cause of 5-FU resistance in Ell3 OE cells. To confirm this
possibility, whether suppression of LCN2 expression in Ell3 OE
cells diminished 5-FU resistance. siLCN2-transfected Ell3 OE cells
were subsequently supplemented with 5-FU; cell viability was
similar to the wild type, which indicated that overexpression of
LCN2 was the main cause of 5-FU resistance (Fig. 2C). Prior to 5-FU treatment, Ell3 OE
cells were pretreated with EGCG, which is a chemical inhibitor of
LCN2 activity, and drug resistance was significantly decreased
(data not shown). These results suggested that LCN2 expression
induced 5-FU drug resistance in Ell3 OE cells.
Wnt signaling is associated with 5-FU
drug resistance in Ell3 OE cells
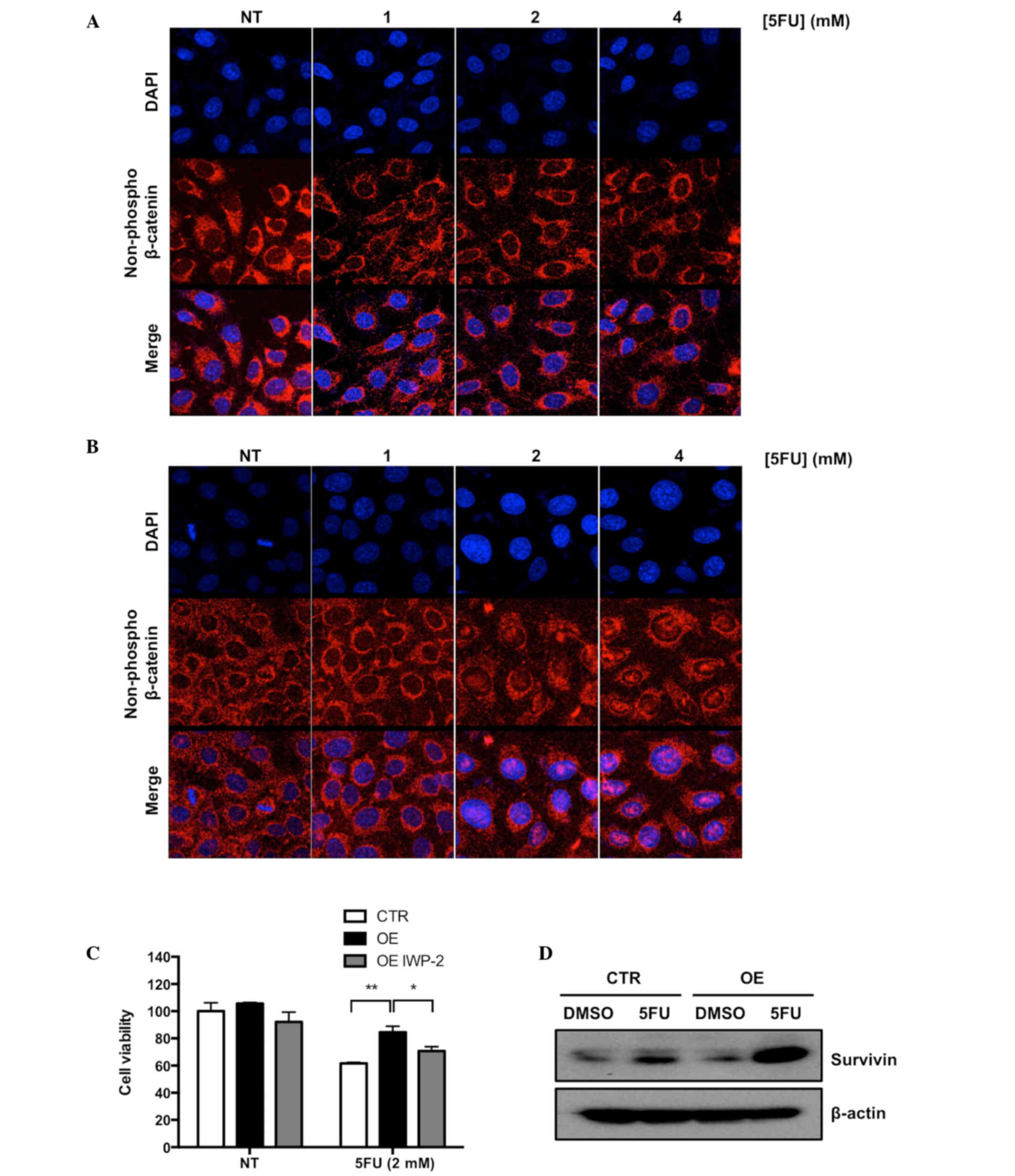
Activation of the Wnt signaling pathway induced
accumulation of non-phosphorylated β-catenin in the nucleus of a
cell (14). To confirm that the Wnt
signaling pathway was activated in Ell3 OE cells upon 5-FU
treatment, nuclear localization of non-phosphorylated β-catenin was
analyzed via immunocytochemical (ICC) staining. As shown in
Fig. 3A and B, non-phosphorylated
β-catenin accumulated in the nucleus of Ell3 OE after treatment
with 2 and 4 mM 5-FU, whereas β-catenin in control cells was
detected in the cytosol under the same conditions. To further
elucidate the role of Wnt signaling in the resistance of Ell3 OE
cells to 5-FU, the effect of IWP-2, which is an inhibitor of Wnt
signaling, on the resistance of Ell3 OE cells to 5-FU was
investigated. As hypothesized, cell viability of Ell3 OE after 5-FU
treatment was significantly decreased in the presence of IWP-2
(Fig. 3C).
Activation of Wnt signaling was associated with
enhanced expression of survivin, a member of the inhibitor of
apoptosis protein family (24).
Therefore, whether survivin expression was increased in Ell3 OE
cells was examined in the presence or absence of 5-FU. As shown in
Fig. 3D, survivin was markedly
increased in Ell3 OE cells treated with 5-FU, but not in control
cells. These results indicated that Wnt signaling was activated in
Ell3 OE cells after 5-FU treatment and supported the possibility
that drug resistance to 5-FU was mediated via the inhibition of
apoptosis-related protein activity.
Discussion
The present study investigated the drug resistant
mechanism of Ell3 OE upon 5-FU treatment. The findings showed that
LCN2 expression in Ell3 OE was higher than control and LCN2
expression was increased after 5-FU treatment in the Ell3 OE
groups, as compared with the control. Elevated cell viability in
Ell3 OE was decreased by treatment with siLCN2 and the LCN2
chemical inhibitor, EGCG. Expression of LCN2 has been reported to
be associated with the anticancer drug resistance of several
cancers, including renal cell carcinoma and pancreatic duct
adenocarcinomas (30,31), which implies the pivotal role of LCN2
in the drug resistance of cancer cells. Therefore, investigation of
the role of Ell3 in the expression of LCN2 may provide important
insight into the regulatory mechanism of LCN2 expression.
The present findings also showed that Wnt signaling
and survivin expression were enhanced in Ell3 OE cells and that
inhibition of Wnt signaling resulted in the suppression of 5-FU
resistance in Ell3 OE cells. In contrast to LCN2 expression, Wnt
signaling and survivin expression were only increased after 5-FU
treatment. This result suggested that LCN2 expression, which was
activated in Ell3 OE cells in the absence of 5-FU, was not
associated with Wnt signaling. Furthermore, the resistance of Ell3
OE cells to 5-FU was induced independently by LCN2 activity and Wnt
signaling. Since Wnt signaling has a crucial role in tumor
development and drug resistance, understanding the role of Wnt
signaling will aid the development of effective strategies to
overcome chemotherapeutic resistance in various types of
cancer.
Acknowledgements
The present study was supported by the Ministry of
Education, Science, and Technology of the Korean government (grant
nos. 2012M3A9C6050367 and 2015R1A2A2A01003498).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ahn HJ, Kim G and Park KS: Ell3 stimulates
proliferation, drug resistance, and cancer stem cell properties of
breast cancer cells via a MEK/ERK-dependent signaling pathway.
Biochem Biophys Res Commun. 437:557–564. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miller T, Williams K, Johnstone RW and
Shilatifard A: Identification, cloning, expression, and biochemical
characterization of the testis-specific RNA polymerase II
elongation factor ELL3. J Biol Chem. 275:32052–32056. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maki K, Mitani K, Yamagata T, Kurokawa M,
Kanda Y, Yazaki Y and Hirai H: Transcriptional inhibition of p53 by
the MLL/MEN chimeric protein found in myeloid leukemia. Blood.
93:3216–3224. 1999.PubMed/NCBI
|
|
5
|
Johnstone RW, Gerber MA, Landewe T,
Tollefson A, Wold WS and Shilatifard A: Functional analysis of the
leukemia protein ELL: Evidence for a role in the regulation of cell
growth and survival. Mol Cell Biol. 21:1672–1681. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin C, Garruss AS, Luo Z, Guo F and
Shilatifard A: The RNA Pol II elongation factor Ell3 marks
enhancers in ES cells and primes future gene activation. Cell.
152:144–156. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iannetti A, Pacifico F, Acquaviva R,
Lavorgna A, Crescenzi E, Vascotto C, Tell G, Salzano AM, Scaloni A,
Vuttariello E, et al: The neutrophil gelatinase-associated
lipocalin (NGAL), a NF-kappaB-regulated gene, is a survival factor
for thyroid neoplastic cells. Proc Natl Acad Sci USA.
105:14058–14063. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang H, Xu L, Xiao D, Xie J, Zeng H, Wang
Z, Zhang X, Niu Y, Shen Z, Shen J, et al: Upregulation of
neutrophil gelatinase-associated lipocalin in oesophageal squamous
cell carcinoma: Significant correlation with cell differentiation
and tumour invasion. J Clin Pathol. 60:555–561. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leng X, Ding T, Lin H, Wang Y, Hu L, Hu J,
Feig B, Zhang W, Pusztai L, Symmans WF, et al: Inhibition of
lipocalin 2 impairs breast tumorigenesis and metastasis. Cancer
Res. 69:8579–8584. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang J, McNeish B, Butterfield C and Moses
MA: Lipocalin 2 is a novel regulator of angiogenesis in human
breast cancer. FASEB J. 27:45–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gruvberger S, Ringnér M, Chen Y, Panavally
S, Saal LH, Borg A, Fernö M, Peterson C and Meltzer PS: Estrogen
receptor status in breast cancer is associated with remarkably
distinct gene expression patterns. Cancer Res. 61:5979–5984.
2001.PubMed/NCBI
|
|
12
|
Bauer M, Eickhoff JC, Gould MN, Mundhenke
C, Maass N and Friedl A: Neutrophil gelatinase-associated lipocalin
(NGAL) is a predictor of poor prognosis in human primary breast
cancer. Breast Cancer Res Treat. 108:389–397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tong Z, Wu X, Ovcharenko D, Zhu J, Chen CS
and Kehrer JP: Neutrophil gelatinase-associated lipocalin as a
survival factor. Biochem J. 391:441–448. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Giles RH, van Es JH and Clevers H: Caught
up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta.
1653:1–24. 2003.PubMed/NCBI
|
|
15
|
van Noort M, Meeldijk J, van Der Zee R,
Destree O and Clevers H: Wnt signaling controls the phosphorylation
status of beta-catenin. J Biol Chem. 277:17901–17905. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matsuda Y, Schlange T, Oakeley EJ, Boulay
A and Hynes NE: WNT signaling enhances breast cancer cell motility
and blockade of the WNT pathway by sFRP1 suppresses MDA-MB-231
xenograft growth. Breast Cancer Res. 11:R322009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Many AM and Brown AM: Both canonical and
non-canonical Wnt signaling independently promote stem cell growth
in mammospheres. PLoS One. 9:e1018002014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, Zhang X, Huang J and Dong Q: Wnt
signaling regulation of stem-like properties in human lung
adenocarcinoma cell lines. Med Oncol. 32:1572015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cui J, Jiang W, Wang S, Wang L and Xie K:
Role of Wnt/β-catenin signaling in drug resistance of pancreatic
cancer. Curr Pharm Des. 18:2464–2471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chikazawa N, Tanaka H, Tasaka T, Nakamura
M, Tanaka M, Onishi H and Katano M: Inhibition of Wnt signaling
pathway decreases chemotherapy-resistant side-population colon
cancer cells. Anticancer Res. 30:2041–2048. 2010.PubMed/NCBI
|
|
21
|
Pećina-Slaus N: Wnt signal transduction
pathway and apoptosis: A review. Cancer Cell Int. 10:222010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
You L, He B, Xu Z, Uematsu K, Mazieres J,
Fujii N, Mikami I, Reguart N, McIntosh JK, Kashani-Sabet M, et al:
An anti-Wnt-2 monoclonal antibody induces apoptosis in malignant
melanoma cells and inhibits tumor growth. Cancer Res. 64:5385–5389.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mita AC, Mita MM, Nawrocki ST and Giles
FJ: Survivin: Key regulator of mitosis and apoptosis and novel
target for cancer therapeutics. Clin Cancer Res. 14:5000–5005.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fodde R and Brabletz T: Wnt/beta-catenin
signaling in cancer stemness and malignant behavior. Curr Opin Cell
Biol. 19:150–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim PJ, Plescia J, Clevers H, Fearon ER
and Altieri DC: Survivin and molecular pathogenesis of colorectal
cancer. Lancet. 362:205–209. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schimittgen TD: Analysis of
relative gene expression data using real-time quantitative PDCR and
the 2 delta delta method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chappell WH, Abrams SL, Franklin RA,
LaHair MM, Montalto G, Cervello M, Martelli AM, Nicoletti F,
Candido S, Libra M, et al: Ectopic NGAL expression can alter
sensitivity of breast cancer cells to EGFR, Bcl-2, CaM-K inhibitors
and the natural plant product berberine. Cell Cycle. 11:4447–4461.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ogata H, Goto S, Sato K, Fujibuchi W, Bono
H and Kanehisa M: KEGG: Kyoto encyclopedia of genes and genomes.
Nucleic Acids Res. 27:29–34. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu DS, Wu CL, Ping SY, Huang YL and Shen
KH: NGAL can alternately mediate sunitinib resistance in renal cell
carcinoma. J Urol. 192:559–566. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Leung L, Radulovich N, Zhu CQ, Organ S,
Bandarchi B, Pintilie M, To C, Panchal D and Tsao MS: Lipocalin2
promotes invasion, tumorigenicity and gemcitabine resistance in
pancreatic ductal adenocarcinoma. PLoS One. 7:e466772012.
View Article : Google Scholar : PubMed/NCBI
|