Introduction
Lung cancer is one of the most common malignant
tumors and health hazards worldwide. It is also one of the world's
fastest rising cancers in terms of morbidity and mortality
(1). The majority of lung cancer
patients present with a late-stage disease when they are diagnosed,
which is not curable by current therapies. Cancer is an extremely
complex process, and a variety of genes are involved in the
completion of this multi-step process (2). Preventing the invasion and metastasis of
lung cancer, which affects failure treatment and mortality in
patients with lung cancer, is currently the key to the treatment of
lung cancer (3). Despite the fact
that numerous novel therapeutic interventions have been performed,
lung cancer remains a serious healthcare problem, and its incidence
and mortality have continuously increased during the last decade
(4). Therefore, an effective and safe
method for the treatment of lung cancer is required.
Endothelins (ETs), a class of peptides produced by
endothelial cells, have a strong vasoconstrictor effect (5). ETs are composed of 21 amino acids, and
four isomers (peptide isoforms), namely ET-1, ET-2, ET-3 and ET-4,
have been identified in the ET family (6). Since their discovery by Yanagisawa et
al in 1988 (7), multiple studies
have demonstrated that ETs not only are able to strengthen the
contraction of vascular and cardiac muscle as well as promote the
neuroendocrine function, but are also powerful pro-growth and cell
differentiation factors (8). ETs have
a cell growth factor-like effect that can promote DNA synthesis,
proto-oncogene expression and cell proliferation. In addition, ETs
can induce angiogenesis during tumor growth (6). At present, the majority of studies focus
on ET-1 due to its strong biological activity (9). ETs serve an important role in the
development of tumors, since they can activate proto-oncogenic
genes as well as promote cell division and proliferation in
collaboration with other growth factors (10). In addition, the activation of
oncogenes mediated by ETs promotes cell proliferation and serves an
important role in the development of tumors (6). ET-1 can promote tumor angiogenesis
directly or indirectly via the paracrine route (11). Previous studies have shown that ET-1
is highly expressed in lung cancer and is involved in lung cancer
cell proliferation (12). However,
the association between ETs and tumors' growth and metastasis has
not been fully clarified yet.
Endostar, a modified recombinant human endostatin,
demonstrated a better effect on vasoconstrictivity than endostatin,
and has been widely studied for the treatment of diseases caused by
pathological angiogenesis (13).
Extensive studies revealed that Endostar significantly inhibited
cell proliferation, migration and invasion in a dose-dependent
manner (13). Thus, in the present
study, Endostar was used as a positive control drug during the
experiments.
In the present study, ET-1 was silenced via RNA
interference (RNAi), and the proliferation of A549 cells and the
expression of apoptosis, growth and invasion-associated factors,
including RhoA/C, vascular endothelial growth factor (VEGF),
pigment epithelium-derived factor (PEDF), AKT, E-cadherin and
cyclooxygenase (Cox)-2, were then examined. It was observed that
ET-1 RNAi can inhibit A549 cells proliferation and invasion more
effectively than Endostar, which suggested that ET-1 RNAi may be a
new effective strategy to treat lung cancer.
Materials and methods
Cell culture
The A549 cell line used in the present study was
purchased from the American Type Culture Collection (Manassas, VA,
USA) and was maintained in Dulbecco's modified Eagle's medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc.) at 37°C and 5% CO2.
ET-1 short hairpin RNA (shRNA)
construction
The shRNAs targeting three positions of the human
ET-1 gene (1, 5′-CCATGAGAAACAGCGTCAA-3′; 2,
5′-AAGGCAACAGACCGTGAAA-3′; and 3, 5′-TGCCAATGTGCTAGCCAAA-3′) were
designed by Sangon Biotech Co., Ltd. (Shanghai, China). The
synthesized shRNAs were then ligated into the pCMV-G&NR vector
(Sangon Biotech Co., Ltd.), giving rise to the different
pCMV-G&NR-ET-shRNA constructs. The constructs were then
transfected into A549 cells using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The transfection efficiency was determined by
fluorescence microscopy (Olympus Corporation, Tokyo, Japan).
Reverse transcription
(RT)-quantitative polymerase chain reaction (PCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), and the integrity of
RNA was analyzed by 1% agarose gel electrophoresis (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Gels were visualized using
the Gel Imaging and Analysis System (Syngene, Frederick, MD, USA).
High Capacity cDNA Reverse Transcription kit (Thermo Fisher
Scientific, Inc.) was used for the RT step, and SYBR Green PCR
Master Mix kit (Thermo Fisher Scientific, Inc.) was used for the
amplification step. The sequences of the primers for ET-1 and GAPDH
were as follows: ET-1 forward (F), 5′-GCCTGTCTGAAGCCATAG-3′ and
reverse (R), 5′-GCTGAGAGGTCCATTGTC-3′; and GAPDH F,
5′-CACCCACTCCTCCACCTTTG-3′ and R, 5′-CCACCACCCTGTTGCTGTAG-3′. The
following PCR conditions were used: 95°C for 10 min, followed by 40
cycles of 95°C for 15 sec and 60°C for 45 sec. The messenger RNA
(mRNA) level was analyzed by ABI 7300 Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The results were
analyzed by ABI Prism 7300 SDS software version 1.4 (Applied
Biosystems; Thermo Fisher Scientific, Inc.).
Cell proliferation assay
Cell Counting kit (CKK)-8 assay (Beyotime Institute
of Biotechnology, Haimen, China) was used to detect cell
proliferation according to the manufacturer's protocol. A549 cells
were plated at a density of 2,000 cells/well onto 96-well plates.
Upon incubation, 10 µl CKK-8 reagent was added to the plates, which
were then incubated at 37°C for 4 h. Next, absorbance at 450 nm was
measured using a microplate reader (Bio-Rad Laboratories,
Inc.).
Invasion detection
Invasion assay was performed using Transwell inserts
(EMD Millipore, Billerica, MA, USA) to test the invasion of A549
cells subjected to different treatments. The upper well of the
Transwell (Corning Inc., Corning, NY, USA) was coated with Matrigel
(BD Biosciences, Franklin Lakes, NJ, USA) and incubated at 37°C in
a 5% CO2 for 1 h. The indicated cells were serum starved
for 24 h. Subsequently, 5×104 cells in 500 μl serum-free
DMEM were seeded into the upper well of the Transwell chamber.
Culture medium, supplemented with 10% FBS (750 μl) was added into
the lower well of the chamber. After 48 h incubation, the cells in
the upper well were removed with a cotton swab and the cells that
migrated into the lower well were washed with PBS, fixed in 3.7%
paraformaldehyde and stained with 0.2% crystal violet. Images of
the cells were captured and cell number was counted using an
Olympus CX41RF microscope (Olympus Corporation). The number of
cells that traversed the filter was detected by staining with
crystal violet.
Western blotting
Cells were harvested and washed twice with PBS, and
then lysed for 10 min in a lysis buffer (Beyotime Institute of
Biotechnology) at 4°C. Upon centrifugation (12,000 × g, 10 min at
25°C), the protein concentration was determined using Pierce BCA
Protein Assay kit (Thermo Fisher Scientific, Inc.). Proteins were
separated by 10% SDS-PAGE and electroblotted onto Immobilon
polyvinylidene difluoride membranes (EMD Millipore). The membranes
were then blocked with fat-free milk overnight at 4°C, and
incubated with primary antibodies over night at 4°C. Primary
antibodies against VEGF (ab46154), PEDF (ab115489), RhoC (ab54837)
and Cox-2 (ab15191) were purchased from Abcam (Cambridge, MA, USA).
Primary antibodies against E-cadherin (Sc-9989) and RhoA (Sc-179)
were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA), while antibodies for AKT (9272S), phosphorylated (p)-AKT
(4058S) and GAPDH (5471) were purchased from CST Biological
Reagents Co., Ltd. (Shanghai, China). The dilutions of the
antibodies were as follows: Anti-VEGF, 1:1,000; anti-PEDF, 1:1,000;
anti-AKT, 1:1,000; anti-p-AKT, 1:1,000; anti-RhoC, 1:500;
anti-Cox-2, 1:400; anti-E-cadherin, 1:100; anti-RhoA, 1:100; and
anti-GAPDH, 1:1,500. The secondary antibodies anti-rabbit
immunoglobulin (Ig)G (A0208; 1:1,000; Beyotime Institute of
Biotechnology) and anti-mouse IgG (A0216; 1:1,000; Beyotime
Institute of Biotechnology) were used to detect the primary
antibodies. Membranes were incubated with secondary antibodies for
1 h at 37°C, and visualized with enhanced chemiluminescence (EMD
Millipore) according to the manufacturer's protocol. Signals were
quantified by densitometry (Quantity One software, version 4.62;
Bio-Rad Laboratories, Inc.). GAPDH was used as a control.
In vivo tumor-inhibitory
experiments
A total of 12 BALB/c male nude mice (4–5 weeks of
age and ~20 g of weight; housed at 25°C on a 12 h light/dark cycle,
with access to food and water ad libitum) were purchased
from the Second Military Medical University (Shanghai, China) and
were randomly divided into two groups (6 mice/group). In total, 6
nude mice were injected subcutaneously with vector-transfected A549
cells (2×106), while another 6 mice were injected
subcutaneously with ET-1-silenced A549 cells (2×106).
The tumors were formed 1–2 weeks later, and the tumor size was
determined every 3–4 days after tumor formation. Tumor volume (V)
was measured and calculated using the following formula: V
(mm3) = 0.5 × larger diameter × smaller
diameter2. At 43 days following injection, nude mice
were sacrificed following cervical dislocation. Once the nude mice
were sacrificed, the tumors were dissected and their weights were
measured. The experiment was performed under the Zhongshan Hospital
Affiliated to Xiamen University's (Xiamen, China) guidelines for
animal experiments. The study was approved by the ethics committee
of Zhongshan Hospital Affiliated to Xiamen University.
Statistical analyses
All data are presented as the mean ± standard
deviation of at least three experiments. Statistical significance
was determined by analysis of variance using SPSS 17.0 software
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
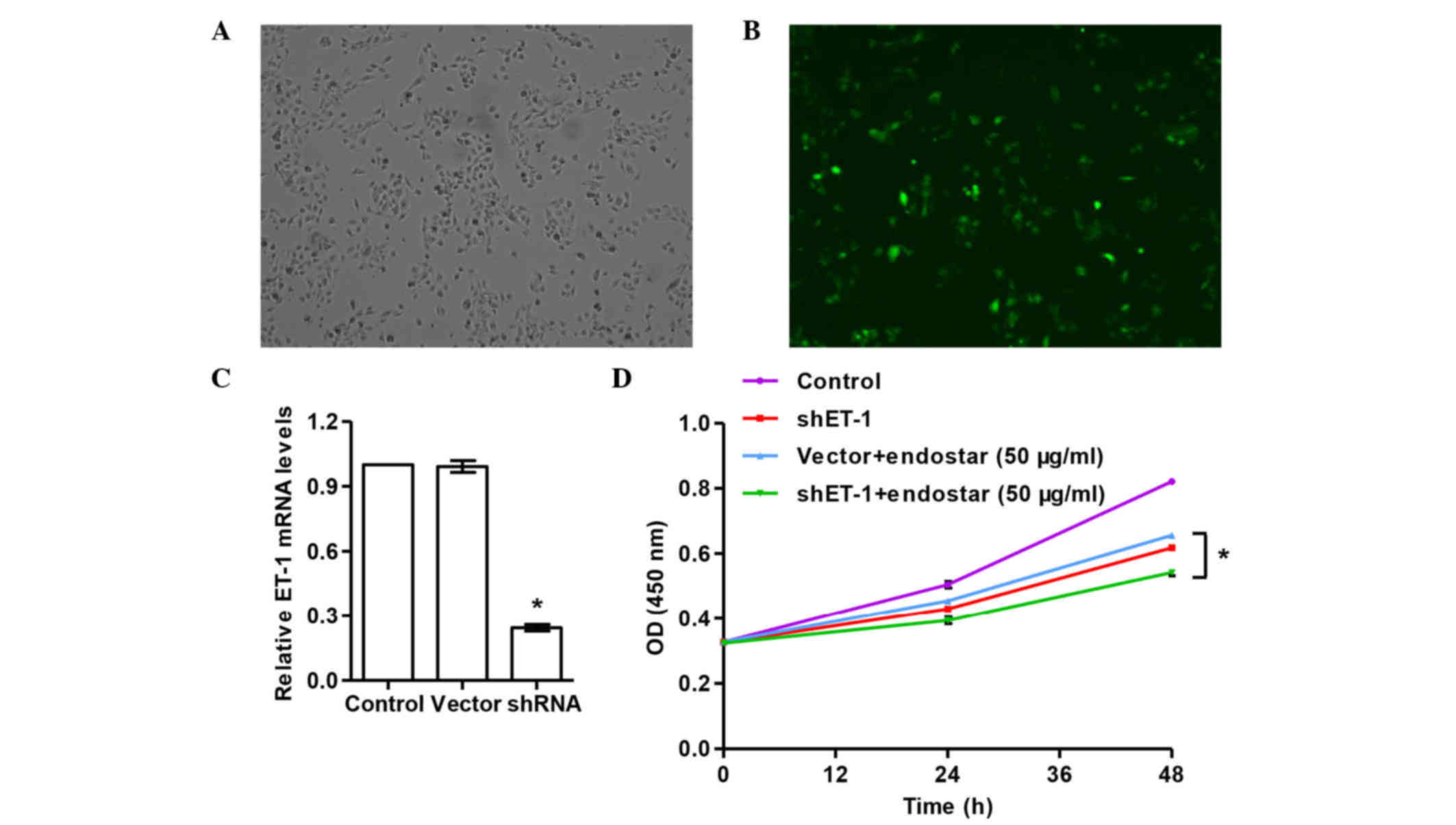
Establishment of shET-1 A549
cells
To investigate the function of ET-1 in lung cancer,
the expression of ET-1 was suppressed by RNAi in A549 cells. Three
shRNAs targeting human ET-1 were designed, cloned into the
pCMV-G&NR vector and transfected into A549 cells. The
transfection efficiency was 53% (transfection efficiency = number
of cells with green fluorescence/total cells), as evaluated by
fluorescence microscopy (Fig. 1A and
B). The third fragment, 5′-TGCCAATGTGCTAGCCAAA-3′ (targeting
position 321–339), was the most efficient shRNA for silencing ET-1
(Fig. 1C). shRNAs 1, 2 and 3
significantly decreased mRNA expression of ET-1 by 35.0, 42.8 and
75.4%, respectively. These data suggested that the shRNAs designed
in the present study had a significant interference effect on the
expression of ET-1, and that ET-1-silenced A549 cells (transfected
with shRNA 3) were successfully established and could be used for
further analysis.
Effect of ET-1 RNAi on the
proliferation of A549 cells
To determine the effects of ET-1 RNAi with/without
Endostar on the proliferation capability of A549 cells, CCK-8 assay
was performed. The results of CCK-8 assay (Fig. 1D) revealed that both Endostar and ET-1
RNAi could suppress the proliferation of A549 cells. In addition,
the combined use of Endostar and ET-1 shRNA exhibited a stronger
inhibition of the proliferation of A549 cells compared with that
caused by Endostar or ET-1 shRNA separately (P<0.05). These
results indicated that the downregulation of ET-1 can decrease the
growth capacity of A549 cells, and such effect can be augmented by
Endostar treatment.
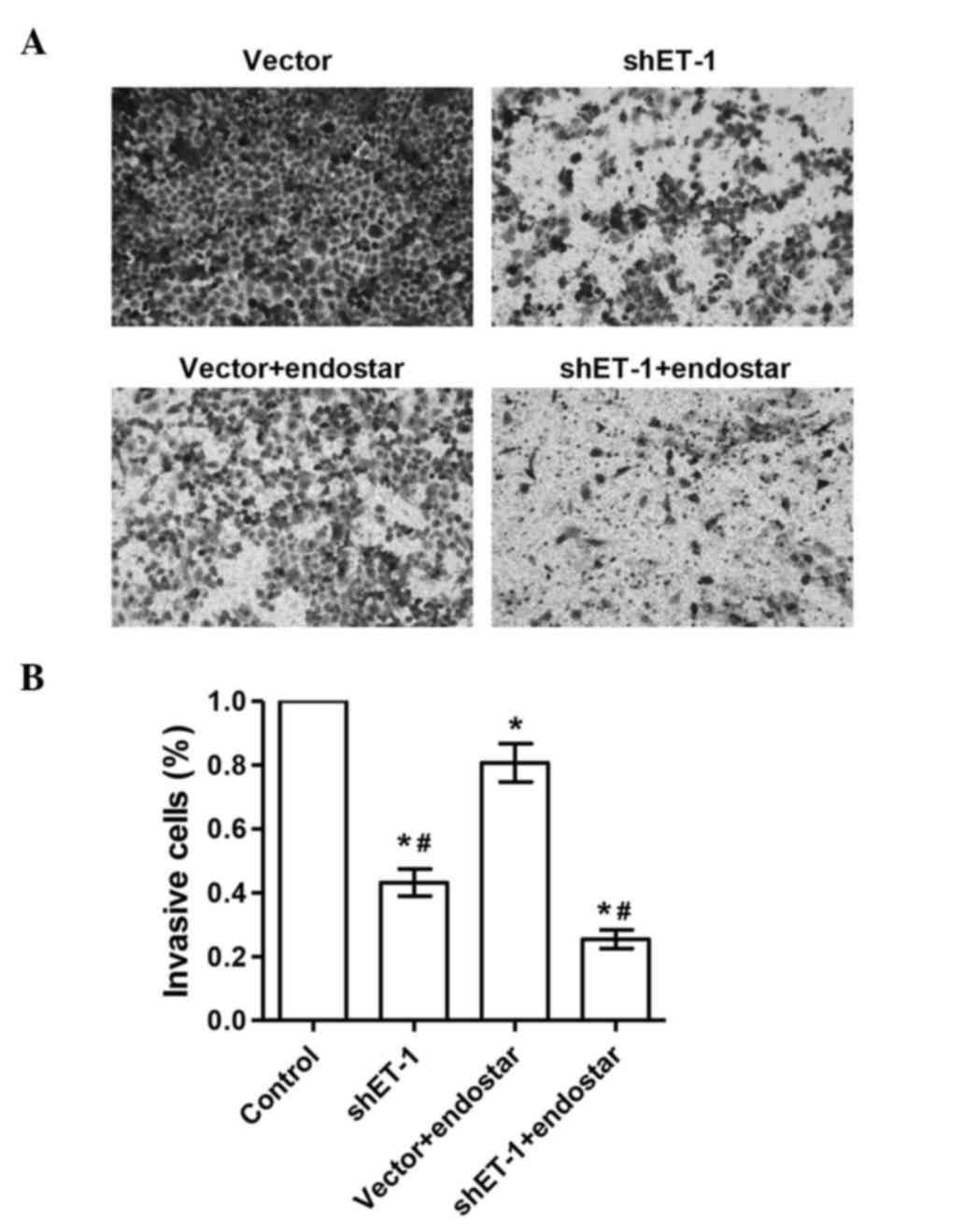
The invasive ability of A549 cells is
suppressed by ET-1 RNAi
Invasive capacity is essential for the malignant
biological behavior of tumors. The present study investigated
whether ET-1 RNAi affected the invasive ability of A549 cells. In
contrast to control cells, ET-1 RNAi and Endostar could
significantly reduce the cell invasive ability of A549 cells. The
number of invaded cells in the ET-1 shRNA + Endostar group was
significantly decreased compared with that in the ET-1 shRNA or
Endostar groups (P<0.05) (Fig. 2).
The results indicated that the downregulation of ET-1 decreased the
invasive ability of A549 cells, which could be potentiated by
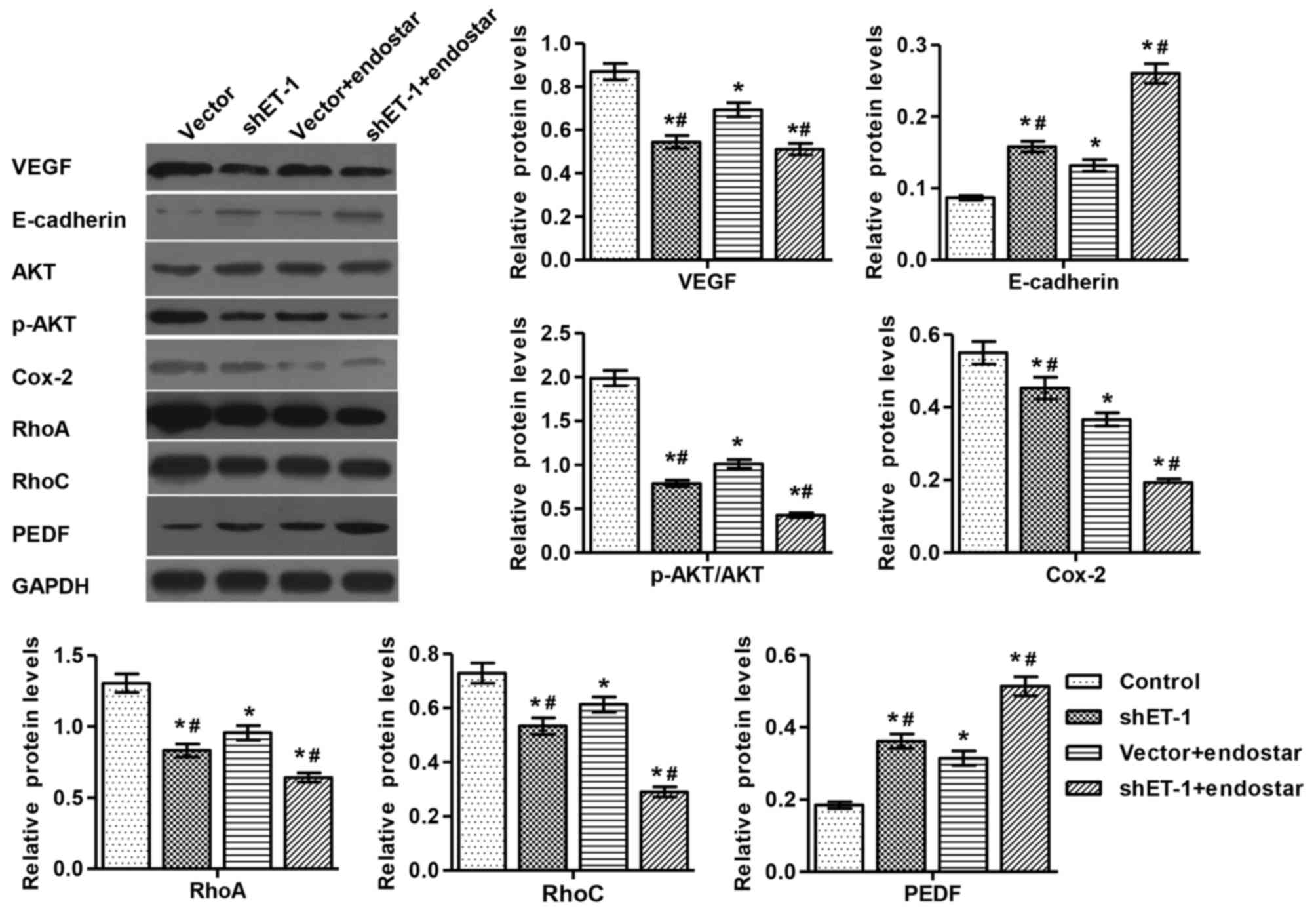
Endostar treatment. In addition, the expression of RhoA/C, VEGF,
PEDF, AKT, E-cadherin and Cox-2 proteins was detected by western
blotting (Fig. 3).
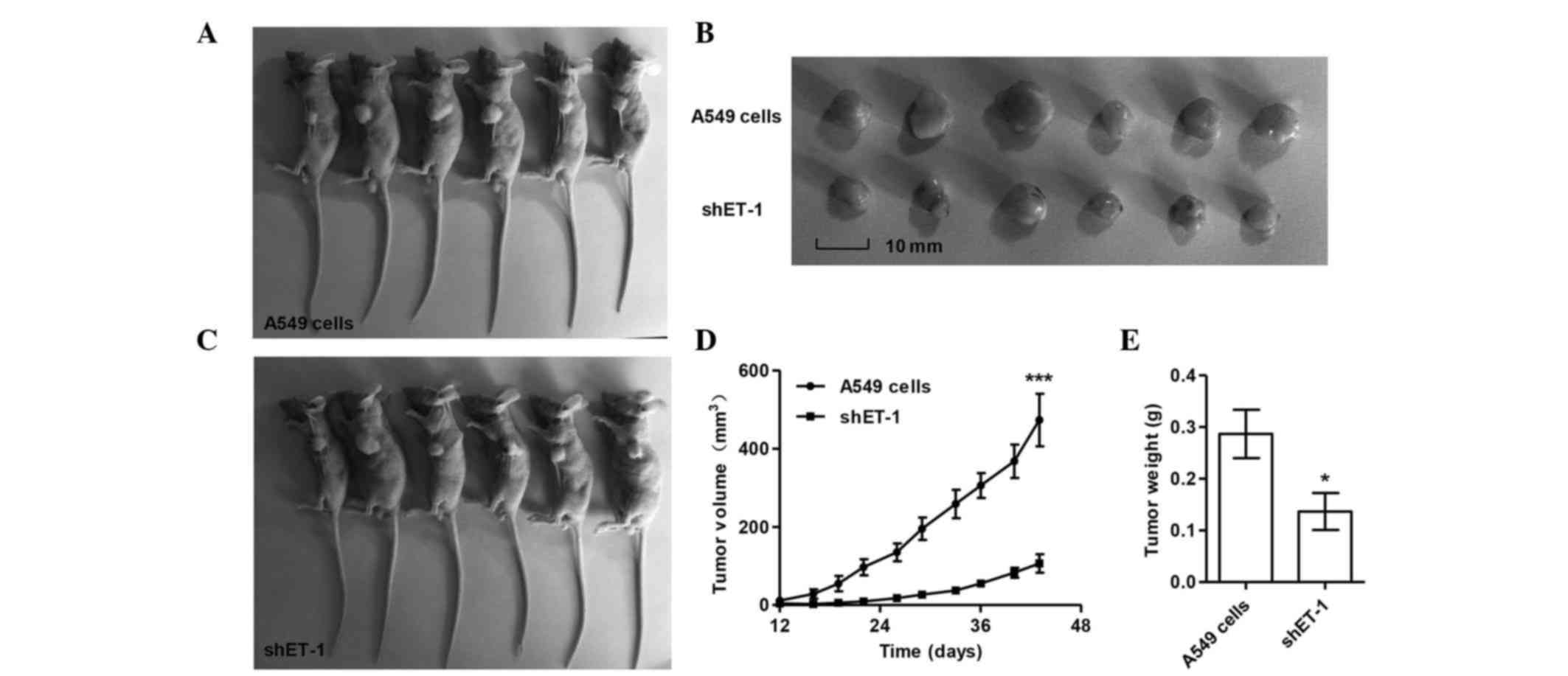
Inhibitory effect of ET-1 RNAi on the
growth of human lung carcinoma xenografts in nude mice
Next, it was determined whether knocking down ET-1
in lung cancer cells could reduce tumor growth in vivo. A549
cells transfected with control or ET-1-shRNA were subcutaneously
injected in athymic nude mice, and tumor volumes were measured for
45 days. As shown in Fig. 4, the
ET-1-shRNA-treated tumors grew slower than the control tumors
(P<0.05). These data indicated that ET-1 RNAi can effectively
inhibit tumor growth in vivo.
Discussion
Despite the fact that numerous novel therapeutic
interventions have been performed, lung cancer remains a serious
healthcare problem (3). The growth,
division and DNA synthesis of normal lung cells are regulated
through the orderly cell cycle. If this order is disrupted, tumors
may be eventually induced (14). In
addition, the invasion and metastasis of lung cancer are extremely
complex processes, where multiple genes and steps are involved
(15). Lung cancer invasion and
metastasis seriously affect the efficacy and prognosis of patients
with cancer who are undergoing treatment, and are also the main
causes of patients' treatment failure and mortality. Therefore,
preventing the invasion and metastasis of lung cancer is currently
key in lung cancer treatment. At present, a great progress has been
achieved for the prevention and treatment of early lung cancer;
however, the treatment effect and prognosis remain poor (3). Thus, to prevent and cure lung cancer,
tumor pathogenesis must be further explored, and more effective
targeted therapies must be developed.
Since ETs were identified, it has been verified that
ETs not only perform the function of vasoconstriction, but are also
closely connected with the growth of tumors. Numerous studies
conducted on different types of tumor tissue, observed that ETs
exist in malignant tumor tissues (8,16).
Furthermore, ET-1 levels in plasma exhibited a visible difference
prior and subsequent to treatment, and ET-1 content was
significantly decreased following treatment (17). In addition, previous studies have
suggested that ETs can activate proto-oncogenic genes, promote cell
division and proliferation in collaboration with other growth
factors, and serve an important role in the development of tumors.
In recent years, the association between ETs and tumor growth and
metastasis is receiving great attention, but this association has
not been fully elucidated yet.
The present study detected VEGF, PEDF, Cox-2, AKT,
p-AKT, E-cadherin, RhoA and RhoC, which are associated with tumor
apoptosis, growth, invasion and metastasis (18–20). The
results revealed that the expression of all the above factors was
increased or decreased when shET-1 and Endostar were present or
absent. In the current study, the expression of VEGF was decreased
when ET-1 was silenced. VEGF is the most effective pro-angiogenic
growth factor, and serves an important role in cancer angiogenesis,
tumor growth and metastasis. ET-1 can promote the processes of
invasion and metastasis via activation of hypoxia-inducible
factor-1α and VEGF in melanoma. Melanoma can also promote the
expression of VEGF mRNA and the secretion of VEGF via ET-1. Thus,
there is an indirect synergism between ET-1 and VEGF (21,22).
Matsuura et al (23) indicated
that mutual stimulation of ET-1 and VEGF resulted in increased
endothelial cell proliferation, migration and invasion of the
basement membrane, suggesting that ET-1 induced endothelial cell
proliferation, invasion and tumor angiogenesis through stimulation
of the secretion of VEGF (24). That
study confirmed that ET-1 stimulates the secretion of VEGF and
endothelial cell proliferation, invasion which induced by VEGF, and
indirectly effects on tumor angiogenesis and provides a
microenvironment for tumor growth (24). The present results are consistent with
those from previous studies.
E-cadherin serves an important role in selective
cell aggregation. Previous studies have shown that ET-1 can reduce
the expression of E-cadherin, and influence the invasion and
metastasis of tumors (25). In the
present study, E-cadherin was increased in the ET-1 RNAi group,
which suggested that ET-1 RNAi could promote the invasion of lung
cancer. The serine/threonine protein kinase AKT and activated AKT,
which is phosphorylated, are important in regulating apoptosis,
angiogenesis and the cell cycle (26–28).
Generally, the extent of activation of the PI3K/AKT signaling
pathway is determined based on the phosphorylation level of AKT,
which is a key signaling protein in the PI3K signaling pathway
(26,29,30). It
has been reported that the apoptosis of ovarian carcinoma cells
induced by paclitaxel can be reverted by ET-1 through upregulating
the activity of AKT (31). As a
synthesis rate-limiting enzyme of prostaglandin (PG), which is an
important inflammatory mediator, Cox-2 can reduce endothelial cell
apoptosis (32). Despite the fact
that Cox-2 cannot be detected in the majority of normal tissues and
cells, when an inflammatory response happens in vascular
endothelial and smooth muscle cells, Cox-2 may be induced by
cytokines, ETs and PGs (33). Cox-2
participates in a variety of pathological and physiological
processes. Cox-2 induces tumor angiogenesis and invasion by
promoting cell proliferation and inhibiting cell apoptosis, thus
participating in the development of tumors (34,35).
Previous studies have reported that Cox-2 can induce the expression
of several vascular factors such as ET-1 and transforming growth
factor-β, while ET-1 can also upregulate Cox-2 expression. Thereby,
it forms a positive feedback network, which could co-stimulate
tumor angiogenesis (36). In the
present study, p-Akt/Akt and Cox-2 expression exhibited a decreased
trend when ET-1 was silenced. All these data suggested that the
interference of ET-1 was effective for the treatment of lung
cancer.
Rho is a type of guanosine-5′-triphosphate
(GTP)-binding protein with GTPase enzymatic activity. Rho has been
associated with tumorigenesis, invasion and metastasis, and has
been observed to exhibit a high expression in malignancy (37). Previous studies suggested that the
RhoC gene was involved in the occurrence and development of liver
cancer, and that a close association existed between the high
expression of RhoC and the phenotype of hepatocellular carcinoma
regarding cell invasion and metastasis (38). Previous studies demonstrated that
RhoA/C was highly expressed in a variety of malignancies,
participated in the adhesion and migration of tumor cells, and
markedly promoted the occurrence and development of tumors
(39–42). As a novel tumor marker, Rho can be
applied to distinguish between benign and malignant lesions, and
may help to assess the ability of tumor metastasis and prognosis
(43). This has potentially important
clinical value for cancer prevention. Rho was observed to
participate in the movement and angiogenesis of endothelial cells
induced by VEGF through Rho-associated protein kinase signal
transduction pathways, thus promoting tumor angiogenesis and
assisting tumor cells to establish distant metastases through the
vascular endothelia (44). Previous
studies speculated that a potential mechanism to inhibit tumor
angiogenesis may be to inhibit VEGF expression by enhancing P53 (a
tumor-suppressor gene) expression (45). Other studies indicated that Rho
protein was involved in regulating tumor angiogenesis (46). A study has revealed that there are
complex interactions between Rho protein and E-cadherin (47). Overexpression of Rho proteins may
promote the metastasis of tumor cells through undermining
intercellular adhesion (48). The
present study noticed that RhoA/C was expressed at lower levels in
the ET-1 RNAi group than in the empty vector group. Thus, it could
be speculated that ET-1 RNAi can inhibit the expression of RhoA/C.
However, the mechanism involved remains unclear.
The present data also revealed that the PEDF level
was decreased when ET-1 RNAi was applied to A549 cells. PEDF was an
effective factor in inhibiting angiogenesis, having a
neuroprotective effect, inducing tumor apoptosis, and inhibiting
tumor cells invasion and metastasis. However, it has been
demonstrated that PEDF cannot affect the AKT signaling pathway
(49). Mahtabifard et al
(50) reported that PEDF can inhibit
angiogenesis in breast cancer. Takenaka et al (51) reported that PEDF can induce the
apoptosis of MG63 (osteosarcoma cells) and inhibit the expression
of VEGF, thus inhibiting tumor angiogenesis. Zhang et al
(52) reported that PEDF expression
was decreased in non-small cell lung cancer. PEDF level is
inversely proportional to microvessel density in tumor tissue, and
PEDF mRNA level is inversely proportional to the size of the tumor.
The low level of PEDF in lung cancer suggests a poor prognosis
(53,54). Abe et al (55) reported that PEDF serves an important
role in inhibiting tumor proliferation and tumor angiogenesis
processes, and deletion of the PEDF gene may be associated with the
malignant differentiation of normal cells. However, the association
between malignant tumors and the PEDF gene remains unclear.
Although there are no studies indicating that Rho
and PEDF have any direct connection with ET-1, it can be speculated
that Rho and PEDF may be indirectly regulated in the present study.
Compared with their expression in the vector group, the expression
of the tumor-associated factors examined in the present study
exhibited significant changes. These results indicated that ET-1
RNAi may have a potential therapeutic effect on the treatment of
lung cancer. Furthermore, it was observed that the therapeutic
effect was improved when both ET-1 RNAi and Endostar acted on the
cells, and this result was also verified in in vivo
experiments.
References
|
1
|
Stanzani F, de Moraes Paisani D, de
Oliveira A, de Souza RC, Perfeito JAJ and Faresin SM: Morbidity,
mortality, and categorization of the risk of perioperative
complications in lung cancer patients. J Bras Pneumol. 40:21–19.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wong YH, Chen RH and Chen BS: Core and
specific network markers of carcinogenesis from multiple cancer
samples. J Theor Biol. 362:17–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reno TA, Kim JY and Raz DJ: Triptolide
inhibits lung cancer cell migration, invasion, and metastasis. Ann
Thorac Surg. 100:1917–1824. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Justilien V and Fields AP: Utility and
applications of orthotopic models of human non-small cell lung
cancer (NSCLC) for the evaluation of novel and emerging cancer
therapeutics. Curr Protoc Pharmacol. 62:14.27.1–14.27.17. 2013.
|
|
5
|
Feldstein C and Romero C: Role of
endothelins in hypertension. Am J Ther. 14:147–153. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hynynen MM and Khalil RA: The vascular
endothelin system in hypertension-recent patents and discoveries.
Recent Pat Cardiov Drug Discov. 1:952006. View Article : Google Scholar
|
|
7
|
Yanagisawa M, Kurihara H, Kimura S, Tomobe
Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K and Masaki T: A novel
potent vasoconstrictor peptide produced by vascular endothelial
cells. Nature. 332:411–415. 1988. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dworkin S, Simkin J, Darido C, Partridge
DD, Georgy SR, Caddy J, Wilanowski T, Lieschke GJ, Doggett K, Heath
JK and Jane SM: Grainyhead-like 3 regulation of endothelin-1 in the
pharyngeal endoderm is critical for growth and development of the
craniofacial skeleton. Mech Dev. 133:77–90. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Grimshaw MJ: Endothelins and
hypoxia-inducible factor in cancer. Endocr Relat Cancer.
14:233–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schrey MP, Patel KV and Tezapsidis N:
Bombesin and glucocorticoids stimulate human breast cancer cells to
produce endothelin, a paracrine mitogen for breast stromal cells.
Cancer Res. 52:1786–1790. 1992.PubMed/NCBI
|
|
11
|
Knowles J, Loizidou M and Taylor I:
Endothelin-1 and angiogenesis in cancer. Curr Vasc Pharmacol.
3:309–314. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang WM, Zhou J and Ye QJ: Endothelin-1
enhances proliferation of lung cancer cells by increasing
intracellular free Ca2+. Life Sci. 82:764–771. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu X, Mao W, Chen Q, Zhuang Q, Wang L, Dai
J, Wang H and Huang Z: Endostar, a modified recombinant human
endostatin, suppresses angiogenesis through inhibition of
Wnt/β-catenin signaling pathway. Plos One. 9:e1074632014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tamura K: Development of cell-cycle
checkpoint therapy for solid tumors. Jpn J Clin Oncol.
45:1097–1192. 2015.PubMed/NCBI
|
|
15
|
Gutschner T, Hämmerle M, Eissmann M, Hsu
J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Gross M, et al:
The noncoding RNA MALAT1 is a critical regulator of the metastasis
phenotype of lung cancer cells. Cancer Res. 73:1180–1189. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rosanò L, Spinella F and Bagnato A:
Endothelin 1 in cancer: Biological implications and therapeutic
opportunities. Nat Rev Cancer. 13:637–651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fagan KA, McMurtry IF and Rodman DM: Role
of endothelin-1 in lung disease. Respir Res. 2:90–101. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fu L, Chen W, Guo W, Wang J, Tian Y, Shi
D, Zhang X, Qiu H, Xiao X, Kang T, et al: Berberine targets
AP-2/hTERT, NF-κB/COX-2, HIF-1α/VEGF and cytochrome-c/caspase
signaling to suppress human cancer cell growth. PLoS One.
8:e692402013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lau MT, Klausen C and Leung PCK:
E-cadherin inhibits tumor cell growth by suppressing PI3K/Akt
signaling via β-catenin-Egr1-mediated PTEN expression. Oncogene.
30:2753–2766. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ridley AJ: RhoA, RhoB and RhoC have
different roles in cancer cell migration. J Microsc. 251:242–249.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Patterson DM, Gao D, Trahan DN, Johnson
BA, Ludwig A, Barbieri E, Chen Z, Diaz-Miron J, Vassilev L, Shohet
JM and Kim ES: Effect of MDM2 and vascular endothelial growth
factor inhibition on tumor angiogenesis and metastasis in
neuroblastoma. Angiogenesis. 14:255–266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Spinella F, Rosanò L, Di Castro V,
Decandia S, Nicotra MR, Natali PG and Bagnato A: Endothelin-1 and
endothelin-3 promote invasive behavior via hypoxia-inducible
factor-1alpha in human melanoma cells. Cancer Res. 67:1725–1734.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Matsuura A, Yamochi W, Ki Kawashima,
Hirata S and Yokoyama M: Stimulatory interaction between vascular
endothelial growth factor and endothelin-1 on each gene expression.
Hypertension. 32:89–95. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Salani D, Di Castro V, Nicotra MR, Rosanò
L, Tecce R, Venuti A, Natali PG and Bagnato A: Role of endothelin-1
in neovascularization of ovarian carcinoma. Am J Pathol.
157:1537–1547. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rosanò L, Spinella F, Di Castro V, Nicotra
MR, Dedhar S, de Herreros AG, Natali PG and Bagnato A: Endothelin-1
promotes epithelial-to-mesenchymal transition in human ovarian
cancer cells. Cancer Res. 65:11649–11657. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu G, Zhang W, Bertram P, Zheng XF and
McLeod H: Pharmacogenomic profiling of the PI3K/PTEN-AKT-mTOR
pathway in common human tumors. Int J Oncol. 24:893–900.
2004.PubMed/NCBI
|
|
27
|
Beckner ME, Gobbel GT, Abounader R,
Burovic F, Agostino NR, Laterra J and Pollack IF: Glycolytic glioma
cells with active glycogen synthase are sensitive to PTEN and
inhibitors of PI3K and gluconeogenesis. Lab Invest. 85:1457–1470.
2005.PubMed/NCBI
|
|
28
|
Thomas WD, Zhang XD, Franco AV, Nguyen T
and Hersey P: TNF-related apoptosis-inducing ligand-induced
apoptosis of melanoma is associated with changes in mitochondrial
membrane potential and perinuclear clustering of mitochondria. J
Immunol. 165:5612–5620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ruth MC, Xu Y, Maxwell IH, Ahn NG, Norris
DA and Shellman YG: RhoC promotes human melanoma invasion in a
PI3K/Akt-dependent pathway. J Invest Dermatol. 126:862–868. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Blanco-Aparicio C, Pequeño B, Moneo V,
Romero L, Leal JF, Velasco J, Fominaya J and Carnero A: Inhibition
of phosphatidylinositol-3-kinase synergizes with gemcitabine in
low-passage tumor cell lines correlating with Bax translocation to
the mitochondria. Anti-Cancer Drug. 16:977–987. 2005. View Article : Google Scholar
|
|
31
|
Del Bufalo D, Di Castro V, Biroccio A,
Varmi M, Salani D, Rosanò L, Trisciuoglio D, Spinella F and Bagnato
A: Endothelin-1 protects ovarian carcinoma cells against
paclitaxel-induced apoptosis: Requirement for Akt activation. Mol
Pharmacol. 61:524–532. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Leahy KM, Ornberg RL, Wang Y, Zweifel BS,
Koki AT and Masferrer JL: Cyclooxygenase-2 inhibition by celecoxib
reduces proliferation and induces apoptosis in angiogenic
endothelial cells in vivo. Cancer Res. 62:625–631. 2002.PubMed/NCBI
|
|
33
|
Hsieh HL, Sun CC, Wang TS and Yang CM:
PKC-delta/c-Src-mediated EGF receptor transactivation regulates
thrombin-induced COX-2 expression and PGE(2) production in rat
vascular smooth muscle cells. Biochim Biophys Acta. 1783:1563–1575.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Appleby SB, Ristimäki A, Neilson K, Narko
K and Hla T: Structure of the human cyclo-oxygenase-2 gene. Biochem
J. 302:723–727. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morita I, Schindler M, Regier MK, Otto JC,
Hori T, DeWitt DL and Smith WL: Different intracellular locations
for prostaglandin endoperoxide H synthase-1 and −2. J Biol Chem.
270:10902–10908. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Murata H, Kawano S, Tsuji S, Tsuji M,
Sawaoka H, Kimura Y, Shiozaki H and Hori M: Cyclooxygenase-2
overexpression enhances lymphatic invasion and metastasis in human
gastric carcinoma. Am J Gastroenterol. 94:451–455. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Narumiya S, Tanji M and Ishizaki T: Rho
signaling, ROCK and mDia1, in transformation, metastasis and
invasion. Cancer Metastasis Rev. 28:65–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wong CM and Ng IO: Molecular pathogenesis
of hepatocellular carcinoma. Liver Int. 28:160–174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang M, Wang XJ and Liu BR: Effect of
shRNA targeted against RhoA on proliferation and migration of human
colonic cancer cells. Int J Clin Exp Pathol. 8:7040–7044.
2015.PubMed/NCBI
|
|
40
|
He X, Qian Y, Cai H and Wang Z: The effect
of RhoC siRNA on the invasiveness and proliferation of human
cervical cancer cell line SiHa cells. J Huazhong Univ Sci Technolog
Med Sci. 28:665–669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hakem A, Sanchez-Sweatman O, You-Ten A,
Duncan G, Wakeham A, Khokha R and Mak TW: RhoC is dispensable for
embryogenesis and tumor initiation but essential for metastasis.
Genes Dev. 19:1974–1999. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li NF, Gemenetzidis E, Marshall FJ, Davies
D, Yu Y, Frese K, Froeling FE, Woolf AK, Feakins RM, Naito Y, et
al: RhoC interacts with integrin α5β1 and enhances its trafficking
in migrating pancreatic carcinoma cells. PLoS One. 8:0e815752013.
View Article : Google Scholar
|
|
43
|
Kleer CG, van Golen KL, Zhang Y, Wu ZF,
Rubin MA and Merajver SD: Characterization of RhoC expression in
benign and malignant breast disease: a potential new marker for
small breast carcinomas with metastatic ability. Am J Pathol.
160:579–584. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pillé JY, Denoyelle C, Varet J, Bertrand
JR, Soria J, Opolon P, Lu H, Pritchard LL, Vannier JP, Malvy C, et
al: Anti-RhoA and anti-RhoC siRNAs inhibit the proliferation and
invasiveness of MDA-MB-231 breast cancer cells in vitro and in
vivo. Mol Ther. 11:267–274. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yu YF, Zhang Y, Shen N, Zhang RY and Lu
XQ: Effect of VEGF, P53 and telomerase on angiogenesis of gastric
carcinoma tissue. Asian Pac J Trop Med. 7:293–296. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
van Golen KL, Wu ZF, Qiao XT, Bao L and
Merajver SD: RhoC GTPase overexpression modulates induction of
angiogenic factors in breast cells. Neoplasia. 2:418–425. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fukata M and Kaibuchi K: Rho-family
GTPases in cadherin-mediated cell-cell adhesion. Nat Rev Mol Cell
Bio. 2:887–897. 2001. View Article : Google Scholar
|
|
48
|
Kidera Y, Tsubaki M, Yamazoe Y, Shoji K,
Nakamura H, Ogaki M, Satou T, Itoh T, Isozaki M, Kaneko J, et al:
Reduction of lung metastasis, cell invasion, and adhesion in mouse
melanoma by statin-induced blockade of the Rho/Rho-associated
coiled-coil-containing protein kinase pathway. J Exp Clin Cancer
Res. 29:1272010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bernard A, Gao-Li J, Franco CA, Bouceba T,
Huet A and Li Z: Laminin receptor involvement in the
anti-angiogenic activity of pigment epithelium-derived factor. J
Biol Chem. 284:10480–10490. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mahtabifard A, Merritt RE, Yamada RE,
Crystal RG and Korst RJ: In vivo gene transfer of pigment
epithelium-derived factor inhibits tumor growth in syngeneic murine
models of thoracic malignancies. J Thorac Cardiov Surg. 126:28–38.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Takenaka K, Yamagishi S, Jinnouchi Y,
Nakamura K, Matsui T and Imaizumi T: Pigment epithelium-derived
factor (PEDF)-induced apoptosis and inhibition of vascular
endothelial growth factor (VEGF) expression in MG63 human
osteosarcoma cells. Life Sci. 77:3231–3241. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang L, Chen J, Ke Y, Mansel RE and Jiang
WG: Expression of pigment epithelial derived factor is reduced in
non-small cell lung cancer and is linked to clinical outcome. Int J
Mol Med. 17:937–944. 2006.PubMed/NCBI
|
|
53
|
Huang X, Chen L, Fu G, Xu H and Zhang X:
Decreased expression of pigment epithelium-derived factor and
increased microvascular density in ovarian endometriotic lesions in
women with endometriosis. Eur J Obstet Gynecol Reprod Biol.
165:104–109. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen J, Ye L, Zhang L and Jiang WG: The
molecular impact of pigment epithelium-derived factor, PEDF, on
lung cancer cells and the clinical significance. Int J Oncol.
35:159–166. 2009.PubMed/NCBI
|
|
55
|
Abe R, Fujita Y and Yamagishi S:
Angiogenesis and metastasis inhibitors for the treatment of
malignant melanoma. Mini Rev Med Chem. 7:649–661. 2007. View Article : Google Scholar : PubMed/NCBI
|