Introduction
At present, tyrosine kinase inhibitors are often
used for the treatment of chronic myeloid leukaemia (CML). However,
certain patients with CML develop the acute phase of this disease
due to drug resistance (1). To
develop a rational treatment schedule and minimize drug resistance
in the treatment of CML, additional investigation with respect to
the pathogenic mechanism is required.
The tyrosine phosphatase SHP1 is a key negative
regulator of intracellular signalling. Numerous studies have
revealed that the expression of SHP1 is low in malignant lymphoma,
acute leukaemia, multiple myeloma and other haematological
malignancies, including myelodysplastic syndrome. However, the SHP1
gene was predominantly observed to be in a highly methylated state.
Therefore, it was hypothesised that the methylation of the SHP1
gene serves an important role in the pathogenesis of malignant
haematological diseases (2).
DNA methylation is an important epigenetic
modification. DNA methyltransferases serve a role in gene
methylation. At present, there are 3 known DNA methyltransferases
(3): DNA methyltransferase 1 (DNMTl),
DNMT2 and DNMT3a/b.
EZH2 is a member of the polycomb group (PcG) gene
family. Enhancer of zeste homolog 2 (EZH2) has been identified as
key epigenetic regulator involved in transcriptional repression in
haematological diseases (4). In
mantle cell lymphoma, EZH2 has been identified to function in the
regulation of homeobox (HOX) genes, and EZH2 activity may lead to
high methylation levels of the HOX gene promoter region and
extended gene silencing (5–7). This modification is associated with the
onset of mantle cell lymphoma.
At present, there is limited research on the
associations between DNMT1 and EZH2 activity and SHP1 methylation
in CML. The present study investigated the association between
these factors and SHP1 methylation via chromatin
immunoprecipitation (ChIP), to identify the roles of DNMT1 and EZH2
in the process of blast crisis in CML.
Materials and methods
Patients
A total of 60 patients with CML treated at the
Affiliated Hospital of Hebei University (Baoding, China) from March
2014 to June 2015 were included in the present study. Among these
patients, 35 were male and 25 were female, and the median age of
the patients was 46 years with a range of 29–65 years. A total of
10 healthy donors were included as a control group. Subsequent to
informed consent being obtained from all of the patients,
mononuclear cells were collected by bone marrow aspiration. This
study was approved by the ethics committee of the Affiliated
Hospital of Hebei University (Baoding, China).
Cell culture and treatment
The CML cell line K562 (Beijing Institute for Cancer
Research, Beijing, China) was maintained in the laboratory of the
Department of Hematology of the Second Hospital of Hebei Medical
University (Shijiazhuang, China). The cells were cultured in
RPMI-1640 medium supplemented with 100 U/ml penicillin, 100 U/ml
streptomycin and 10% foetal bovine serum. The cell lines were
passaged every 2 to 3 days. The K562 cells were seeded at a density
of 1×106 cells/ml in 25 cm2 culture flasks,
then treated with 5-aza-2′-deoxycytidine (decitabine; Sigma
Aldrich; Merck KGaA, Darmstadt, Germany) or the inhibitor
3-deazaneplanocin A (DZNep; Sigma-Aldrich; Merck KGaA)
Subsequently, the cells were harvested and used for western blot
analysis, reverse transcription quantitative polymerase chain
reaction (RT-qPCR), methylation-specific (MSP) PCR and ChIP.
Western blot analysis
The protein samples were separated on a 7.5–15%
SDS-PAGE gel, and transferred to a polyvinylidene fluoride
membrane. Subsequent to blocking of the non-specific binding sites
with 5% non-fat milk in TBS-Tween 20 (T; 20 mmol/l Tris-HCl, pH
7.4, 150 mmol/l NaCl and 0.1% T) for 60 min, the membranes were
incubated overnight at 4°C, with primary antibodies at 1:1,000
dilution. Anti-DNMT1 (#ab13537) and anti-EZH2 (#ab3748) antibodies
were purchased from Abcam (Cambridge, UK) and an anti-SHP1
(#sc52885) antibody was purchased from Santa Cruz Biotechnology,
Inc., (Dallas, TX, USA). The membranes were then probed with a
horseradish peroxidase-conjugated secondary antibody (#sc2789;
dilution, 1:5,000; Santa Cruz Biotechnology, Inc.) for 60 min at
37°C. Subsequent to washing 3 times with TBS-T, the membranes were
developed using an enhanced chemiluminescence detection system
(LI-COR Biosciences, Lincoln, NE, USA).
RT-qPCR
The total RNA from the CML patient samples and K562
cell lines was extracted using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the protocol of the
manufacturer. Then, complementary (c) DNA was synthesized using an
All-in-one™ First-Strand cDNA Synthesis kit (GeneCopoeia
Inc., Rockville, MD, USA) according to the manufacturer's
protocol.
qPCR was then performed using an ABI7500 real-time
PCR detection system (Applied Biosystems; Thermo Fisher Scientific,
Inc. Waltham, MA, USA) with gene-specific primers. A β-actin cDNA
fragment was used as an internal control. The sequences of all
primers used in the RT-qPCR are provided in Table I. The PCR reaction contained 12 µl of
Platinum SYBR-Green qPCR SuperMix-UDG (Invitrogen; Thermo Fisher
Scientific, Inc.), 1 µl cDNA and 0.5 µl of the forward and reverse
primers. The PCR protocol was: 95°C for 2 min, followed by 40
cycles of 95°C for 15 sec, 60°C for 30 sec and 68°C for 45 sec. The
procedure was repeated 3 times and the 2−ΔΔCq method
(8) was performed to quantify the
gene expression level relative to the β-actin cDNA.
 | Table I.Primers for reverse transcription
quantitative polymerase chain reaction amplification. |
Table I.
Primers for reverse transcription
quantitative polymerase chain reaction amplification.
| Primers | Sequence |
|---|
| SHP1 |
|
Sense |
5′-CACCATCATCCACCTCAAGT-3′ |
|
Antisense |
5′-TCTCAGCACAAGAAACGTC-3′ |
| DNMT1 |
|
Sense |
5′-TACCTGGACGACCCTGACCTC-3′ |
|
Antisense |
5′-CGTTGGCATCAAAGATGGACA-3′ |
| EZH2 |
|
Sense |
5′-TTCATGCAACACCCAACACT-3′ |
|
Antisense |
5′-GGGCCTGCTACTGTTATTGG-3′ |
| β-actin |
|
Sense |
5′-TGACGTGGACATCCGCAAAG-3′ |
|
Antisense |
5′-CTGGAAGGTGGACAGCGAGG-3′ |
MSP
Genomic DNA from K562 cells and patients with CML
isolated using the Rapid DNA Extraction kit (Bo Maide Biotechnology
Co., Ltd., Beijing, China) was modified, via bisulphite treatment
with a DNA methylation modification kit (Zymo Research Corp.,
Irvine, CA, USA). The genomic DNA was then amplified by PCR, using
2 sets of SHP1 promoter-specific primer pairs that recognized
either a methylated or an unmethylated CpG sequence. The sequences
of all primers used in the MSP PCR are provided in Table II. The PCR reaction mixes contained
2.5 µl 10X Reaction Buffer, 1 µl extracted DNA, 1 µl of 5 µM
forward and reverse primers, 2 µl of a 2.5 mM dNTP mix, 2 µl
MgCl2 and 0.5 U of DNA polymerase (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The thermocycler settings were as
follows: 95°C for 10 min, followed by 38 cycles of 95°C for 30 sec,
54°C for 30 sec and 72°C for 30 sec. The reaction products were
analysed by electrophoresis with an Invitrogen DNA ladder (Thermo
Fisher Scientific, Inc.) and 10,000X GelRed™ Nucleic
Acid Gel Stain (Biotium, Inc., Hayward, CA, USA).
| Table II.Primers for methylation-specific
polymerase chain reaction. |
Table II.
Primers for methylation-specific
polymerase chain reaction.
| Primers | Sequence | Length |
|---|
| M-MSP |
|
Sense |
5′-GAACGTTATTATAGTATAGCGTTC-3′ |
|
|
Antisense |
5′-TCACGCATACGAACCCAAACG-3′ | 159 bp |
| U-MSP |
|
Sense |
5′-GTGAATGTTATTATAGTATAGTGTTTGG-3′ |
|
| Antisense |
5′-TTCACACATACAAACCCAAACAAT-3′ | 162 bp |
ChIP
The ChIP assay was performed using a
SimpleChIP® Enzymatic Chromatin Immunoprecipitation kit
(Cell Signaling Technology, Inc., Danvers, MA, USA) and antibodies
targeting DNMT1, EZH2 and rabbit IgG, as according to the
manufacturer's protocol. The K562 cells were seeded at a density of
1×106 cells/ml in 25 cm2 culture flasks, then
incubated alone, with decitabine (50 µM) or with DZNep (10 µM) and
grown to a final count of 5–10×107 cells for each ChIP
experiment. Formaldehyde was added to the cells to a final
concentration of 1% and the cells were re-suspended in SDS lysis
buffer and immunoprecipitated. The resulting complexes were
pelleted with protein A agarose and eluted with elution buffer
containing 50 mM Tris-HCl, 10 mM EDTA and 1% SDS. Histone-DNA
crosslinks were reversed in 5 M sodium chloride, and DNA was
recovered via phenol/chloroform extraction. The immunoprecipitated
DNA and input samples were analysed by PCR. The following primers
for the SHP1 gene promoter were used: Forward,
5′-ATGATAAAGATAGCCCCTGTT-3′; reverse, 5′-TCATCGAGTGAGTCCTGCTG-3′.
The PCR mixes contained 2 µl DNA, 1 µl of 5µM forward and reverse
primers, 2 µl of a 2.5 mM dNTP mix, 2 µl MgCl2 and 0.5 U
of DNA polymerase (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The thermocycler settings were as follows: 94°C for 10 min,
followed by 32 cycles of 94°C for 30 sec, 59°C for 30 sec and 72°C
for 30 sec. The reaction products were then analyzed by
electrophoresis as previously described.
Statistical analysis
The values were presented as the mean ± standard
deviation. Significant differences were assessed using SAS 9.1.3
statistical software (SAS Institute, Inc., Cary, NC, USA). First,
normality and variance homogeneity tests of the count data were
performed. The data were then subjected to analysis of variance or
the independent-samples t test. The clinical variables were
analysed using the χ2 test and Fisher's exact test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Expression of SHP1 and the methylation
of the promoter region of SHP1 in patients with CML
The present study detected the protein expression of
SHP1 in the mononuclear cells from the bone marrow of patients with
CML by western blot analysis. The results demonstrated that the
protein expression of SHP1 increased in patients with chronic phase
CML (CML-CP) and normal control (NC) subjects compared to patients
with accelerated phase CML (CML-A) and patients with blast phase
CML (CML-BP; P<0.05). There was no significant difference in the
levels of SHP1 protein expression between NC subjects and patients
with CML-CP (P>0.05) or between CML-AP and patients with CML-BP,
as demonstrated in Fig. 1A.
Methylation of SHP1 was detected in 7/33 patients with CML-CP,
positive rate of 21.2%. However, methylation of the SHP1 gene was
detected in all patients with advanced stage CML, including
patients with CML-AP and CML-BP. The positive rate for SHP1 gene
methylation in patients with advanced stage CML was significantly
higher compared with patients with CML-CP (P<0.01).
Specifically, 4 patients with CML-AP (40%) and 8 patients with
BP-CML (47%) exhibited complete SHP1 promoter methylation, and this
difference was not statistically significant (P>0.05), as
illustrated in Fig. 1B.
High expression of EZH2 and DNMT1 in
K562 cells and in patients with advanced CML
RT-qPCR analysis revealed that the relative mRNA
levels of DNMT1 were 3.54±1.23, 1.31±0.18, 1.75±0.53, 2.76±0.78,
and 3.05±1.23 in the samples from K562 cells, NC donors, and
patients with CML-CP, CML-AP and CML-BP, respectively. The mRNA
levels of DNMT1 were significantly higher in the samples from K562
cells and patients with CML-AP and CML-BP than in those from NC
subjects and patients with CML-CP (P<0.05). No significant
difference in the DNMT1 mRNA levels between K562 cells and patients
with CML-AP or CML-BP or between NC subjects and patients with
CML-CP was observed (P>0.05), as demonstrated in Table III.
| Table III.Relative expression of DNMT1 and EZH2
in patients with CML and normal control. |
Table III.
Relative expression of DNMT1 and EZH2
in patients with CML and normal control.
| Groups | n | Relative levels of
EZH2 RNA | Relative levels of
DNMT1 RNA |
|---|
| NC | 10 | 1.12±0.08 | 1.31±0.18 |
| CML-CP | 33 | 1.25±0.33 | 1.75±0.53 |
| CML-AP | 10 | 1.99±0.78 | 2.76±0.78 |
| CML-BP | 17 | 2.15±1.01 | 3.05±1.23 |
RT-qPCR analysis demonstrated that the relative mRNA
levels of EZH2 were 2.54±1.23, 1.12±0.08, 1.25±0.33, 1.99±0.78, and
2.15±1.01 in the samples from the K562 cells, NC donors, and
patients with CML-CP, CML-AP, and CML-BP, respectively. The mRNA
levels of EZH2 were significantly higher in samples from K562 cells
and patients with CML-AP and CML-BP compared with the samples from
NC subjects and patients with CML-CP (P<0.05). No significant
difference in the EZH2 mRNA levels between K562 cells and patients
with CML-AP or CML-BP or between NC subjects and patients with
CML-CP was observed (P>0.05), as demonstrated in Table III.
DNMT1 and SHP1 expression prior and
subsequent to treatment with decitabine
RT-qPCR analysis illustrated that the relative mRNA
levels of DNMT1 and SHP1 in K562 cells were 3.54±1.23 and
0.18±0.08, respectively. Treatment with decitabine led to a
decrease in the DNMT1 mRNA level to 1.55±0.61 (P<0.05), but
significantly increased the SHP1 expression level to 1.08±0.20
(P<0.05), as demonstrated in Fig.
2A.
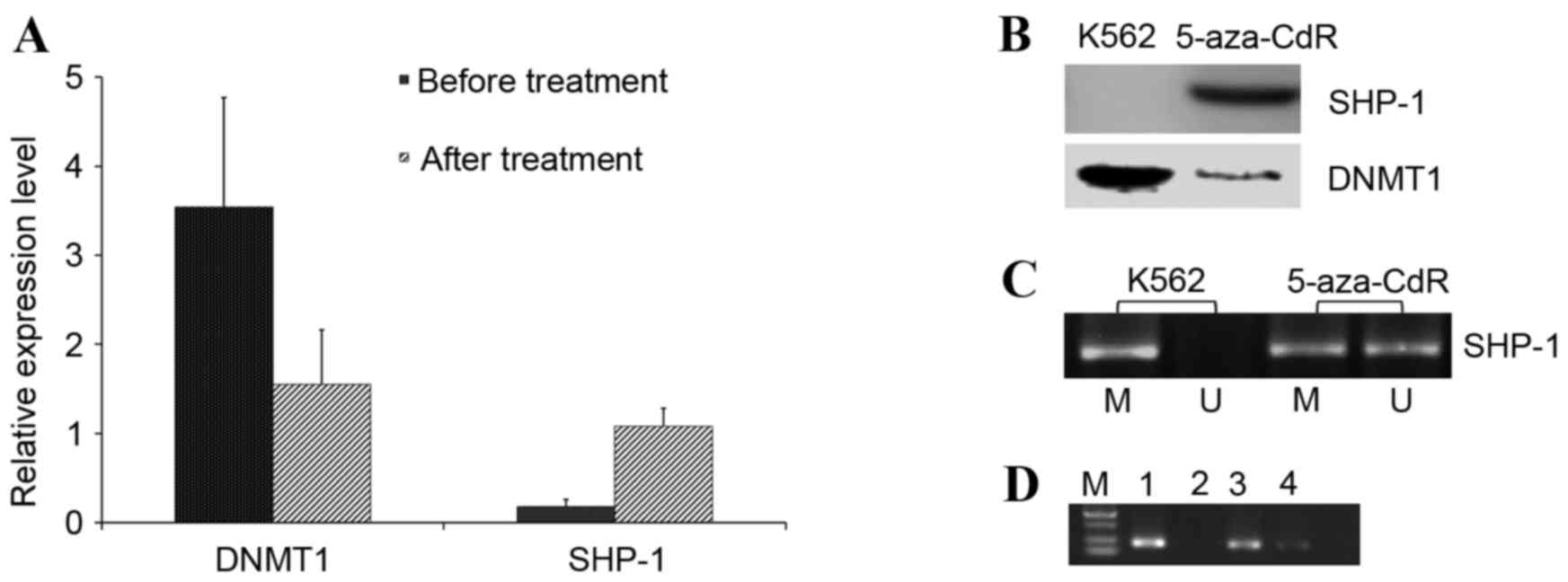 | Figure 2.Effect of decitabine on K562 cells.
(A) Relative expression level of DNMT1 and SHP1 mRNA. Error bars
represent standard deviation. (B) A western blot to demonstrate the
relative expression of SHP1 and DNMT1 proteins. (C) Extent of CpG
methylation of the SHP1 promoter, as determined with PCR using
methylation-specific primers. (D) The extent of DNMT1 binding to
the SHP-1 promoter. Cell lysates were examined in the chromatin
immunoprecipitation assay. U, unmethylated-specific primers; (C) M,
methylated-specific primers; (D) M, marker; 1, input; 2, negative
control; 3, prior to treatment with decitabine; 4, subsequent to
treatment with 5-aza-2′-deoxycytidine (decitabine); decitabine,
5-aza-2′-deoxycytidine; SHP1, Src homology 2 domain-containing
phosphatase-1; DNMT1, DNA methyltransferase 1. |
A similar pattern of the levels of SHP1 and DNMT1
expression was observed at the protein level, as demonstrated in
Fig. 2B. In addition, subsequent to
the application of decitabine, the methylation level of the SHP1
gene was decreased in the K562 cells, from complete methylation to
partial methylation, as illustrated in Fig. 2C.
EZH2 and SHP1 expression prior and
subsequent to treatment with DZNep
RT-qPCR analysis demonstrated that the relative mRNA
levels of EZH2 and SHP1 in K562 cells were 2.54±1.23 and 0.18±0.08,
respectively. Treatment with DZNep led to a decrease in the EZH2
mRNA level to 1.80±0.62 (P<0.05) but a significant increase in
the relative mRNA level of SHP1 to 0.75±0.18 (P<0.05), as
illustrated in Fig. 3A. A similar
pattern of SHP1 and EZH2 expression was observed at the protein
level, as demonstrated in Fig. 3B.
Furthermore, treatment with DZNep led to the progressive
demethylation of SHP1, as illustrated in Fig. 3C. Overall, these data suggested that
decitabine and DZNep successfully reduced the level of CpG
hypermethylation, leading to a re-expression of SHP1 mRNA and
protein.
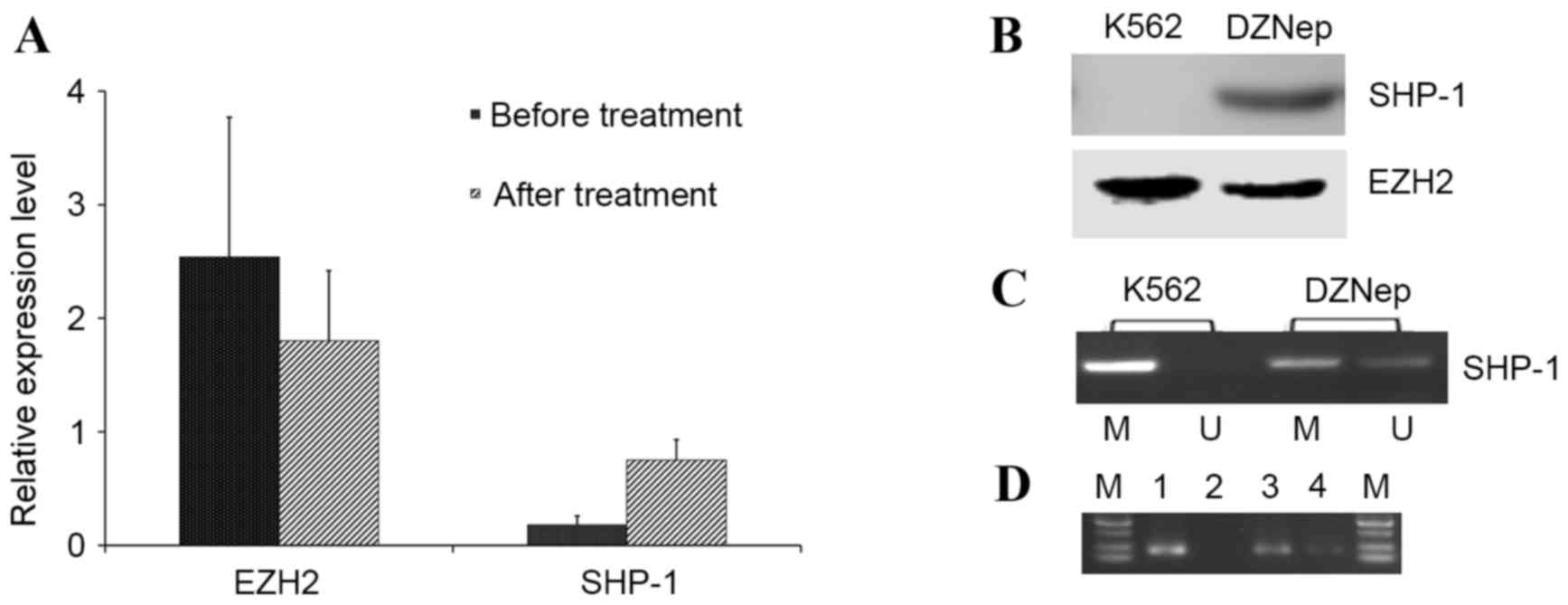 | Figure 3.Effect of DZNep on K562 cells. (A)
Relative expression level of EZH2 and SHP1 mRNA. Error bars
represent standard deviation. (B) A western blot to demonstrate the
relative expression of SHP1 and EZH2 proteins. (C) Extent of CpG
methylation of the SHP1 promoter, as determined with PCR using
methylation-specific primers. (D) The extent of EZH2 binding to the
SHP-1 promoter. Cell lysates were examined with a chromatin
immunoprecipitation assay. U, unmethylated-specific primers; (C) M,
methylated-specific primers; (D) M, marker; 1, input; 2, negative
control; 3, prior to treatment with DZNep; 4, subsequent to
treatment with DZNep. DZNep, 3-deazaneplanocin A; SHP1, Src
homology 2 domain-containing phosphatase-1; EZH2, enhancer of zeste
homolog 2. |
DNMT1 bound to the SHP1 gene promoter
region of K562 cells by ChIP
Subsequent to the addition of decitabine, the
interaction between DNMT1 and the SHP1 promoter region was
weakened. To demonstrate that DNMT1 is functionally involved in the
methylation of the SHP1 gene promoter and the inhibition of SHP1
gene expression, ChIP assays using anti-DNMT1 antibodies were
performed, and DNMT1 was revealed to be enriched in the SHP1
promoter region of the K562 cells. Next, drug intervention
experiments on K562 cells using the DNA methyltransferase inhibitor
decitabine were carried out. The expression of SHP1 increased in
the K562 cell line subsequent to decitabine treatment. Under these
conditions, the interaction between DNMT1 and the SHP1 gene
promoter region decreased based on ChIP, as illustrated in Fig. 2D. The methylation of the SHP1 promoter
region also decreased subsequent to decitabine treatment.
EZH2 bound to the SHP1 gene promoter
region in K562 cells by CHIP
Subsequent to the addition of DZNep, the interaction
between EZH2 and the SHP1 promoter region was weakened. To identify
the epigenetic control of SHP1 in K562 cells, the role of EZH2 in
the regulation of SHP1 expression was investigated. To evaluate DNA
methylation, ChIP assays were performed. In K562 cells, the SHP1
core region displayed enriched binding to EZH2. To confirm that
EZH2 regulates SHP1 expression, K562 cells were treated with DZNep.
Treatment with DZNep was revealed to increase SHP1 expression, and
the results of the ChIP assay demonstrated reduced binding
efficiency of EZH2 to the SHP1 promoter in the DZNep-treated cells,
as illustrated in Fig. 3D. In
addition, DZNep treatment led to decreased SHP1 methylation.
Discussion
CML is one of the most common types of malignant
haematological tumor. At present, imatinib is the first-line drug
for the treatment of CML. However, certain patients cannot afford
long-term imatinib therapy or are resistant to second-generation
kinase inhibitors. Therefore, it is necessary to explore novel
treatment approaches from the perspective of CML pathogenesis.
Methylation in the promoter regions of tumor
suppressor genes leads to tumor formation (9,10). The
tyrosine phosphatase SHP1 is a key negative regulator of
intracellular signalling and is hypothesised to be a tumor
suppressor gene. The methylation status of the SHP1 promoter is
associated with gene dormancy, which is an important mechanism in
the development of lymphoma and leukaemia. SHP1 protein expression
was completely absent or partially reduced in the majority of types
of lymphoma and leukaemia cell lines, such as YS2, IWA3, NK,
TomJim, MTI, EDS, and ATLIK (11,12). The
present study demonstrated the lack of SHP1 expression in K562
cells. However, the SHP1 gene promoter was highly methylated. The
degree of SHP1 promoter methylation increased with decreasing SHP1
expression in patients with CML-BP. In contrast, the degree of SHP1
promoter methylation decreased with increasing SHP1 expression in
patients with CML-CP. These data suggested that the SHP1 gene
methylation status in patients with CML-BP may be associated with
the disease progression of CML.
Decitabine, a methyltransferase inhibitor, has been
applied to other haematological diseases, such as myelodysplastic
syndromes (MDS) and acute myelogenous leukaemia (AML). Therefore,
the present study investigated the activity of methyltransferases
in CML. The expression of DNMT1 was revealed to be significantly
increased in patients with CML-BP and in K562 cells. The expression
of DNMT1 decreased subsequent to the application of decitabine.
Additionally, the degree of SHP1 promoter methylation decreased
with increasing SHP1 expression. Thus, it was hypothesised that
DNMT1 may be associated with SHP1 gene methylation and CML disease
progression. In the present study, based on ChIP assays, it was
revealed that DNMT1 binds to the SHP1 promoter region under control
conditions and that the interaction between DNMT1 and the SHP1 gene
promoter region and the methylation of SHP1 significantly decreased
following inhibitor treatment. These results indicated that a
methyltransferase interacts with the SHP1 gene promoter and is
involved in SHP1 gene methylation, thus affecting the blast crisis
of CML. The present study established a theoretical basis for the
treatment of CML with methyltransferase inhibitors.
In determining whether only the DNMT family
participates in the methylation of SHP1, it was demonstrated that
the PcG protein was one of the most important types of epigenetic
regulators involved in the inhibition of gene activity. To support
the future development of additional therapeutic targets for the
treatment of CML, EZH2 was investigated.
EZH2 is a member of the PcG histone
methyltransferase complex, which is a group of proteins associated
with histone methyltransferases that are involved in the inhibition
of transcription. The overexpression of the EZH2 gene is associated
with the progression of malignant tumors and poor prognosis
(13–15).
The EZH2 gene expression level in patients with
high-risk MDS or AML was significantly higher compared with
patients with low-risk MDS and in normal subjects (4). These results suggested that EZH2 serves
a role in the development of myeloid malignancies. Similar to the
observations in other types of malignancy, in mantle cell lymphoma
the chromatin modifier EZH2 is overexpressed in proliferating cells
and is associated with poor outcome (15–17).
EZH2 catalyses the trimethylation at lysine 27 of
histone H3 (H3K27me3) (18), which
serves as an anchorage point for the recruitment of additional PcG
proteins and contributes to the formation of a repressive chromatin
state. Consequently, EZH2 may indirectly inhibit target gene
expression. The majority of these target genes inhibit tumor
formation and regulate stem cell differentiation. Therefore,
silencing these genes may lead to tumor formation. Inhibiting EZH2
activity in mouse models has been demonstrated to completely
inhibit tumor growth (19). According
to the transcriptional inhibition of PcG, there are 1,000 silencing
target genes in human embryonic fibroblasts, which contribute to
embryonic development, including members of the Notch, Hox,
Hedgehog, Wnt, transforming growth factor (TGF) and fibroblast
growth factor (FGF) signalling pathways (17,20).
In solid tumors, there were a number of studies
concerning EZH2. For example, it was reported that MYC and EZH2
function as potent suppressors of levels of macrophage stimulating
1 (MST1) expression in human prostate cancer cells Pharmacological
and RNAi experiments revealed that MYC and EZH2 inhibit the
promoter activity of MST1 and thus silence expression, and that
EZH2 is a mediator of the MYC-induced silencing of MST1 (21).
EZH2 has been reported to directly control DNA
methylation by regulating the activity of DNA methyltransferases in
haematological malignancies. A ChIP study demonstrated that DNMT1,
DNMT3a, DNMT3b and EZH2 bound to the promoter region of MYT1, and
that the number of these interactions correlated with the
methylation status of MYT1. Similar experiments confirmed that
WNT1, KCNA1, and CNR1 were targeted by EZH2 (22).
In addition, it was revealed that EZH2 and DNMT3a
localized to the PTEN promoter region in the EOL-1 cell line, which
is resistant to imatinib, thus, participating in PTEN methylation.
PTEN acts as a negative regulatory gene, as decreasing PTEN
expression may cause cells to become drug-resistant (23). Additionally, in acute lymphoblastic
leukaemia, EZH2 and H3K27me3 were revealed to interact with the
PTEN promoter region, and these interactions are associated with
PTEN gene methylation. In summary, these data indicate that the PcG
family protein EZH2 may be involved in CML pathogenesis by
inhibiting the expression of the tumor suppressor gene PTEN
(24). Therefore, in haematological
diseases, the PTEN, WTY1 and HOX genes may be targets of EZH2. Gene
silencing induced by DNA methylation requires EZH2 and DNA
methylation enzymes (22,25).
At present, a small number of studies have focused
on the association between EZH2 and SHP1 gene methylation. Previous
studies have demonstrated that SHP1 is highly methylated in diffuse
large B cell lymphoma (DLBCL) (26,27). The
role of histone modifications was investigated via ChIP assays,
which demonstrated that the P2 region of SHP1 was associated with
the silencing of the histone mark H3K27me3 in DLBCL cells.
Treatment with DZNep, an inhibitor of EZH2, decreased the levels of
the H3K27me3 mark within the P2 region of SHP1, resulting in
re-expression of SHP1. The previous studies revealed novel
epigenetic mechanisms of SHP1 suppression in DLBCL. However,
additional studies are required to establish the effect of histone
modifications, particularly the EZH2-mediated histone modifications
on SHP1 in CML.
Based on the aforementioned previous data, including
the high methylation of the SHP1 gene in patients with CML, it was
hypothesised that EZH2 also participates in the process of SHP1
gene methylation. In the present study, a high level of methylation
of the SHP1 gene in patients with CML-BP was detected. The
methylation status of SHP1 decreased subsequent to the application
of an inhibitor of EZH2. Therefore, assays were performed on K562
cells prior and subsequent to treatment with an inhibitor of EZH2.
The SHP1 gene promoter was revealed to enrich binding to EZH2.
Subsequent to drug intervention, the interaction of EZH2 with the
promoter of SHP1 weakened, and the methylation status of SHP1
reduced. Therefore, it was hypothesised that EZH2 and DNMT1 are
involved in SHP1 gene methylation, which may be associated with the
progression of CML.
The present study demonstrates that EZH2 and DNMT1
interact with the SHP1 gene promoter and these interactions are
associated with the SHP1 gene methylation status, which may lead to
disease progression. The results of the present study may provide
novel therapeutic targets for CML treatment in the future.
Acknowledgements
The present study would sincerely like to thank Ms.
Yan Qin and Mr. Zheng LS for technical assistance, and Ms. Shi JH
for valuable discussion in the course of the study.
References
|
1
|
Cools J, Maertens C and Marynen P:
Resistance to tyrosine kinase inhibitors: Calling on extra forces.
Drug Resist Updat. 8:119–129. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sato H, Oka T, Shinnou Y, Kondo T, Washio
K, Takano M, Takata K, Morito T, Huang X, Tamura M, et al:
Multi-step aberrant CpG island hyper-methylation is associated with
the progression of adult T-cell Leukemia/lymphoma. Am J Pathol.
176:402–415. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheng X and Blumenthal RM: Mammalian DNA
methyltransferases: A structural perspective. Structure.
16:341–350. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu F, Li X, Wu L, Zhang Q, Yang R, Yang Y,
Zhang Z, He Q and Chang C: Overexpression of the EZH2, RINGl and
BMIl genes is common in myelodysplastic syndromes: Relation to
adverse epigenetic alteration and poor prognostic scoring. Ann
Hematol. 90:643–653. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nagel S, Venturini L, Marquez VE, Meyer C,
Kaufmann M, Scherr M, MacLeod RA and Drexler HG: Polycomb repressor
complex 2 regulates HOXA9 and HOXA10, activating ID2 in NK/T-cell
lines. Mol Cancer. 9:1512010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu X, Gong Y, Yue J, Qiang B, Yuan J and
Peng X: Cooperation between EZH2, NSPc1-mediated histone H2A
ubiquitination and Dnmt1 in HOX gene silencing. Nucleic Acids Res.
36:3590–3599. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kanduri M, Sander B, Ntoufa S,
Papakonstantinou N, Sutton LA, Stamatopoulos K, Kanduri C and
Rosenquist R: A key role for EZH2 in epigenetic silencing of HOX
genes in mantle cell lymphoma. Epigenetics. 8:1280–1288. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qi J, Zhu YQ, Luo J, Tao WH and Zhang JM:
Hypermethylation and regulation of expression of secreted
frizzled-related protein genes in colorectal tumor. Zhonghua Zhong
Liu Za Zhi. 29:842–845. 2007.(In Chinese). PubMed/NCBI
|
|
10
|
Reddy J, Shivapurkar N, Takahashi T,
Parikh G, Stastny V, Echebiri C, Crumrine K, Zöchbauer-Müller S,
Drach J, Zheng Y, et al: Differential methylation of genes that
regulate cytokine signaling in lymphoid and hematopoietic tumors.
Oncogene. 24:732–736. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Migone TS, Cacalano NA, Taylor N, Yi T,
Waldmann TA and Johnston JA: Recruitment of SH2 containing protein
tyrosinephosphatase SHP-1 to the interleukin 2 receptor; loss of
SHP-1 expression in human T-lymphotropic virus type I transformedT
cells. Proc Natl Acad Sci USA. 95:pp. 3845–3850. 1998; View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Uhm KO, Lee ES, Lee YM, Park JS, Kim SJ,
Kim BS, Kim HS and Park SH: Differential methylation pattern of
ID4, SFRP1, and SHP1 between acute myeloid leukemia and chronic
myeloid leukemia. J Korean Med Sci. 24:493–497. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chase A and Cross NC: Aberrations of EZH2
in cancer. Clin Cancer Res. 17:2613–2618. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
He IJ, Cai MY, Xu GL, Li JJ, Weng ZJ, Xu
DZ, Luo GY, Zhu SL and Xie D: Prognostic significance of
overexpression of EZH2 and H3k27me3 proteins in gastric cancer.
Asian Pac J Cancer Prev. 13:3173–3178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McCabe MT, Ott HM, Ganji G, Korenchuk S,
Thompson C, van Aller GS, Liu Y, Graves AP, Pietra A Della III,
Diaz E, et al: EZH2 inhibition as a therapeutic strategy for
lymphoma with EZH2-activating mutations. Nature. 492:108–112. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fiskus W, Wang Y, Sreekumar A, Buckley KM,
Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, et al:
Combined epigenetic therapy with the histone methyltransferase EZH2
inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor
panobinostat against human AML cells. Blood. 114:2733–2743. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Simon JA and Lange CA: Roles of the EZH2
histone methyltransferase in cancer epigenetics. Mutat Res.
647:21–29. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cao R, Tsukada Y and Zhang Y: Role of
Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol
Cell. 20:845–854. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wilson BG, Wang X, Shen X, McKenna ES,
Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH and
Roberts CW: Epigenetic antagonism between polycomb and SWI/SNF
complexes during oncogenic transformation. Cancer Cell. 18:316–328.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bracken AP, Dietrich N, Pasini D, Hansen
KH and Helin K: Genome-wide mapping of polycomb target genes
unravels their roles in cell fate transitions. Genes Dev.
20:1123–1136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kuser-Abali G, Alptekin A and Cinar B:
Overexpression of MYC and EZH2 cooperates to epigenetically silence
MST1 expression. Epigenetics. 9:634–643. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vire E, Brenner C, Deplus R, Blanchon L,
Fraga M, Didelot C, Morey L, van Eynde A, Bernard D, Vanderwinden
JM, et al: The polycomb group protein EZH2 directly controls DNA
methylation. Nature. 439:871–874. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nishioka C, Ikezoe T, Yang J, Udaka K and
Yokoyama A: Imatinib causes epigenetic alterations of PTEN gene via
upregulation of DNA methyltransferases and polycomb group proteins.
Blood Cancer J. 1:e482011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen JH: Epigenetic studies of mesenchymal
and B-cell leukemia. Beijing: Chinese Academy of Medical Sciences,
Peking Union Medical College; 2010
|
|
25
|
Sharma A, Heuck CJ, Fazzari MJ, Mehta J,
Singhal S, Greally JM and Verma A: DNA methylation alterations in
multiple myeloma as a model for epigenetic changes in cancer. Wiley
Interdiscip Rev Syst Biol Med. 2:654–669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Witzig TE, Hu G, Offer SM, Wellik LE, Han
JJ, Stenson MJ, Dogan A, Diasio RB and Gupta M: Epigenetic
mechanisms of protein tyrosine phosphatase 6 suppression in diffuse
large B-cell lymphoma: Implications for epigenetic therapy.
Leukemia. 28:147–154. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen X, Fu R, Wang Y, Song W, Ruan E, Qu
W, Wang H, Wang G, Song J, Wang X, et al: Plasma DNA methylation of
shp1 in patients with diffuse large B cell lymphoma. Zhonghua Yi
Xue Za Zhi. 94:1071–1075. 2014.(In Chinese). PubMed/NCBI
|















