Introduction
Maternal embryonic leucine zipper kinase (MELK) is a
member of the adenosine monophosphate-activated protein kinase
family of kinases. MELK was first cloned from the mouse egg and
preimplantation embryo (1), and is
expressed in progenitor cells and tumor stem cells of aggressive
cancers, including glioblastomas and breast cancers (2–4). MELK
promotes proliferation by phosphorylating cell division cycle 25B
and apoptosis signal-regulating kinase 1 (5,6) and MELK
induces apoptosis through phosphorylation of p53 at the Ser15 site
(7). In a number of studies, the
anti-apoptosis function of MELK was demonstrated through its
regulation of p53 expression or interaction with B cell lymphoma-G
(8,9).
Numerous studies have indicated that MELK is associated with tumor
prognosis; MELK expression is increased in clinical samples of
breast cancer or high-grade prostate cancer (10,11). At
present, numerous studies on MELK have focused on the tumorigenesis
stage, and to the best of our knowledge, there has been no study
investigating the function of MELK in tumor metastasis.
Tumor metastasis is a complex process, involving a
single tumor cell disassociating from the tumor mass, migrating in
the extracellular matrix, intravasating into the blood stream,
extravasating from the blood vessel and colonizing distant organs
(12). Epithelial-mesenchymal
transition (EMT) is an important step for the initiation of tumor
metastasis. When EMT occurs, epithelial tumor cells transition to
mesenchymal-like cells, lose the expression of intercellular
E-cadherin and acquire the expression of N-cadherin and α-smooth
muscle actin, allowing tumor cells to migrate and enter the blood
stream (13). A number of factors
regulate the EMT. Among these, transforming growth factor-β (TGF-β)
signaling is intensively studied. TGF-β signaling can upregulate
the expression of transcription factors including Snail, ZEB and
Twist, which repress the expression of E-cadherin and induce the
expression of N-cadherin and fibronectin (13).
In the present study, the lung cancer A549 cell is
utilized as an EMT model to observe the role of MELK in metastasis.
Knockdown of MELK was identified to promote EMT by upregulation of
TGF-β signaling.
Materials and methods
Cell culture and plasmids
The A549 cell line was bought from American Type
Culture Collection (Manassas, VA, USA), and was cultured in
Dulbecco's modified Eagle's medium/low glucose medium containing
10% fetal bovine serum (HyClone; GE Healthcare Life Sciences,
Logan, UT, USA), 2 mM glutamine, penicillin and streptomycin
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). A549 cells were
treated with TGF-β1 (R&D Systems, Inc., Minneapolis, MN, USA)
at a concentration of 5 ng/ml. The plasmid p3TP reporter gene,
hemagglutinin (HA)-Smad2, HA-Smad3 and HA-Smad4 were stored by the
central laboratory of Henan Provincial People's Hospital
(Zhengzhou, China). The plasmid Flag-MELK was constructed through
polymerase chain reaction (PCR) amplification (upstream primer,
3′-CGGGATCCATGAAAGATTATGATGAACTTC-5′ and downstream primer,
3′-CCCTCGAGTACCTTGCAGCTAGATAGGATG-5′), and ligation to plasmid
pCS4-3Flag.
RNAi interference
The sequences (sh1, AACCCAAGGGTAACAAGGATT and sh2,
CAGGCAAACAATGGAGGAT) for RNAi interference are as previously
described (14). The synthesized
oligonucleotides were annealed and constructed into the pLKO.1
plasmid (Addgene, Inc., Cambridge, MA, USA) (15). Following sequencing, the lentivirus
was packaged in 293T cells using package plasmids psPAX2 and PMD2 G
(Addgene, Inc.). The stable cell lines were built with shMock
(sequence, 5′-CCTAAGGTTAAGTCGCCCTCG-3′), shMELK1 and shMELK2
lentiviruses.
Western blot analysis
The cells were harvested in radioimmunoprecipitation
assay buffer, supplemented with proteinase cocktail (Roche
Diagnostics, Basel, Switzerland), 50 mM sodium orthovanadate and 10
mM sodium fluoride. The protein concentration was determined using
the bicinchoninic acid kit (Thermo Fisher Scientific, Inc.). The
antibodies for western blot analysis were MELK (dilution, 1:200;
cat. no. sc-48035; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA,), E-cadherin (dilution, 1:400; cat. no. sc-7870; Santa Cruz
Biotechnology, Inc.), p-Smad2 (dilution, 1:1,000; cat. no. 3108;
Cell Signaling Technology, Inc., Danvers, MA, USA), Smad2
(dilution, 1:1,000; cat. no. 3122; Cell Signaling Technology, Inc.)
and b-Actin (dilution, 1:2,000; cat. no. sc-7210; Santa Cruz
Biotechnology, Inc.).
Reverse transcription-polymerase chain
reaction (RT-PCR)
The A549 cells were stimulated with TGF-β1 (5 ng/ml)
for different periods of time (1, 3, 6, 12 and 24 h), and the cells
were lysed in TRIzol (Thermo Fisher Scientific, Inc.). The RNA was
extracted following the protocol for TRIzol (16). Following RNA quantification, cDNA was
obtained by reverse-transcription using RT master mix with the gDNA
remover kit (cat. no. FSQ-301; Toyobo Co., Ltd., Osaka, Japan). PCR
was performed using the thunderbird SYBR qPCR mix kit (cat. no.
QPS-201; Toyobo Co., Ltd.) on the iCycler (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) instrument. The 2−ΔΔCq method
was used to analyze the data (17).
Proliferation assay
The shMock, shMELK1, and shMELK2 cells were seeded
onto 96-well plates. A total of 2×103 cells were used in
each well. At 24, 48, 72 and 96 h, the absorbance at 492 nm was
read using a microplate reader (Elx808; Norgen Biotek Corp.,
Thorold, ON, Canada) for the quantification of cell numbers.
Wound healing assay
The shMock and shMELK cells were counted using a
hemocytometer plate under a microscope (DMi1; Leica Microsystems
GmbH, Wetzlar, Germany) using ×20 magnification. A total of
3×105 cells were seeded onto a 6-well plate. Following
overnight cell culture at 37°C, the cells reached confluency. The
200 µl pipette tips were used to scratch the cell layer. Following
scratching, images were captured at 0 h immediately, and TGF-β1 (5
ng/ml) was added to the medium. After 24 h, the cell scratch was
observed, and the images were captured as shown in Fig. 1D. The distance of the scratch in the
images was measured using Photoshop software (Adobe, San Jose, CA,
USA). The experiments were performed in triplicate wells.
Reporter gene assay
The p3TP reporter gene was used to evaluate TGF-β
signaling activity. Briefly, 8×104 cells were seeded
onto a 12-well plate. In total, 1 µg plasmids were transfected into
each well using Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.). After 24 h, transfected cells were treated with
TGF-β1 (5 ng/ml) for another 24 h. The reporter gene activity was
measured using the dual-luciferase reporter assay system (Promega
Corporation, Madison, WI, USA). Triplicate wells for each plasmid
combination were performed.
Immunoprecipitation
For immunoprecipitation, HEK293 cells were seeded
onto 60 mm dishes at a density of 8×105 cells per dish.
Following overnight incubation at 37°C, the plasmid combinations (4
µg per dish) were transfected. After three days, the cells were
harvested in Nonidet-P40 buffer (0.1% NP40). A total of 300 µg of
cell lysate was used for the immunoprecipitation assay. In each
tube, 2 µg of HA antibody (dilution, 1:40; cat. no. sc-7392; Santa
Cruz Biotechnology, Inc.) was added. Following overnight incubation
on an agitator in a cold room, protein A/G beads (cat. no. sc-2003;
Santa Cruz Biotechnology, Inc.) were added to pull down the
immuno-complex. The pellets were washed with washing buffer (0.9%
NaCl including 0.01% NP40) three times, and then loaded onto
SDS-PAGE gel. Flag antibodies (dilution, 1:100; cat. no. F3165;
Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) were used to
detect signals.
Statistical analysis
The reporter gene assay was repeated in triplicate.
The statistical software SPSS v19.0 was used (IBM, Armonk, NY,
USA). Student's t-test was used to analyze differences between
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
MELK is downregulated by TGF-β at the
protein and mRNA level
In order to study the role of MELK in EMT, a A549
lung cancer cell model was employed, which can be induced to
transition between epithelial cells and mesenchymal-like cells, by
TGF-β stimulation. Numerous effectors, including interferon
regulatory factor-1 and thyroid transcription factor-1, are
involved in this process (18,19).
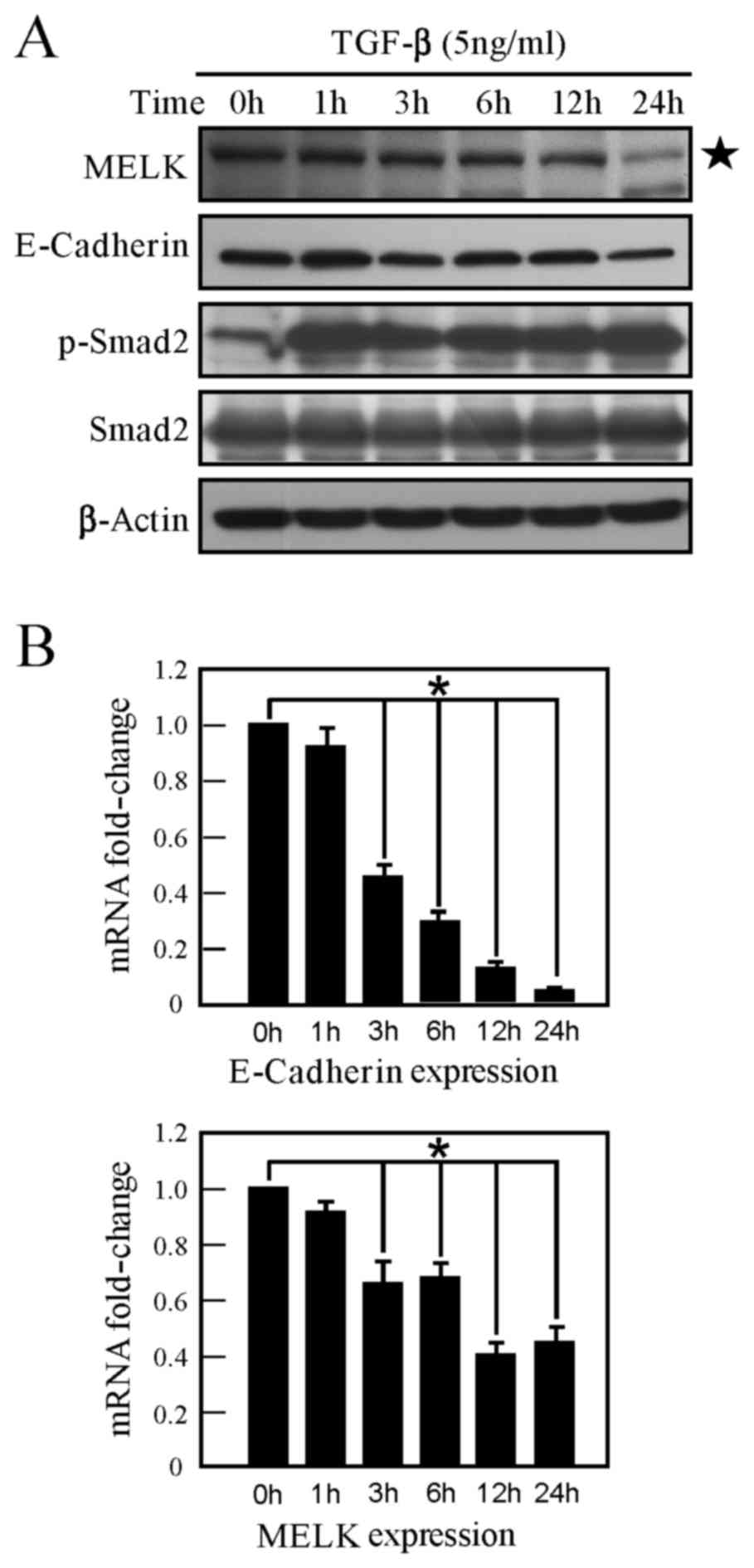
Following TGF-β stimulation of A549 cells for 24 h, it was
identified that TGF-β induces the phosphorylation of Smad2, and the
expression of E-cadherin (a marker of EMT) is decreased (Fig. 2A). The present study identified that
MELK expression is downregulated in A549 cells following TGF-β
stimulation (Fig. 2A). Similarly, the
mRNA level of MELK was examined, and MELK mRNA expression was
revealed to also be inhibited by TGF-β (Fig. 2B). Therefore, TGF-β can inhibit MELK
expression, indicating that MELK may exhibit a number of functions
in TGF-β signaling.
Downregulation of MELK promotes
EMT
Lentivirus-mediated RNA interference was used to
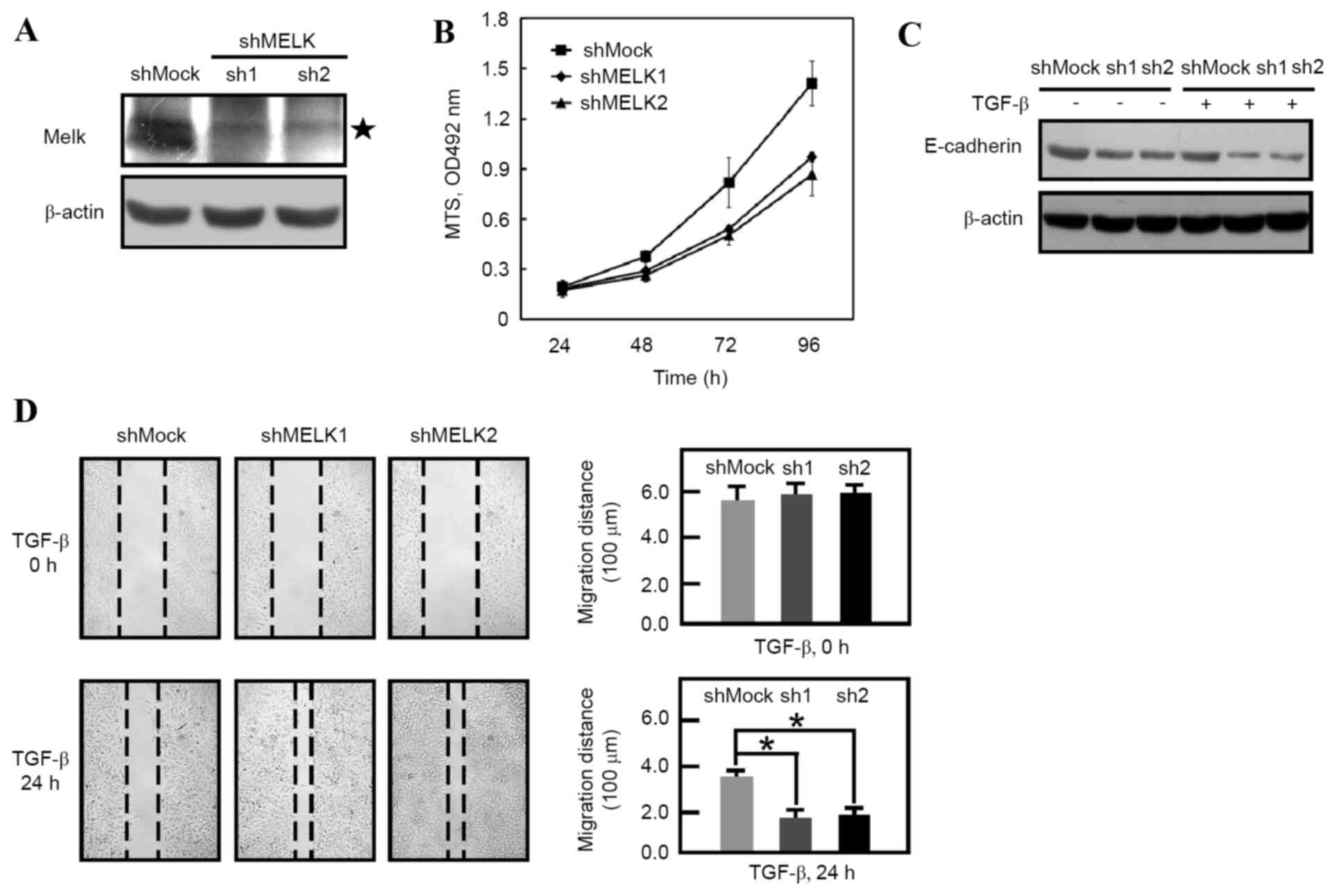
knockdown the expression of MELK. Western blot analysis data
revealed that MELK expression was decreased in MELK knockdown cells
(Fig. 1A). Consistent with a previous
study (18), the knockdown of MELK
inhibited the proliferation of shMELK cells compared with shMock
cells (Fig. 1B). shMock and shMELK
cells were then treated with or without TGF-β for 24 h. It was
identified that the E-cadherin molecules were decreased following
MELK knockdown (Fig. 1C; lanes 1, 2
and 3). Following TGF-β stimulation, it was revealed that
E-cadherin was further downregulated in shMock and shMELK (sh1 and
sh2) cells (Fig. 1C). These results
indicated that knockdown of MELK promotes EMT. Furthermore, a wound
healing experiment was employed in order to observe the migration
of shMock and shMELK cells. In the presence of TGF-β, shMELK (sh1
and sh2) cells were revealed to migrate faster than shMock cells
(Fig. 1D). These data suggested that
MELK is an important molecule in regulating EMT transition.
Knockdown of MELK enhances TGF-β
signaling activity
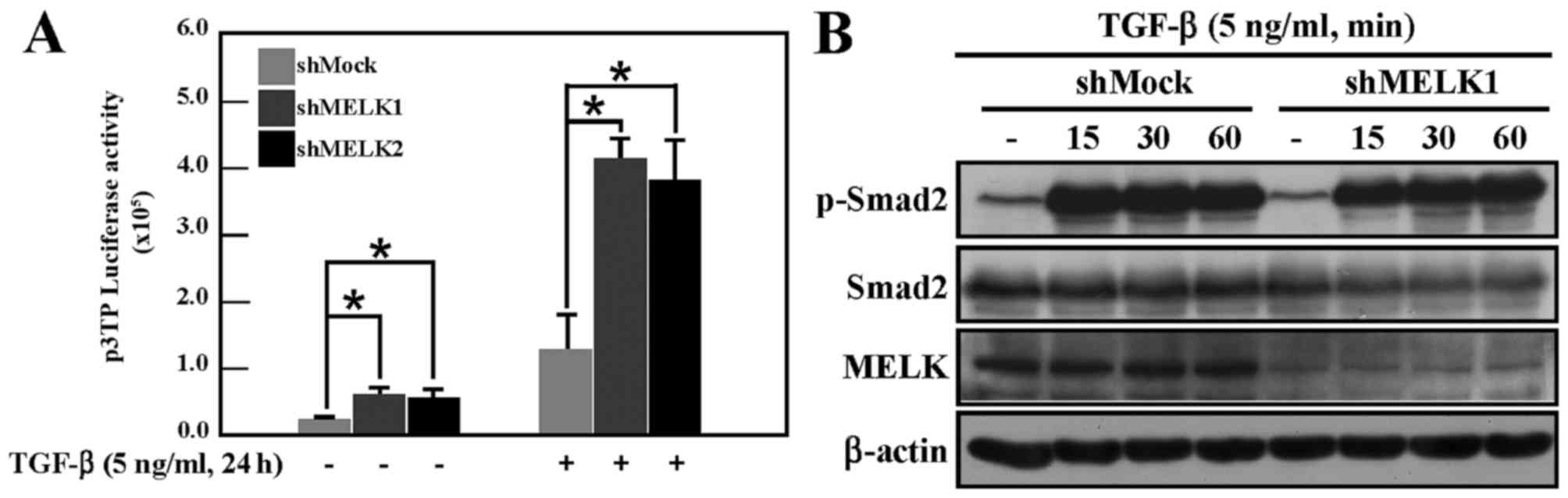
The marked effect of MELK on EMT suggested that MELK
may regulate TGF-β signaling activity. To test this hypothesis, a
luciferase reporter gene assay was performed to analyze TGF-β
signaling in shMock and shMELK cells, using the p3TP reporter gene,
which reflects the activity of TGF-β signaling (20). Under normal culture conditions, the
activity of p3TP in shMELK cells was increased compared with the
activity of shMock cells (Fig. 3A).
Following TGF-β stimulation, the activity in shMELK cells was
markedly increased compared with shMock cells (Fig. 3A). TGF-β signaling was then examined
by detecting the phosphorylation of Smad2 in shMock and shMELK1
cells. Data demonstrated that knockdown of MELK did not affect the
phosphorylation of Smad2 (Fig. 3B).
Smad2 phosphorylation in shMELK2 cells was also examined, and no
difference between shMock and shMELK2 cells was observed (data not
shown). This suggested that knockdown of MELK may affect the
activity of TGF-β signaling in other ways, not through regulating
the phosphorylation of Smad2 protein.
MELK interacts with Smad proteins and
represses the activity of TGF-β signaling
In order to study how MELK regulates TGF-β
signaling, it was firstly examined whether MELK can interact with
Smad proteins. Using immunoprecipitation experiments, MELK was
shown to interact with Smad2, Smad3 and Smad4. When MELK was used
to immunoprecipitate Smad proteins, clear bands of Smad proteins
were produced (Fig. 4A). The reporter
gene assay was also used to analyze whether interaction between
MELK and Smads could regulate TGF-β signaling. As shown in Fig. 4B, overexpression of MELK can inhibit
Smad2 or Smad3-induced reporter gene activity. Therefore, data from
the present study revealed that the interaction between MELK and
Smad proteins repressed TGF-β signaling activity.
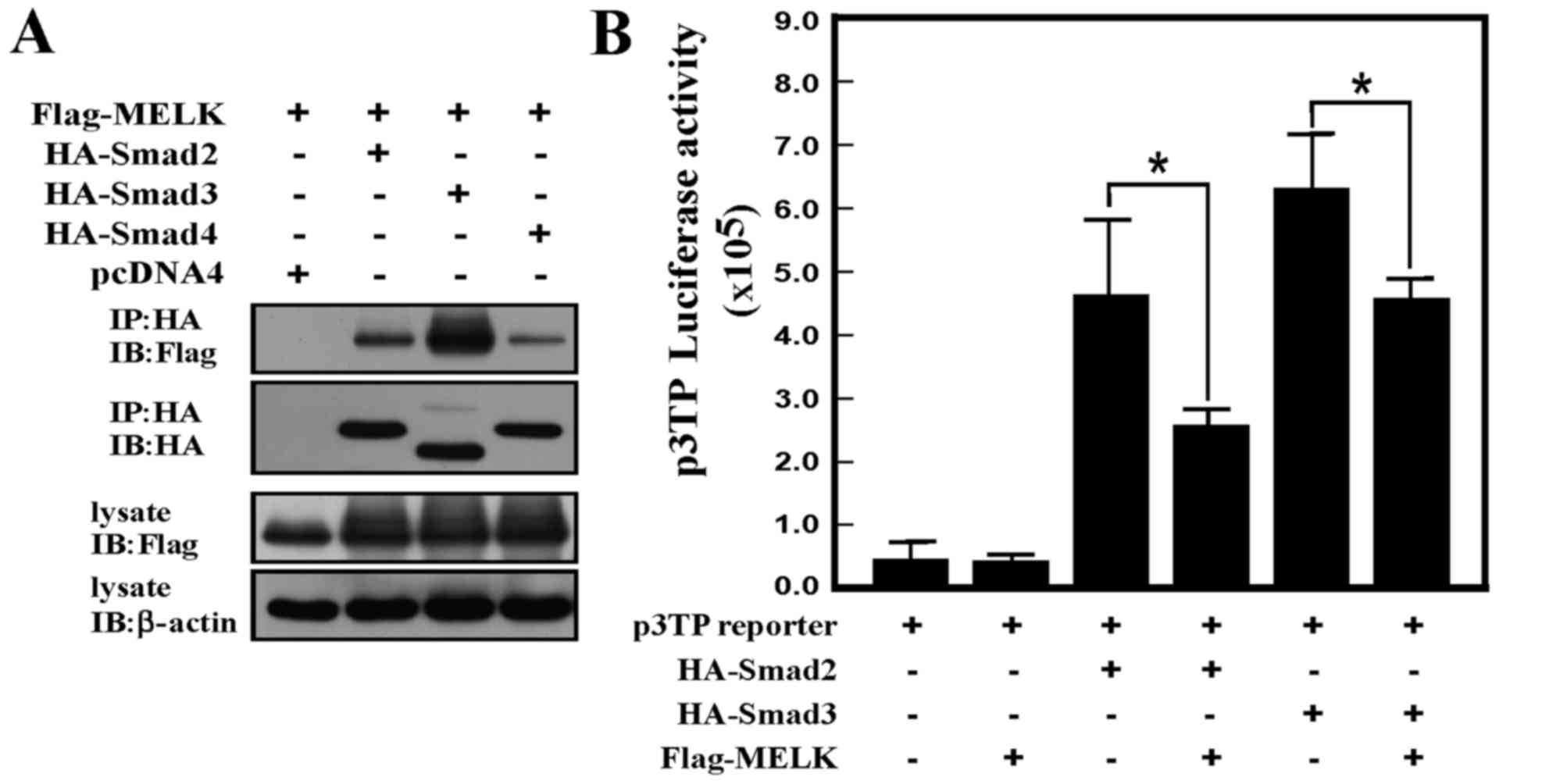 | Figure 4.MELK interacts with the Smad protein
and represses TGF-β signaling activity. (A) MELK interacts with
Smad2, Smad3, or Smad4 protein. The Flag-tagged MELK was
co-transfected with HA-tagged Smad2, Smad3, or Smad4 plasmids. The
HA antibodies were used to immunoprecipitate the immunocomplex, and
the Flag antibodies were used to detect the MELK signals using
western blot analysis. (B) MELK represses Smad2- or Smad3-induced
activity of TGF-β signaling. The p3TP reporter gene was transfected
with different combinations of plasmids. The experiments were
performed in triplicate (*P<0.05). HA, hemagglutinin; TGF-β,
transforming growth factor-β; MELK, murine embryonic leucine zipper
kinase; IP, immunoprecipitation; IB, immnuoblot. |
Discussion
Previous studies on MELK reported the function of
MELK in tumorigenesis (4,14,21). In
the present study, the function of MELK in EMT (the initial step of
metastasis) process was investigated. The results of our study
identified that TGF-β signaling can repress the expression of MELK,
and that MELK can repress TGF-β signaling through interaction with
Smad proteins. In the EMT model of A549 induced using TGF-β, the
knockdown of MELK promoted the cell migration (Fig. 1D).
The data from the present study, firstly disclosed
that MELK is involved in TGF-β signaling. Although knockdown of
MELK did not affect the phosphorylation of Smads, it inhibited
Smad2- and Smad3-induced TGF-β activity (Fig. 4B). MELK may repress TGF-β signaling in
other ways, including the phosphorylation of other sites on Smad
proteins, or affecting the location of Smad protein between the
cytoplasm and nucleus.
A number of studies have already shown the marked
effect of MELK inhibitors in inhibiting tumorigenesis in
vivo (22–24). In these previous papers, the compound
targeting MELK inhibited the proliferation of numerous cancer cell
types, including the cancer stem cells (22,24). In
the current study, the knockdown of MELK in A549 also inhibited
cell proliferation, which is concordant with previous studies.
However, following the observation that MELK affects the process of
EMT, the present study demonstrated that the knockdown of MELK
promotes cell migration in the presence of TGF-β. In the tumor
microenvironment, a number of factors, including TGF-β, are present
and therefore, applying MELK inhibitor may promote the EMT and
metastasis following administration of MELK inhibitors to a
patient. These results require further consideration when
investigating the effect of MELK inhibitor in clinical trials.
Acknowledgements
The present study was funded by the Henan Provincial
Medical Science Gongguan Project (grant no. 112102310266).
References
|
1
|
Heyer BS, Warsowe J, Solter D, Knowles BB
and Ackerman SL: New member of the Snf1/AMPK kinase family, Melk,
is expressed in the mouse egg and preimplantation embryo. Mol
Reprod Dev. 47:148–156. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Joshi K, Banasavadi-Siddegowda Y, Mo X,
Kim SH, Mao P, Kig C, Nardini D, Sobol RW, Chow LM, Kornblum HI, et
al: MELK-dependent FOXM1 phosphorylation is essential for
proliferation of glioma stem cells. Stem Cells. 31:1051–1063. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nakano I, Masterman-Smith M, Saigusa K,
Paucar AA, Horvath S, Shoemaker L, Watanabe M, Negro A, Bajpai R,
Howes A, et al: Maternal embryonic leucine zipper kinase is a key
regulator of the proliferation of malignant brain tumors, including
brain tumor stem cells. J Neurosci Res. 86:48–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hebbard LW, Maurer J, Miller A, Lesperance
J, Hassell J, Oshima RG and Terskikh AV: Maternal embryonic leucine
zipper kinase is upregulated and required in mammary
tumor-initiating cells in vivo. Cancer Res. 70:8863–8873. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mirey G, Chartrain I, Froment C, Quaranta
M, Bouché JP, Monsarrat B, Tassan JP and Ducommun B: CDC25B
phosphorylated by pEg3 localizes to the centrosome and the spindle
poles at mitosis. Cell Cycle. 4:806–811. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jung H, Seong HA and Ha H: Murine protein
serine/threonine kinase 38 activates apoptosis signal-regulating
kinase 1 via Thr 838 phosphorylation. J Biol Chem. 283:34541–34553.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seong HA and Ha H: Murine protein
serine-threonine kinase 38 activates p53 function through Ser15
phosphorylation. J Biol Chem. 287:20797–20810. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gu C, Banasavadi-Siddegowda YK, Joshi K,
Nakamura Y, Kurt H, Gupta S and Nakano I: Tumor-specific activation
of the C-JUN/MELK pathway regulates glioma stem cell growth in a
p53-dependent manner. Stem Cells. 31:870–881. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin ML, Park JH, Nishidate T, Nakamura Y
and Katagiri T: Involvement of maternal embryonic leucine zipper
kinase (MELK) in mammary carcinogenesis through interaction with
Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer
Res. 9:R172007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kuner R, Fälth M, Pressinotti NC, Brase
JC, Puig SB, Metzger J, Gade S, Schäfer G, Bartsch G, Steiner E, et
al: The maternal embryonic leucine zipper kinase (MELK) is
upregulated in high-grade prostate cancer. J Mol Med (Berl).
91:237–248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pickard MR, Green AR, Ellis IO, Caldas C,
Hedge VL, Mourtada-Maarabouni M and Williams GT: Dysregulated
expression of Fau and MELK is associated with poor prognosis in
breast cancer. Breast Cancer Res. 11:R602009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gray D, Jubb AM, Hogue D, Dowd P, Kljavin
N, Yi S, Bai W, Frantz G, Zhang Z, Koeppen H, et al: Maternal
embryonic leucine zipper kinase/murine protein serine-threonine
kinase 38 is a promising therapeutic target for multiple cancers.
Cancer Res. 65:9751–9761. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moffat J, Grueneberg DA, Yang X, Kim SY,
Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK,
et al: A lentiviral RNAi library for human and mouse genes applied
to an arrayed viral high-content screen. Cell. 124:1283–1298. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chomczynski P and Sacchi N: Single Step
Method of RNA Isolation by acid guanidinium
thiocyanate-phenol-chloroform extraction. Anal Biochem.
162:156–159. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shi J, Wang DM, Wang CM, Hu Y, Liu AH,
Zhang YL, Sun B and Song JG: Insulin receptor substrate-1
suppresses transforming growth factor-beta1-mediated
epithelial-mesenchymal transition. Cancer Res. 69:7180–7187. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saito RA, Watabe T, Horiguchi K, Kohyama
T, Saitoh M, Nagase T and Miyazono K: Thyroid transcription
factor-1 inhibits transforming growth factor-beta-mediated
epithelial-to-mesenchymal transition in lung adenocarcinoma cells.
Cancer Res. 69:2783–2791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Datto MB, Frederick JP, Pan L, Borton AJ,
Zhuang Y and Wang XF: Targeted disruption of Smad3 reveals an
essential role in transforming growth factor beta-mediated signal
transduction. Mol Cell Biol. 19:2495–2504. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Lee YM, Baitsch L, Huang A, Xiang
Y, Tong H, Lako A, Von T, Choi C, Lim E, et al: MELK is an
oncogenic kinase essential for mitotic progression in basal-like
breast cancer cells. Elife. 3:e017632014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alachkar H, Mutonga MB, Metzeler KH,
Fulton N, Malnassy G, Herold T, Spiekermann K, Bohlander SK,
Hiddemann W, Matsuo Y, et al: Preclinical efficacy of maternal
embryonic leucine-zipper kinase (MELK) inhibition in acute myeloid
leukemia. Oncotarget. 5:12371–12382. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chung S and Nakamura Y: MELK inhibitor,
novel molecular targeted therapeutics for human cancer stem cells.
Cell Cycle. 12:1655–1656. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Minata M, Gu C, Joshi K, Nakano-Okuno M,
Hong C, Nguyen CH, Kornblum HI, Molla A and Nakano I: Multi-kinase
inhibitor C1 triggers mitotic catastrophe of glioma stem cells
mainly through MELK kinase inhibition. PLoS One. 9:e925462014.
View Article : Google Scholar : PubMed/NCBI
|