Introduction
MicroRNAs (miRs/miRNAs) are short non-coding RNAs
that inhibit specific mRNAs by binding to 3′-untranslated region
(3′-UTRs) of the target mRNA. Depending on the targets they
regulate, they may act as oncogenes or tumor suppressor genes in
various types of cancer (1). There
has been increasing focus on investigating the role of miRNA in the
development of cancer (2–4). The suppressive functions of miRNAs on
mRNA translation make them useful as small molecular drugs for gene
therapy (5). There has been
increasing evidence demonstrating that miRNAs serve essential roles
in tumorigenesis (6–8). Furthermore, they may function as
oncogenes or tumor suppressor genes.
miR-4530 is located on chromosome 19. In our
previous study, the results indicated that miR-4530 was
downregulated in the serum of patients with diabetic retinopathy
(9). Furthermore, it was demonstrated
that miR-4530 was able to promote angiogenesis in endothelial
cells. However, the molecular mechanism of miR-4530 in the
regulation of angiogenesis remains unclear. In the present study,
the role of miR-4530 in the regulation of angiogenesis in breast
carcinoma was investigated.
TargetScan was used to predict targets of human
miR-4530, and vasohibin-1 (VASH1) was identified in the present
study. VASH1 was first reported and named in 2004 by Watanabe et
al (10). Its expression was
demonstrated to be enhanced in endothelial cells (ECs) during
angiogenesis and it inhibited angiogenesis in secreting VASH1 as
part of a negative feedback (11).
In the present study, VASH1 was identified to be one
of the targets of miR-4530 and may be downregulated by miR-4530.
Furthermore, miR-4530 promoted the tube formation of HUVECs and
breast carcinoma angiogenesis. Finally, the cellular function
experiments demonstrated that miR-4530 suppresses breast carcinoma
by affecting MCF-7 and MDA-MB-231 cell proliferation and also
induces apoptosis.
Materials and methods
Cell culture
Human breast carcinoma MDA-MB-231 and MCF-7 cell
lines were purchased from the Institute of Biochemistry and Cell
Biology, Chinese Academy of Sciences (Shanghai, China). Cells were
cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10%
fetal bovine serum (FBS; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) and 100 µg/ml each of penicillin/streptomycin. Human
umbilical vein endothelial cells (HUVECs) and HEK-293T cells were
purchased from the Institute of Biochemistry and Cell Biology,
Chinese Academy of Sciences cultured in RPMI-1640 medium
(Invitrogen; Thermo Fisher Scientific, Inc.) containing 10% FBS and
100 µg/ml penicillin/streptomycin. Cells were maintained at 37°C in
at atmosphere containing 5% CO2 and saturated
humidity.
Construction of plasmids and stable
transfected cell lines
The plasmids pPG/miR/EGFP, pPG-miR4530-EGFP and
pPG-miR4530sponge-EGFP were purchased from Shanghai GenePharma Co.,
Ltd. (Shanghai, China) and were transfected into tumor cells using
Lipofectamine 3000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Blasticidin
(Sigma-Aldrich; Merck KGaA) was used to screen stable cell lines.
miRNA-4530 mimics, inhibitors and their negative control
(nc)-mimics, and -inhibitors were purchased from Shanghai
GenePharma Co., Ltd.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to extract total RNA from cells,
according to the manufacturer's protocol. A total of 1,000 µg RNA
was reverse-transcribed into miRNA-cDNA using All-in-One miRNA
First-Strand cDNA Synthesis kit (GeneCopoeia, Inc., Rockville, MD,
USA) according to the manufacturer's protocol. A total of 1,000 µg
RNA was reverse-transcribed into mRNA-cDNA using PrimeScript RT
Reagent kit with gDNA Eraser (Takara, Dalian, China) according to
the manufacturer's protocol. qPCR was performed to evaluate the
expression levels of miRNAs and mRNA using a SYBR Green PCR kit
(GeneCopoeia, Inc. Rockville, MD, USA) with the Applied Biosystems
StepOnePlus™ Real-Time PCR system (Thermo Fisher Scientific, Inc.,
USA). Human U6 was used as an internal control for measuring miRNA
expression and GAPDH was used as an internal control for measuring
mRNA expression. The expression levels were calculated using the
2−∆∆Cq method (12). The
primers for U6 were provided by GeneCopoeia, Inc. All primers are
detailed in Table I and the
thermocycling conditions are presented in Table II).
 | Table I.Primers for quantitative polymerase
chain reaction. |
Table I.
Primers for quantitative polymerase
chain reaction.
| Primer | Sequence, 5′→3′ |
|---|
| miR universal reverse
primer |
TGCTGTCAACGATACGCTACG |
| miR-4530 forward
primer |
ATCAGGACGGGAGCGAAAA |
| GAPDH forward
primer |
GGAGTCCACTGGCGTCTT |
| GAPDH reverse
primer |
ATCTTGAGGCTGTTGTCATAC |
| VASH1 forward
primer |
AGATCCCCATACCGAGTGTG |
| VASH1 reverse
primer |
GCTTCCAGGCATTTGATTGGC |
| Table II.Thermocycling program for quantitative
polymerase chain reaction. |
Table II.
Thermocycling program for quantitative
polymerase chain reaction.
| Step | Temperature, °C | Duration |
|---|
| 1 | 95 | 10 min |
| 2 | 95 | 10 sec |
| 3 | 55 | 15 sec |
| 4 | 72 | 30 sec |
| Steps 2–4 | – | 40 cycles |
| Melt curve
program | – | 60 min |
Western blotting
Total protein was extracted using
radioimmunoprecipitation buffer containing phenylmethylsulfonyl
fluoride. Lysates were centrifuged at 14,000 × g for 10 min at 4°C
to remove cellular debris and the protein concentration was
determined using bicinchoninic acid protein assay kit (Beyotime
Institute of Biotechnology, Haimen, China) according to the
manufacturer's protocol. A total of 30 µg protein was separated on
by SDS-PAGE (10% gel) and transferred onto polyvinylidene fluoride
membranes (EMD Millipore, Billerica, MA, USA). Following blocking
with milk 2 h at room temperature, primary antibodies directed
against VASH1 (1:1,000 diluted in Beyotime Primary Antibody
Dilution Buffer; #ab199732; Abcam, Cambridge, UK) and GAPDH
(1:1,000 diluted in Beyotime Primary Antibody Dilution Buffer;
#2118S; Cell Signaling Technology, Inc., Danvers, MA, USA) were
incubated with the membranes overnight at 4°C. Subsequently,
horseradish peroxidase-conjugated secondary antibodies (dilution,
1:10,000 in TBST; #A0208; Beyotime Institute of Biotechnology,
Haimen, China) were incubated with membranes for 2 h prior to
washing the membranes for 1 h Band signals were detected using
enhanced chemiluminescence (Beyotime Institute of Biotechnology),
according to the manufacturer's protocol. Western blots were
performed ≥3 times.
Luciferase reporter assay
Firstly, TargetScan was used to predict the
potential targets of miR-4530. In TargetScan Release 6.2 database,
the human species was selected and the microRNA name was entered as
‘has-miR-4530’. The request was submitted and the top potential
targets were chosen. The gene function and the data of preliminary
luciferase reporter assay were evaluated and VASH1 was chosen for
further investigation. Human VASH1 mRNA 3′UTR and a mutation of the
miR-4530 seed sequence were amplified using PCR with plasmid
templates (Shanghai GenePharma Co., Ltd.) and inserted into
pmiR-report plasmids. The sequences were subsequently confirmed. A
total of 25 ng luciferase reporter plasmids and miR-4530 or
miR-negative control (NC) mimics were co-transfected into HEK-293T
cells in 24-well plates using Lipofectamine 3000 with RPMI-1640
medium (Invitrogen; Thermo Fisher Scientific, Inc.). After 24 h of
transfection and incubation at 37°C, the cells were harvested using
the passive lysis buffer from the Dual-Luciferase Reporter Assay
System (Promega Corporation, Madison, WI, USA). The luciferase
signal was measured using a Tecan M1000 microplate reader (Thermo
Fisher Scientific, Inc. The Renilla luciferase signal was
used as an internal control and the firefly luciferase signal
corresponded to the expression of firefly luciferase.
Colony formation assays
The cells were counted and seeded into a 6-well
plate at a density of 500 cells/well. Cells were cultured for 10
days and medium was replaced with fresh DMEM every 2 days.
Subsequently, cells were washed twice with PBS and fixed with 4%
paraformaldehyde for 15 min at room temperature. Finally, cells
were stained with 0.1% crystal violet (Beyotime Institute of
Biotechnology) for 15 min at room temperature and washed with
double-distilled water. The colony formation assay was performed in
triplicate and images were captured using a digital camera.
Cell proliferation assays
A total of 3.5×103 stable transfected
cells were seeded into 96-well plates and the medium was replaced
with fresh DMEM every 2 days. After 24, 48, 72 and 96 h of
incubation, cell proliferation was detected using a Cell Counting
kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). At each time point, the medium was replaced with fresh DMEM
and the cells were incubated for 1 h with 10 µl CCK-8 solution.
Subsequently, all plates were scanned at 450 nm using a microplate
reader. Each experiment was performed independently three
times.
Cell cycle and cell apoptosis
analysis
For the cell cycle assay, stably transfected cells
were collected by centrifugation after 70–80% confluency was
achieved and fixed in 70% ethanol at 4°C overnight. The MDA-MB-231
cells were washed twice with 4°C 1X PBS and then resuspended in 160
µl 0.5 mg/ml RNase A (Nanjing KeyGen Biotech Co., Ltd., Nanjing,
China) at 37°C for 30 min. Cells were subsequently stained with 50
µmol/l propidium iodide (KeyGen Biotech Co., Ltd.) in the dark for
1 h at 4°C and analyzed using flow cytometry (BD Biosciences,
Franklin Lakes, NJ, USA). For analysis of cell apoptosis, stably
transfected cells were collected by centrifugation at 2,500 × g for
5 min at 4°C, washed and stained using the annexin
V-allophycocyanin (APC)/propidium iodide kit (KeyGen Biotech Co.,
Ltd.) according to the manufacturer's protocol. The cells were
analyzed immediately using flow cytometry (BD Biosciences). Data
were analyzed using BD Accuri™ C6 software (BD Biosciences).
Tube formation assay
HUVECs and stably transfected MCF-7 cells growing in
6-well plates were serum-starved in RPMI-1640 medium for 24 h at
37°C. Frozen (−20°C) Growth factor-reduced Matrigel (BD
Biosciences) was melted in 4°C overnight; subsequently, 50 µl
Matrigel was spread across the bottom of the wells of 96-well
plates. Following solidification of Matrigel at 37°C, the
supernatant medium of the MCF-7 cells was centrifuged (10,000 × g
for 5 min at 4°C) and collected as tumor cell-conditioned medium
(TCM). The supernatant of the HUVECs was discarded and the cells
were suspended in TCM. A total of 8×104/well HUVECs were
seeded on the Matrigel-coated wells in 100 µl TMC and incubated at
37°C for 10 h. The angiogenic activity was evaluated as the number
of tubular structures in each well.
In vivo growth assay
Nude mice were purchased from Shanghai SLAC
Laboratory Animal Co., Ltd. (Shanghai, China) and raised in the
Laboratory Animal Center of Wenzhou Medical University (Wenzhou,
China). In total 18 female nude mice (weight, 20–25 g) were kept in
light/dark cycles with sufficient food and water at 20–26°C with a
humidity of 40–70% in a specific pathogen-free laboratory animal
room. A total of 1×107 stably transfected MDA-MB-231
cells were subcutaneously injected into the foreleg armpit of
2-month-old female nude mice (n=6/group). Tumor size was measured
using a Vernier caliper every 5 days. After 10 weeks, all mice were
sacrificed by cervical dislocation and dissected. Tumor tissues
were removed for analysis. Animal experimentation was approved by
the Laboratory Animal Ethics Committee of Wenzhou Medical
University.
Statistics
SPSS v17.0 software (SPSS, Inc., Chicago, IL, USA)
was used to perform statistical analysis. Statistical significance
was calculated by using the two-tailed Student's t-test. All
experiments were performed ≥3 times and data were expressed as mean
± standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
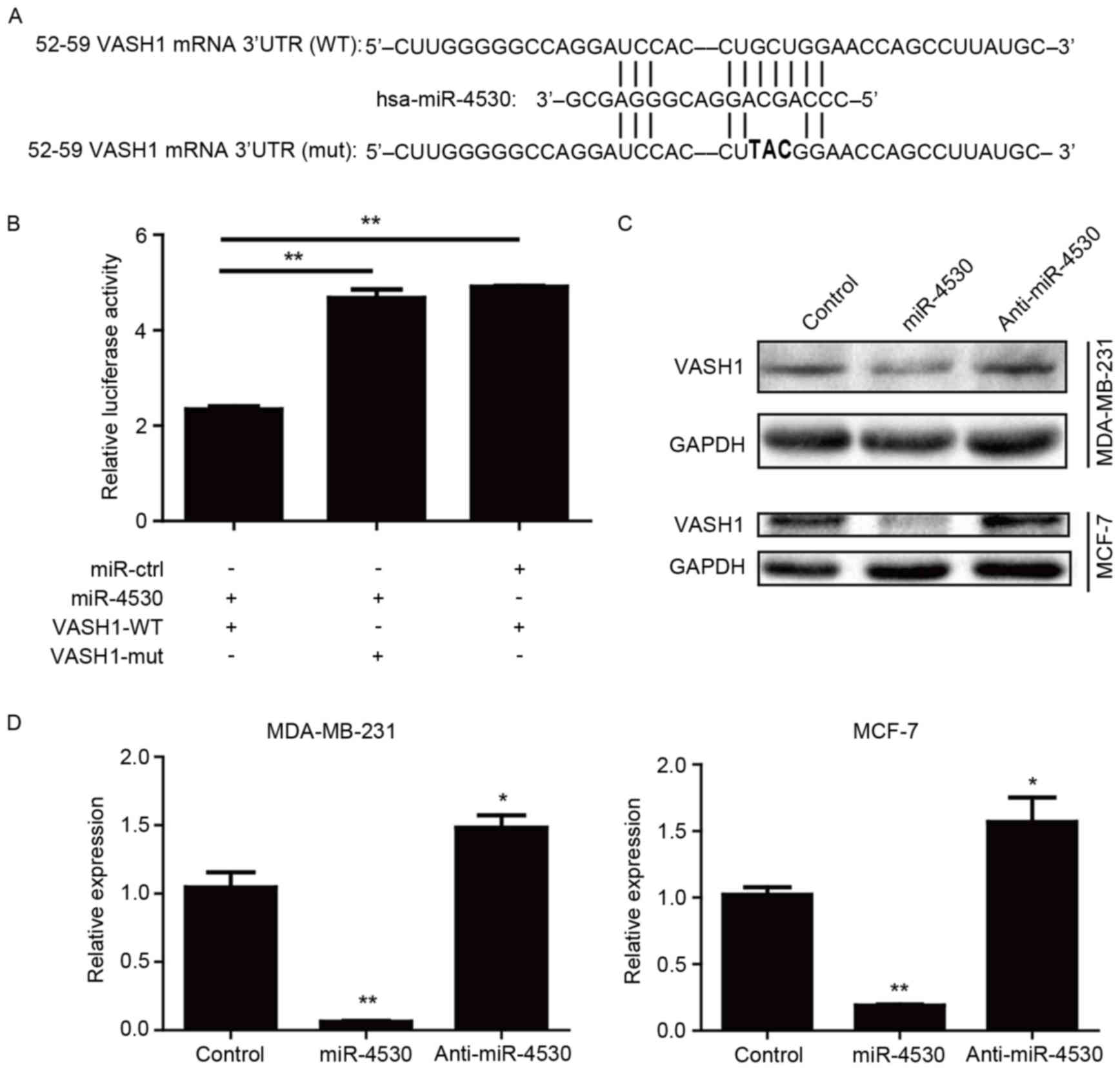
miR-4530 downregulates VASH1
expression through post-transcriptional repression
The miRNA database TargetScan was utilized to
identify the potential targets and the binding site of miR-4530.
TargetScan predicted VASH1 as one of the miR-4530 targets with high
complementarity. The binding site was located between bases 52 and
59 of the VASH1 3′-UTR (Fig. 1A).
Luciferase reporter assays were performed on HEK-293T cells to
confirm the association between miR-4530 and VASH1. The results
demonstrated that miR-4530 significantly suppressed the firefly
luciferase signal compared with the control group and that this
inhibitory effect was significantly reversed when the binding site
for miR-4530 was mutated (Fig.
1B).
To verify that the suppression of VASH1 expression
by miR-4530 does occur in breast carcinoma cell lines, stably
transfected MCF-7 and MDA-MB-231 cell lines were constructed.
Stably transfected miR-4530 precursor, anti-miR-4530 and empty
plasmid groups were produced for each cell line. The expression of
miR-4530 in each group was confirmed using qPCR and western blot
analyses. The protein expression of VASH1 in the cells with
miR-4530 overexpression was downregulated (Fig. 1C), and the mRNA expression was
significantly downregulated (Fig. 1D)
compared with the corresponding empty plasmid control groups in the
two breast carcinoma cell lines. Furthermore, the protein
expression of VASH1 in the anti-miR-4530 groups was upregulated
(Fig. 1C), and the mRNA expression
was significantly upregulated (Fig.
1D) compared with the corresponding empty plasmid control
groups. These results suggest that miR-4530 inhibits VASH1
expression through post-transcriptional repression.
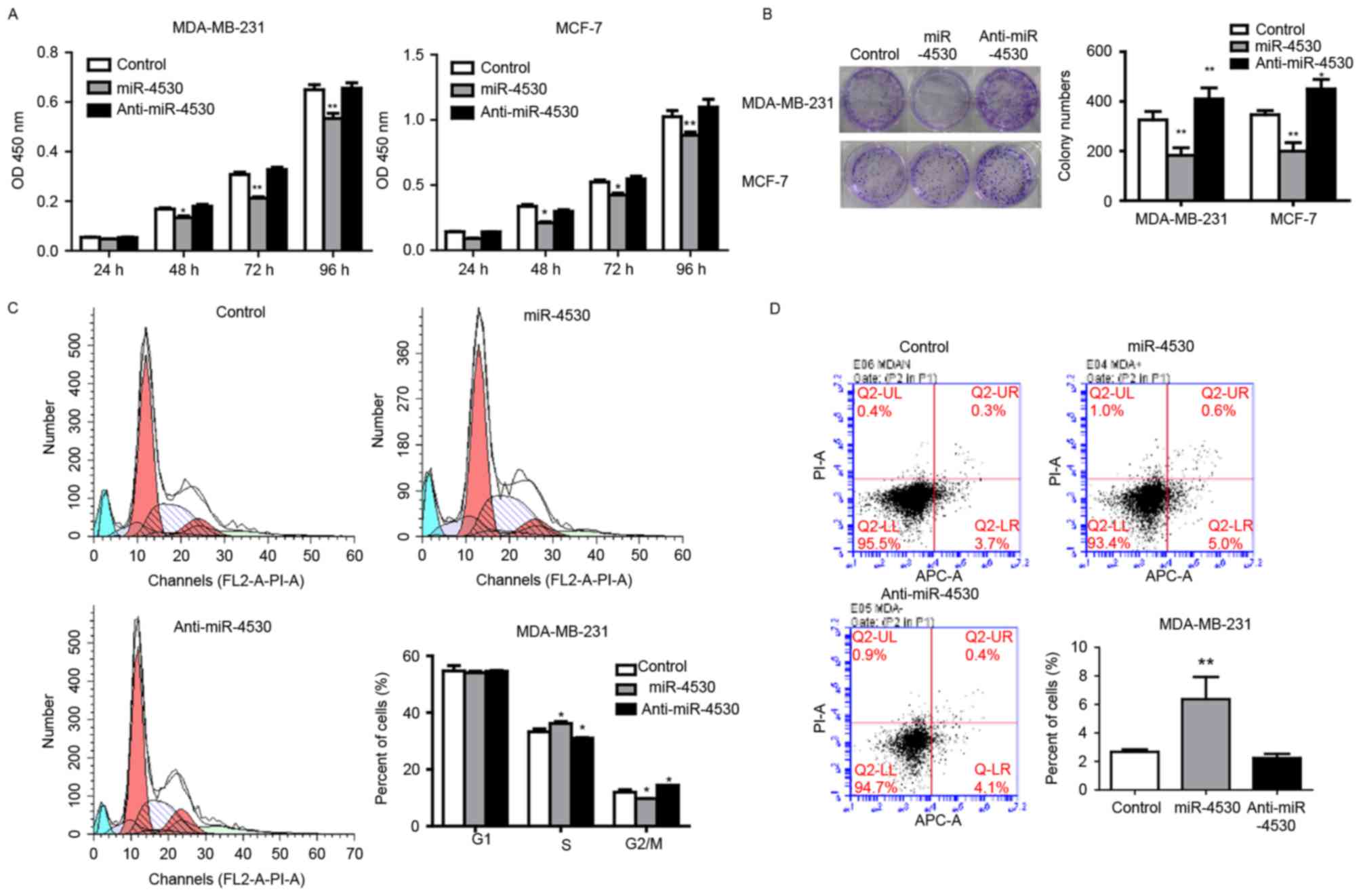
Overexpression of miR-4530 results in
the inhibition of cell proliferation, colony formation and induces
apoptosis
To investigate the role of miR-4530 in breast
carcinoma cells, the cell proliferation rate of stably transfected
MDA-MB-231 and MCF-7 cells was determined using CCK-8 and colony
formation assays. The results of the CCK-8 assay demonstrated that
the proliferation rate of MDA-MB-231 and MCF-7 cells was
significantly repressed from 48 h onwards following overexpression
of miR-4530 compared with the control group (Fig. 2A). Furthermore, the ability of colony
formation was significantly decreased following overexpression of
miR-4530, and significantly increased following the knockdown of
miR-4530 compared with the control groups in the two cell lines
(Fig. 2B).
To determine whether cell cycle arrest of cancer
cells contributed to the repressive effects of miR-4530 on cell
proliferation, the percentage of cells in each stage of the cell
cycle were analyzed in stably transfected breast carcinoma cells.
The result revealed that the overexpression of miR-4530 cells
caused cell cycle arrest in S phase. The proportion of S-phase
MDA-MB-231 cells in the miR-4530 overexpression, control and
miR-4530 knockdown groups were 36.83, 32.27 and 30.77%,
respectively (Fig. 2C). Furthermore,
the differences in the percentage of cells in the S phase in the
overexpression and knockdown groups were significant compared with
the control group. These results suggest that the overexpression of
miR-4530 inhibits breast carcinoma cell proliferation by inhibiting
the S/G2 transition. In addition, cell apoptosis rates
were detected using flow cytometry. Stably transfected MDA-MB-231
cells were stained using the Annexin V-APC/propidium iodide kit.
The results demonstrated that miR-4530 expression significantly
increased the percentage of apoptotic MDA-MB-231 cells compared
with the negative control group (Fig.
2D). This suggests that miR-4530 affects apoptotic signaling
pathways.
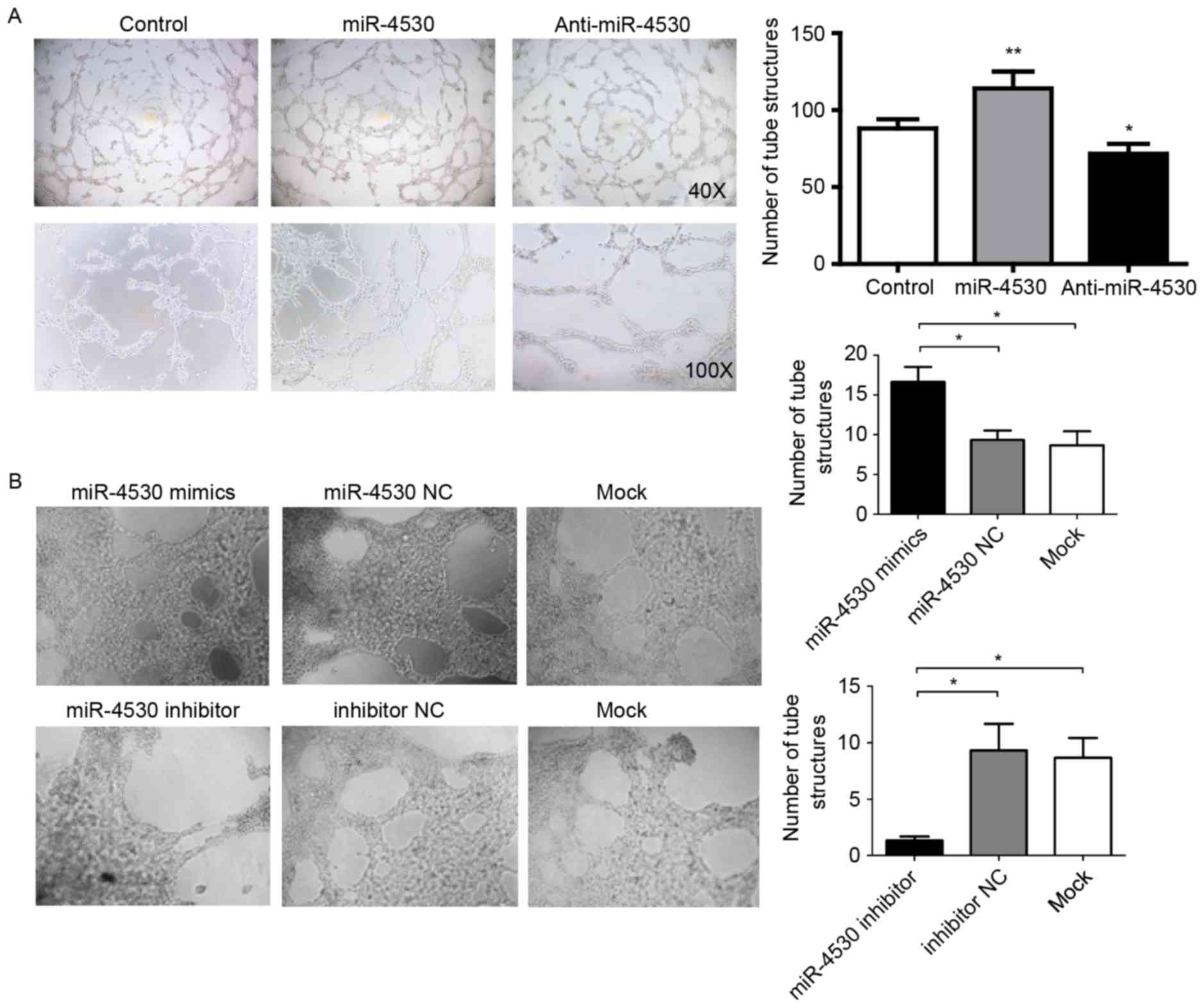
miR-4530 promotes tube formation of
HUVECs and breast carcinoma angiogenesis by negatively regulating
VASH1 expression
VASH1 has been identified as an inhibitor of
angiogenesis in endothelial cells (11). Therefore, it is plausible that
miR-4530 may promote angiogenesis in breast carcinoma cells. To
investigate this hypothesis, a tube formation assay was performed
to confirm whether miR-4530 promotes angiogenesis in breast
carcinoma. The results demonstrated that the TCM from the MCF-7
cells stably transfected with the miR-4530 precursor significantly
promoted HUVECs to form tube structures compared with TCM from
cells stably transfected with the empty vector (Fig. 3A).
As expected, the TCM from the anti-miR-4530 group
significantly suppressed HUVECs from forming tube structures
compared with the control group (Fig.
3A). In addition, tube formation assays were performed on
HUVECs transfected with miRNA-4530 mimics, inhibitors or their
negative controls. The results were similar to previous results,
whereby the mimics significantly increased and the inhibitor
significantly decreased the number of tube structures formed
compared with the mock-transfected control group (Fig. 3B). It was revealed that miR-4530
mimic-transfected HUVECs exhibited a significant increase in their
capability to form tube structures in vitro on Matrigel
(Fig. 3B), with a 50% increase in
tube structures compared with miR-4530 NC- or mock-transfected
HUVECs. The HUVECs transfected with the miR-4530 inhibitors
exhibited significant suppression of angiogenesis in vitro,
with a 75% decrease in tube structures on Matrigel compared with
miR-4530 inhibitors NC- or mock-transfected HUVECs (Fig. 3B).
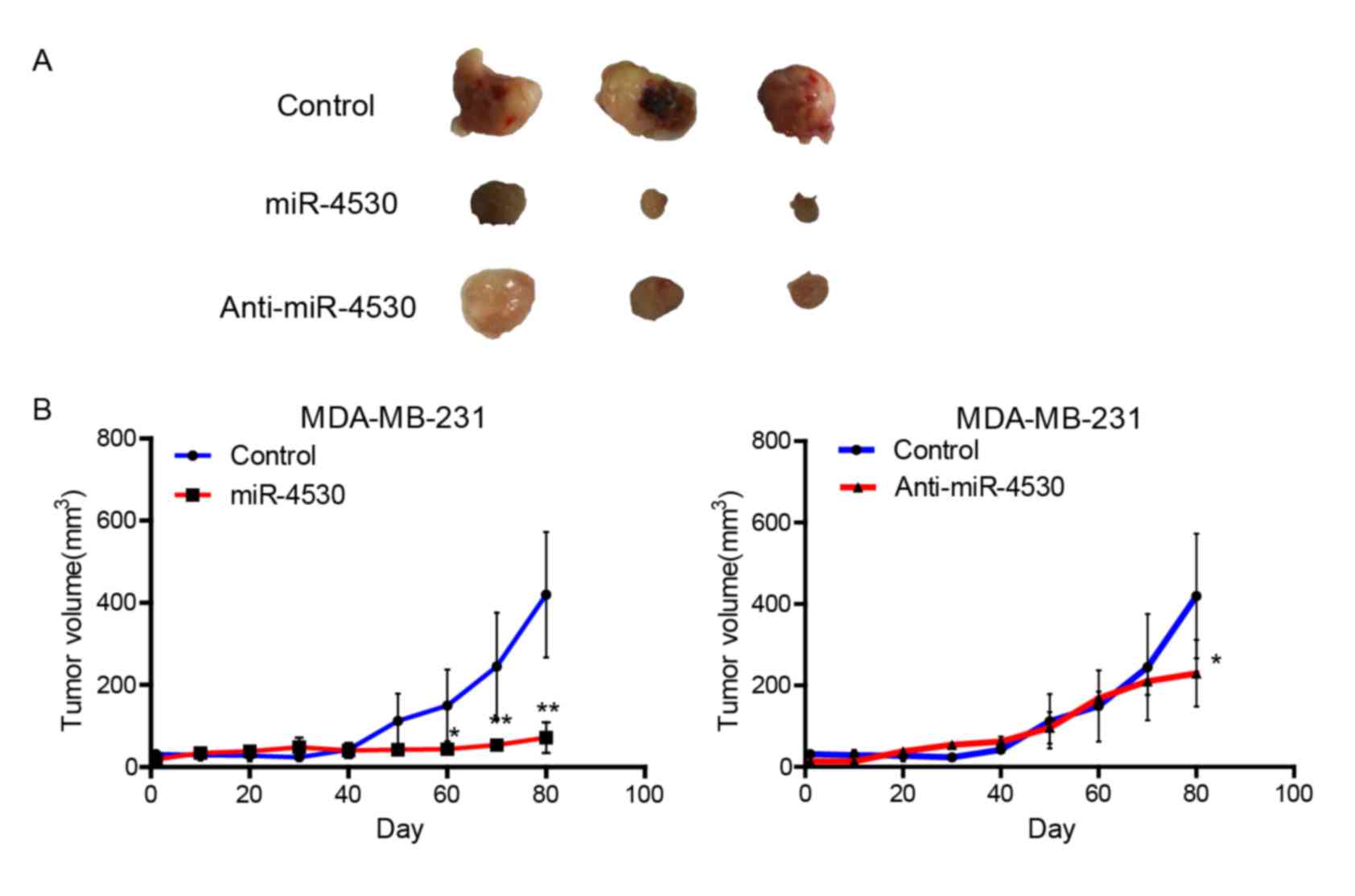
miR-4530 suppresses tumor growth in
mice
The nude mice xenograft model was applied to further
investigate the effect of miR-4530 on tumorigenicity. MDA-MB-231
cells stably transfected with the miR-4530 precursor, anti-miR-4530
or empty plasmids were subcutaneously injected into 2-month-old
female nude mice and the tumor volume was monitored for evaluating
the effect of miR-4530 on tumor growth. As presented in Fig. 4A, stable transfection of miR-4530
precursor into MDA-MB-231 cells resulted in a decreased growth rate
and significantly decreased tumor size of subcutaneous xenograft
tumors by day 60 in nude mice, when compared with the control group
(Fig. 4B). Furthermore, cells
transfected with anti-miR-4530 exhibited a significant decrease in
tumor size by day 80 compared with the control group. The miR-4530
precursor group demonstrated a larger decrease in tumor volume
compared with the anti-miR-4530 group. Therefore, cells stably
transfected with the miR-4530 precursor exhibited poor survival
rates and low tumorigenicity. These results indicate that
upregulation of miR-4530 expression is disadvantageous for
tumorigenicity.
Discussion
In our previous study, the results revealed that
miR-4530 was upregulated in the serum of patients with diabetic
retinopathy (9). Diabetic retinopathy
is a complication of diabetes that involves microvessel damage in
the retina (13–15). The results of the present study
demonstrated that the overexpression and knockdown of miR-4530
regulates angiogenesis. Therefore, this feature may be utilized in
suppressing tumor angiogenesis clinically. Angiogenesis serves an
essential role in tumor growth and metastasis, and in healthy human
development (16). Therefore, in the
present study, the effect of miR-4530 on angiogenesis in human
breast cancer was investigated.
VASH1 is a novel endogenous angiogenesis inhibitor
involved in the negative feedback regulation of angiogenesis
(11,17,18). It
may be a promising candidate for antiangiogenic therapy; however,
the molecular mechanism underlying the antiangiogenic activity of
VASH1 remains unclear. VASH1 is upregulated in numerous types of
tumorous tissue and exhibits therapeutic efficacy in tumor growth
(19–22), proliferative retinopathy (23,24) and
atherosclerosis (25). Furthermore,
VASH1 has been demonstrated to not cause any damage to healthy
blood vessels in mice (26),
suggesting that it is safer and has a limited number of side
effects compared with traditional antiangiogenic drugs.
The results of the present study suggest that VASH1
is a direct target gene of miR-4530 in breast carcinoma cells. The
overexpression of miR-4530 in breast carcinoma cells resulted in
significantly decreased mRNA and protein expression of VASH1 in
breast carcinoma cells compared with the control group.
Furthermore, it was demonstrated that miR-4530 suppressed breast
carcinoma MCF-7 and MDA-MB-231 cell proliferation and induced
apoptosis. Miyashita et al (27) reported that the knockdown of VASH1 in
ECs induced premature senescence and that those ECs were easily
killed by cellular stressors, which may also occur in tumor cells.
The knockdown of miR-4530 resulted in an increase in the expression
of VASH1 but did not suppress or promote breast carcinoma cell
proliferation. These results are consistent with the results of a
previous study whereby VASH1 was demonstrated to suppress tumor
angiogenesis but not the proliferation of the tumor cells (19). Additionally, the results of the
present study suggest that miR-4530 promotes angiogenesis in breast
carcinoma. The knockdown of miR-4530 resulted in the suppression of
angiogenesis as indicated by the tube formation assays. This may be
attributed to the antiangiogenic activity of VASH1.
In vivo assays were also performed in the
present study using a xenograft mouse model. The tumor sizes in the
miR-4530 overexpression group were significantly decreased with
poor survival of cells compared with the control group, which may
be attributed to the suppression of cell proliferation and
apoptosis. Subcutaneous xenograft tumors were ischemic in the early
stages. The majority of tumor cells may have been killed due to
starvation. Tumor growth rates in the anti-miR-4530 group appeared
to decrease when tumor sizes were ~200 mm3, which may be
attributed to a decreased number of microvessels.
VASH1, first identified as an angiogenesis inhibitor
in endothelial cells, has now been identified in various other
types of cell, including cancer cells (20,28–30).
Tamaki et al (22) reported
that VASH1 expression in human breast lesions is associated with
neovascularization through a compensatory mechanism where
antiangiogenesis may negatively regulate intratumoral angiogenesis
in human breast cancer. Invasive breast carcinoma or higher nuclear
and histological grades of human breast carcinoma exhibited
relatively higher VASH1 expression compared with that in ductal
carcinoma in situ (31).
However, the function and mechanism of VASH1 in cancer cells have
rarely been reported.
To the best of our knowledge, the present study is
the first to investigate the association between VASH1 and
miR-4530, and determine the biological characteristics of VASH1 in
human breast carcinoma cells. The results of the present study
suggest that VASH1 and miR-4530 are potential therapeutic targets
for the treatment of breast cancer particularly with regard to
antiangiogenesis therapy.
Acknowledgements
The present study was supported by the Program for
Zhejiang Leading Team of Science and Technology Innovation (grant
no. 2010R50048).
References
|
1
|
Shenouda SK and Alahari SK: MicroRNA
function in cancer: Oncogene or a tumor suppressor? Cancer
Metastasis Rev. 28:369–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jovanovic M and Hengartner MO: miRNAs and
apoptosis: RNAs to die for. Oncogene. 25:6176–6187. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kent OA and Mendell JT: A small piece in
the cancer puzzle: microRNAs as tumor suppressors and oncogenes.
Oncogene. 25:6188–6196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Volinia S, Calin GA, Liu CG, Ambs S,
Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et
al: A microRNA expression signature of human solid tumors defines
cancer gene targets. Proc Natl Acad Sci USA. 103:pp. 2257–2261.
2006; View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang G and Yin B: The advance of
application for microRNAs in cancer gene therapy. Biomed
Pharmacother. 68:137–142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou X, Xia Y, Li L and Zhang G: MiR-101
inhibits cell growth and tumorigenesis of Helicobacter pylori
related gastric cancer by repression of SOCS2. Cancer Biol Ther.
16:160–169. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang XF, Li KK, Gao L, Li SZ, Chen K,
Zhang JB, Wang D, Tu RF, Zhang JX, Tao KX, et al: miR-191 promotes
tumorigenesis of human colorectal cancer through targeting C/EBPβ.
Oncotarget. 6:4144–4158. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shen S, Guo X, Yan H, Lu Y, Ji X, Li L,
Liang T, Zhou D, Feng XH, Zhao JC, et al: A miR-130a-YAP positive
feedback loop promotes organ size and tumorigenesis. Cell Res.
25:997–1012. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ding L, Ai D, Wu R, Zang T, Jing L, Lu J
and Zhong L: Identification of the differential expression of serum
microRNA in type 2 diabetes. Biosci Biotechnol Biochem. 80:461–465.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Watanabe K, Hasegawa Y, Yamashita H,
Shimizu K, Ding Y, Abe M, Ohta H, Imagawa K, Hojo K, Maki H, et al:
Vasohibin as an endothelium-derived negative feedback regulator of
angiogenesis. J Clin Invest. 114:898–907. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kimura H, Miyashita H, Suzuki Y, Kobayashi
M, Watanabe K, Sonoda H, Ohta H, Fujiwara T, Shimosegawa T and Sato
Y: Distinctive localization and opposed roles of vasohibin-1 and
vasohibin-2 in the regulation of angiogenesis. Blood.
113:4810–4818. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using quantitative PCR and the
2-(Delta DeltaC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Du Y, Veenstra A, Palczewski K and Kern
TS: Photoreceptor cells are major contributors to diabetes-induced
oxidative stress and local inflammation in the retina. Proc Natl
Acad Sci USA. 110:pp. 16586–16591. 2013; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang J and Kern TS: Inflammation in
diabetic retinopathy. Prog Retin Eye Res. 30:343–358. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hanahan D and Folkman J: Patterns and
emerging mechanisms of the angiogenic switch during tumorigenesis.
Cell. 86:353–364. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kerbel RS: Vasohibin: The feedback on a
new inhibitor of angiogenesis. J Clin Invest. 114:884–886. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shimizu K, Watanabe K, Yamashita H, Abe M,
Yoshimatsu H, Ohta H, Sonoda H and Sato Y: Gene regulation of a
novel angiogenesis inhibitor, vasohibin, in endothelial cells.
Biochem Biophys Res Commun. 327:700–706. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li D, Zhou K, Wang S, Shi Z and Yang Z:
Recombinant adenovirus encoding vasohibin prevents tumor
angiogenesis and inhibits tumor growth. Cancer Sci. 101:448–452.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hosaka T, Kimura H, Heishi T, Suzuki Y,
Miyashita H, Ohta H, Sonoda H, Moriya T, Suzuki S, Kondo T and Sato
Y: Vasohibin-1 expression in endothelium of tumor blood vessels
regulates angiogenesis. Am J Pathol. 175:430–439. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yoshinaga K, Ito K, Moriya T, Nagase S,
Takano T, Niikura H, Yaegashi N and Sato Y: Expression of vasohibin
as a novel endothelium-derived angiogenesis inhibitor in
endometrial cancer. Cancer Sci. 99:914–919. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tamaki K, Moriya T and Sato Y, Nagase S,
Takano T, Niikura H, Yaegashi N and Sato Y: Vasohibin-1 in human
breast carcinoma: A potential negative feedback regulator of
angiogenesis. Cancer Sci. 100:88–94. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sato H, Abe T, Wakusawa R, Asai N,
Kunikata H, Ohta H, Sonoda H, Sato Y and Nishida K: Vitreous levels
of vasohibin-1 and vascular endothelial growth factor in patients
with proliferative diabetic retinopathy. Diabetologia. 52:359–361.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shen J, Yang X, Xiao WH, Hackett SF, Sato
Y and Campochiaro PA: Vasohibin is up-regulated by VEGF in the
retina and suppresses VEGF receptor 2 and retinal
neovascularization. FASEB J. 20:723–725. 2006.PubMed/NCBI
|
|
25
|
Yamashita H, Abe M, Watanabe K, Shimizu K,
Moriya T, Sato A, Satomi S, Ohta H, Sonoda H and Sato Y: Vasohibin
prevents arterial neointimal formation through angiogenesis
inhibition. Biochem Biophys Res Commun. 345:919–925. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Heishi T, Hosaka T, Suzuki Y, Miyashita H,
Oike Y, Takahashi T, Nakamura T, Arioka S, Mitsuda Y, Takakura T,
et al: Endogenous angiogenesis inhibitor vasohibin1 exhibits
broad-spectrum antilymphangiogenic activity and suppresses lymph
node metastasis. Am J Pathol. 176:1950–1958. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miyashita H, Watanabe T, Hayashi H, Suzuki
Y, Nakamura T, Ito S, Ono M, Hoshikawa Y, Okada Y, Kondo T and Sato
Y: Angiogenesis inhibitor vasohibin-1 enhances stress resistance of
endothelial cells via induction of SOD2 and SIRT1. PLoS One.
7:e464592012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sato Y: Is vasohibin-1 for more than
angiogenesis inhibition? J Biochem. 149:229–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yan Y, Shen Z, Ye Y, Jiang K, Zhang H,
Shen C, Mustonen H, Puolakkainen P and Wang S: A novel molecular
marker of prognosis in colorectal cancer: Vasohibin-1. Med Oncol.
31:8162014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu S, Han B, Zhang Q, Dou J, Wang F, Lin
W, Sun Y and Peng G: Vasohibin-1 suppresses colon cancer.
Oncotarget. 6:7880–7898. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tamaki K, Sasano H, Maruo Y, Takahashi Y,
Miyashita M, Moriya T, Sato Y, Hirakawa H, Tamaki N, Watanabe M, et
al: Vasohibin-1 as a potential predictor of aggressive behavior of
ductal carcinoma in situ of the breast. Cancer Sci. 101:1051–1058.
2010. View Article : Google Scholar : PubMed/NCBI
|