Introduction
Lung cancer is the leading cause of
cancer-associated mortality worldwide (1,2). A total
of one-third of patients with lung cancer initially present at an
advanced stage, when only palliative therapies are typically
available (1,2). Although complete resection of the lung
is the only known curative therapy in stages I–III of this disease,
a number of patients do experience relapse following surgery
(3). A total of one-third of patients
with lung cancer survive for a number of years following diagnosis
(3). Recently, there has been a
paradigm shift in lung cancer therapeutics, from the use of
conventional cytotoxic drugs to the use of variable
molecular-targeted therapeutics, including gefitinib, erlotinib and
crizotinib (4).
The epidermal growth factor receptor (EGFR) is a
member of the transmembrane tyrosine kinase receptor family and its
expression is elevated in a number of solid tumors. Initially, the
overexpression of EGFR was demonstrated to be a strong prognostic
indicator in head and neck, ovarian, cervical, bladder and
esophageal cancer (5). Numerous
trials aimed at identifying the benefits of administering anti-EGFR
therapies to patients with cancer have been conducted (6,7). There are
currently two types of therapeutics available for inhibiting EGFR:
One is a monoclonal antibody against EGFR and the other includes
small-molecule inhibitors of EGFR tyrosine kinase (4). Gefitinib, trade name Iressa®,
is a small molecule that inhibits EGFR tyrosine kinase activity,
and is the first molecular-targeted agent registered to treat
advanced non-small cell lung cancer (NSCLC). Gefitinib trials have
demonstrated that there is an overall response rate of 10–20% in
patients with advanced NSCLC, which could be as high as 70% in
patients with EGFR gene mutations when used as a first-line
treatment (8,9). There are two biomarkers, the presence of
the EGFR gene mutations and an increase in gene copy number, known
to be potent predictors of the response to gefitinib (10,11). The
presence of mutations in the EGFR gene is a major determinant of
the gefitinib response; these mutations are small deletions
affecting amino acids 747–750, or are point mutations (12–15).
There are four obstacles involved in the resistance
to gefitinib that have recently been identified (16,17). The
first is the activation of alternative tyrosine kinase receptors
that bypass the EGFR pathway. The second is the constitutive
activation of signaling pathways downstream of the EGFR pathway,
including the phosphoinositide 3-kinase/protein kinase B (Akt)
pathway or proto-oncogene tyrosine-protein kinase Src family
kinases, which induce resistance. The third is the
ligand-independent activation of EGFR and the associated resistance
to therapy. The fourth is a secondary EGFR mutation which changes
the sensitivity to EGFR inhibitors. Efforts to overcome these
complications involved in the resistance to gefitinib include the
use of tyrosine kinase inhibitors that target other molecules;
however, many limitations remain (18).
In the case of other molecular-targeted
therapeutics, such as imatinib mesylate, changing the redox status
with L-ascorbic acid restores drug sensitivity in
imatinib-resistant cell lines. Previous studies have revealed that
oxidative stress increases phosphorylation of the EGFR in
keratinocytes (19–21). The antioxidant activity of L-ascorbic
acid (vitamin C, water-soluble L-ascorbic acid) is achieved
primarily via its ability to donate electrons and therefore
function as a reducing agent (22).
Cell signaling activities and the expression of associated
molecules are sensitive to exogenous and intracellular redox
status, and L-ascorbic acid serves a role in downregulating the
expression of certain signaling molecules, including nuclear
factor-κB, activator protein-1, Fos proto-oncogene and Jun
proto-oncogene, and in regulating apoptosis (23).
There is considerable debate about whether
L-ascorbic acid serves a therapeutic role in cancer. Recent reports
have established that L-ascorbic acid also acts as a pro-oxidant
and, depending on the dose, cell type and ability to stimulate
apoptosis, may kill cancer cells (24). In human colon cancer cell lines,
L-ascorbic acid functions as a potent antioxidant and blocks the
chemotherapy-mediated induction of apoptosis (25). These results support the hypothesis
that antioxidants may protect cancer cells from the free radical
damage induced by chemotherapy. In addition to oxidation,
L-ascorbic acid may serve a modulatory role in cellular
phosphorylation-dephosphorylation events (20,26–28).
However, the mechanisms underlying these effects are currently
unclear. Clinical trials of antioxidant plus chemotherapy regimens
in patients with cancer are uncommon, due to concerns over
inhibiting the effect of chemotherapy via decreasing the production
of reactive oxygen species (ROS) (25). However, EGFR tyrosine kinase
inhibitors exhibit a different mechanism of action when
administered with cytotoxic chemotherapy to kill cancer cells
(29).
In the present study, the EGFR tyrosine kinase
inhibitor gefitinib was used as an apoptotic stimulus in lung
cancer cell lines, and L-ascorbic acid was used as an adjuvant, in
order to kill cancer cells. The aims of the present study were to
evaluate whether L-ascorbic acid when co-administered with
gefitinib therapy serves an additive or synergistic
tumor-inhibitory role, and to elucidate the mechanisms underlying
its effects in NSCLC cell lines.
Materials and methods
Cell lines
Three human NSCLC cell lines, A549, Calu-3 and
HCC827, were obtained from the American Type Culture Collection
(Manassas, VA, USA). HCC827 possesses an acquired mutation in the
EGFR tyrosine kinase domain (E746-A750 deletion), and the other two
cell lines contain a wild-type EGFR tyrosine kinase domain. The
cells were cultured in complete RPMI-1640 (Thermo Fisher
Scientific, Inc., Waltham, MA USA) supplemented with 2 mM
L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin and 10%
heat-inactivated fetal bovine serum (Thermo Fisher Scientific,
Inc.), and were used for experiments whilst in the log phase of
growth.
Reagents
Gefitinib (ZD1839; trade name Iressa™) was kindly
provided by AstraZeneca Pharmaceuticals (Macclesfield, UK) and was
dissolved in dimethyl sulfoxide at a stock concentration 10 mM and
stored at −20°C for in vitro experiments. L-ascorbic acid
(sodium salt) was purchased from Sigma-Aldrich (St. Louis, MO,
USA). For the flow cytometric analysis, a phycoerythrin-conjugated
mouse anti-human EGFR antibody was purchased from BD Pharmingen
(San Diego, CA, USA). For the western blot analysis, antibodies
against EGFR (cat. no. 4267S; dilution, 1:1,000), phosphorylated
(p)-EGFR [Tyr845 (cat. no. 6963S; dilution, 1:1,000), Tyr992 (cat.
no. 2235S; dilution, 1:1,000) and Tyr1068 (cat. no. 2234S;
dilution, 1:1,000)], Akt (cat. no. 9272S; dilution, 1:1,000), p-Akt
(cat. no. 4060S; dilution, 1:1,000), signal transducer and
activator of transcription 3 (Stat3; cat. no. 9139S; dilution,
1:1,000) and p-Stat3 (cat. no. 9145S; dilution, 1:1,000) were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Antibodies against extracellular signal-related kinase (Erk; cat.
no. sc-94; dilution, 1:1,000), p-Erk (cat. no. sc-7383; dilution,
1:1,000) and β-actin (cat. no. sc-47778; dilution, 1:2,000) were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Trypan blue and 7-aminoactinomycin D were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Cell proliferation and viability
assays
A total of 8×105 cells from each cell
line were cultured in T25 culture flasks at 37°C in a 5%
CO2 humidified incubator, with or without dissolved
L-ascorbic acid and/or gefitinib in PBS at 0, 20 and 40 mM
gefitinib in A549 cells, 0.0.5 and 1.0 mM in Calu-3 cells, 0, 2.5
and 5.0 µM in HCC827 cells, and 0 and 0.5 mM L-ascorbic acid in
A549 cells, 0, 2.5 and 5.0 mM in Calu-3 cells, and 0, 0.5 and 1.0
mM in HCC827 cells. The cells were pretreated with L-ascorbic acid
for 1 h and then treated with gefitinib for 48 h at room
temperature. The number of cells and the viability of the cells
were determined using a Trypan blue dye exclusion assay. An
alternative method to monitor cell proliferation, the
AlamarBlue® assay (cat. no. BUF012A; Bio-Rad Antibodies,
Oxford, UK), was also used. AlamarBlue® is a redox
indicator that is reduced by reactions innate to cellular
metabolism (30). Thus, it provides
an indirect measure of the number of viable cells. The cells
(5×103) were seeded onto a 96-well plate and then
treated with the aforementioned doses of L-ascorbic acid and/or
gefitinib for 48 h at 37°C. AlamarBlue® (10% v/v in
medium) was subsequently added to the cells, the cells were
incubated for 6 h at 37°C and fluorescence was measured at 530 nm
excitation and 590 nm emission wavelengths in a spectrofluorometer
(Fluoroskan Ascent™ FL; Labsystems Diagnostics Oy, Vantaa,
Finland). The results are expressed as a percentage relative to the
total cell number, and the groups that were not treated with
L-ascorbic acid and gefitinib were used as control group.
Detection of intracellular ROS and
cell cycle analysis
Cells from the three lung cancer cell lines were
seeded into a 96-well plate at a density of 1×104
cells/well and incubated with 50 mM
2′,7′-dichlorodihydrofluorescein diacetate (Sigma-Aldrich; Merck
KGaA) for 5, 10, 15, 20, 25 and 30 min at 37°C in a 5%
CO2 humidified incubator. The cells were then analyzed
using a CytoFluor 2350 plate reader (EMD Millipore, Billerica, MA,
USA) with the excitation wavelength set at 485 nm and the emission
wavelength at 530 nm. For cell cycle analydsis, cells were
pretreated with L-ascorbic acid in complete medium for 1 h and then
treated with gefitinib for 48 h at 37°C. The cells were then
trypsinized, washed twice with cold PBS and centrifuged for 5 min
with 200 × g at room temperature. The pellet was resuspended in 1
ml cold PBS and 4 ml cold ethanol for 30 min at 4°C. The cells were
centrifuged at 200 × g for 5 min, and the pellet was washed twice
with cold PBS, suspended in 500 µl propidium iodide staining
solution (BD Pharmingen) and analyzed by flow cytometry BD
FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA).
Western blotting
The cells were lysed and the proteins were extracted
in a buffer containing 50 mM Tris-HCl (pH 7.4), 1% NP-40, 0.25%
sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 50 mM NaF, 1 mM sodium
orthovanadate, 1 mM phenylmethylsulfonyl fluoride and a protease
inhibitor cocktail (Sigma-Aldrich; Merck KGaA). The protein
concentration was measured using a Bio-Rad Protein Assay kit
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Equal amounts of
protein (20–30 µg) were resolved using 8% (for EGFR and p-EGFR) or
12% [for Akt, p-Akt (Ser473), Erk, p-Erk, Stat3, p-Stat3 and
β-actin] SDS-PAGE and transferred onto nitrocellulose membranes.
The membranes were blocked with 5% nonfat milk and 0.1%
Tween-20-PBS for 1 h at RT with gentle agitation, washed with 0.1%
Tween 20-PBS, and then exposed to the relevant primary antibody for
1 h at room temperature. The primary antibodies were diluted
1:1,000 in 0.1% Tween-20-PBS; the antibodies included anti-EGFR
(cat. no. 4267S), anti-p-EGFR (Tyr845; cat. no. 6963S), anti-p-EGFR
(Tyr992; cat. no. 2235S), anti-p-EGFR (Tyr1068; cat. no. 2234S),
anti-Akt (cat. no. 9272S) and anti-p-Akt (Ser473; cat. no. 4060S)
from Cell Signaling Technology, Inc.; anti-Erk (cat. no. sc-94) and
anti-p-Erk (cat. no. sc-7383) from Santa Cruz Biotechnology, Inc.
Subsequent to washing, the blots were exposed to a
biotin-conjugated rabbit developed anti-mouse (cat. no. sc-358919;
dilution, 1:1,000; Santa Cruz Biotechnology, or mouse developed
anti-rabbit antibody (cat. no. sc-2491; dilution, 1:1,000; Santa
Cruz Biotechnology,) for 1 h at room temperature. The membranes
were then washed, incubated with a 1:5,000 dilution of
streptavidin-horseradish peroxidase at room temperature, and the
immunoreactive proteins were visualized using an ECL detection
system (GE Healthcare Life Sciences, Chalfont, UK).
Statistical analysis
The results are presented as the mean, with error
bars representing the standard deviation. Statistical analyses were
performed using SPSS v.18 (SPSS, Inc., Chicago, IL, USA).
Non-parametric tests (Mann-Whitney U test) were used to evaluate
significant differences among the continuous variables and the
Kruskal-Wallis test was used to assess the statistical
significance. For multiple comparisons, MedCalc®
software for Windows (v.16.1) was used. (MedCalc®
Software, Mariakerke, Belgium) P<0.05 was considered to indicate
a statistically significant difference.
Results
Increased expression of EGFR is
induced by L-ascorbic acid or gefitinib in NSCNC cell lines
Cell surface EGFR protein expression was observed in
NSCLC cell lines treated with or without L-ascorbic acid and/or
gefitinib. When L-ascorbic acid and gefitinib were administered
together, a 91.8% of cells expressed surface EGFR proteins, as
compared with 35.3% of untreated Calu-3 cells. Treatment with
gefitinib alone increased surface EGFR expression in Calu-3 cells,
compared with treatment with L-ascorbic acid alone (88.9 vs. 64.5%
of cells; P<0.05). However, co-treatment with gefitinib and
L-ascorbic acid resulted in no significant increase in EGFR
expression, compared with treatment with gefitinib alone (91.8 vs.
88.9% of cells). In untreated A549 cells, 60.4% of cells expressed
surface EGFR protein; this percentage was higher compared with that
in untreated Calu-3 cells. The two agents demonstrated a tendency
to increase EGFR expression in A549 cells; however, L-ascorbic acid
exhibited no synergistic or additional effect on EGFR expression
when co-administered with gefitinib. By contrast, L-ascorbic acid
and gefitinib administered in combination decreased EGFR expression
levels in A549 cells: 63.3% of cells co-treated with the two agents
expressed the EGFR protein, compared with 80.5% of cells treated
with L-ascorbic acid alone and 72.4% treated with gefitinib alone,
although no significance was observed.
Effects of gefitinib and L-ascorbic
acid on cancer cell growth and proliferation
A549 is a lung cancer cell line that is resistant to
EGFR tyrosine kinase inhibitors. Proliferation of these cells was
inhibited with high doses of gefitinib; however, relatively low
doses of L-ascorbic acid (0.5 mM) administered with gefitinib
inhibited cell proliferation to a greater extent (P=0.046; Fig. 1A). The addition of 0.5 mM L-ascorbic
acid to 20 µM gefitinib inhibited cellular proliferation by >32%
(85.6±5.4 vs. 52.7±7.3% of the total number of cells; P=0.046). The
inhibitory effect of gefitinib on Calu-3 cells, which exhibit the
wild-type EGFR but are sensitive to gefitinib, was ~60% of that
observed in the control Calu-3 cells. The additional L-ascorbic
acid significantly inhibited the proliferation of Calu-3 tumor
cells at each concentration level of gefitinib (overall, P=0.027,
no gefitinib; P=0.039, 0.5 µM gefitinib; and P=0.127, 1 µM
gefitinib, respectively; Fig. 1B).
The additional significant inhibitory effect of the L-ascorbic acid
was also observed in gefitinib-sensitive HCC827 cells at each
gefitinib concentration (overall, P=0.066, P=0.039 and P=0.027,
respectively; Fig. 1C). The
AlamarBlue assay used to measure growth inhibition and the Trypan
blue dye exclusion assay demonstrated a similar pattern of
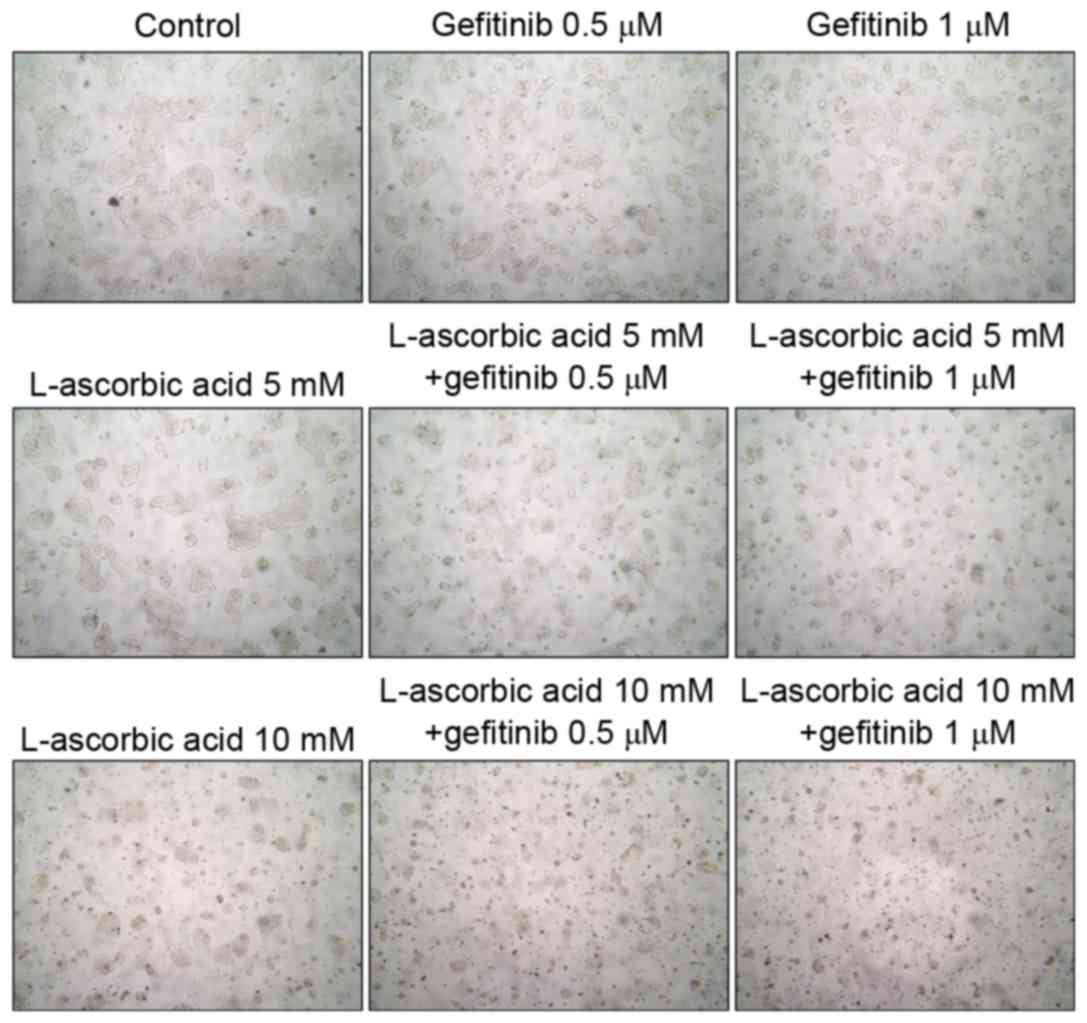
inhibition (data not presented). Microscopic examination of Calu-3
cells revealed that they experienced growth inhibition (Fig. 2). Similar results were observed in all
three cell lines examined.
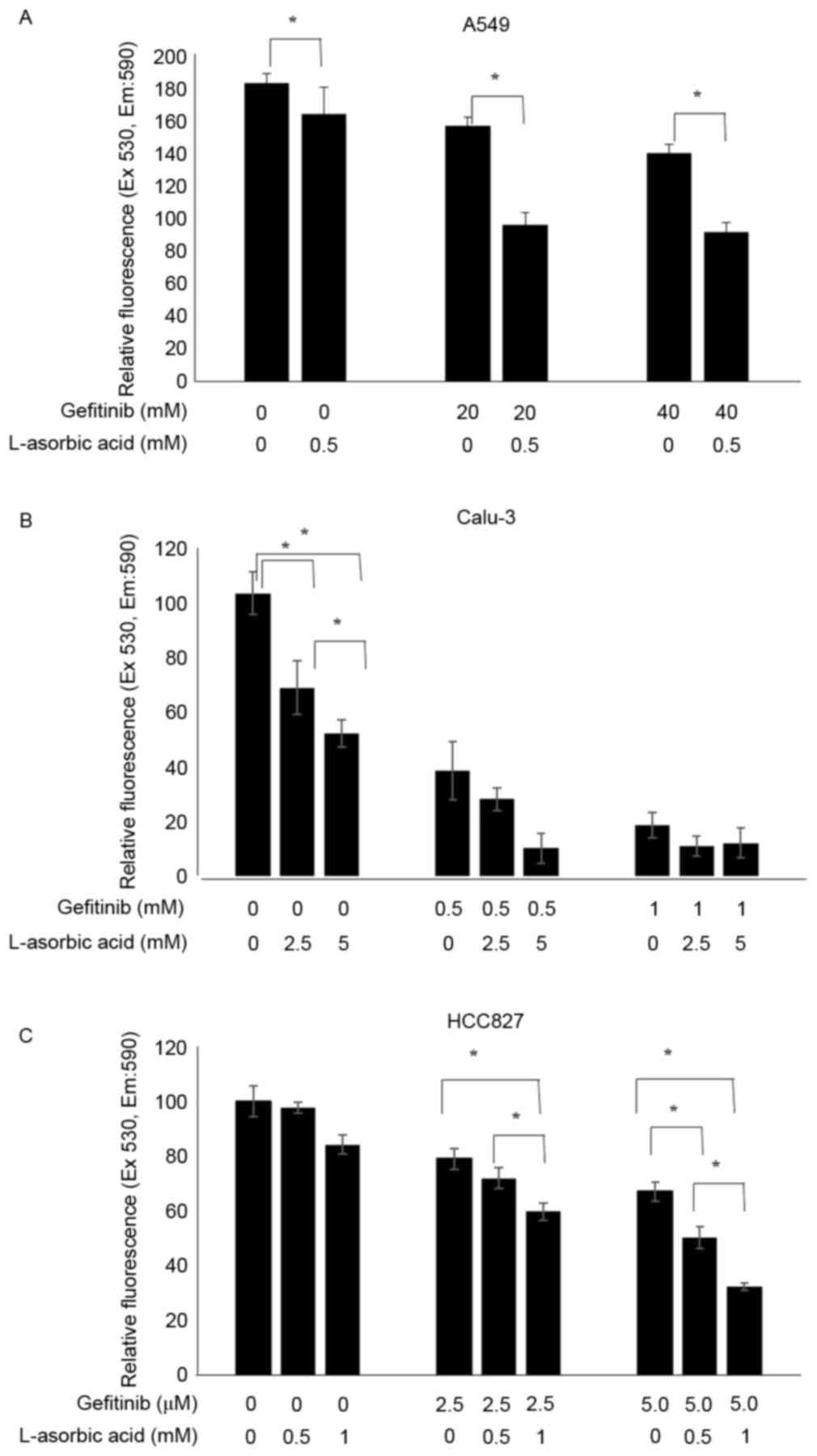 | Figure 1.Increased antiproliferative activity
of gefitinib with L-ascorbic acid in non-small cell lung cancer
cell lines. A total of 1×106 cells were incubated with
various concentrations of L-ascorbic acid and/or gefitinib, and the
cytotoxic effects were assessed using an AlamarBlue assay. Cells
were pretreated with L-ascorbic acid for 1 h, followed by gefitinib
treatment for 48 h. Cell viabilities are expressed as the ratio of
the absorbance of treated cells to control cells. Independent
experiments were performed in triplicate and the data are presented
as the mean ± standard deviation. (A) A549 cells exposed to 0, 20
and 40 µM gefitinib and 0 and 0.5 mM L-ascorbic acid. (B) Calu-3
cells exposed to 0, 0.5 and 1 µM gefitinib and 0, 2.5 and 5 mM
L-ascorbic acid. (C) HCC827 cells exposed to 0, 2.5 and 5.0 nM
gefitinib and 0, 0.5 and 1 mM L-ascorbic acid. *P<0.05,
statistically significant difference between the groups. Ex,
excitation; Em, emission. |
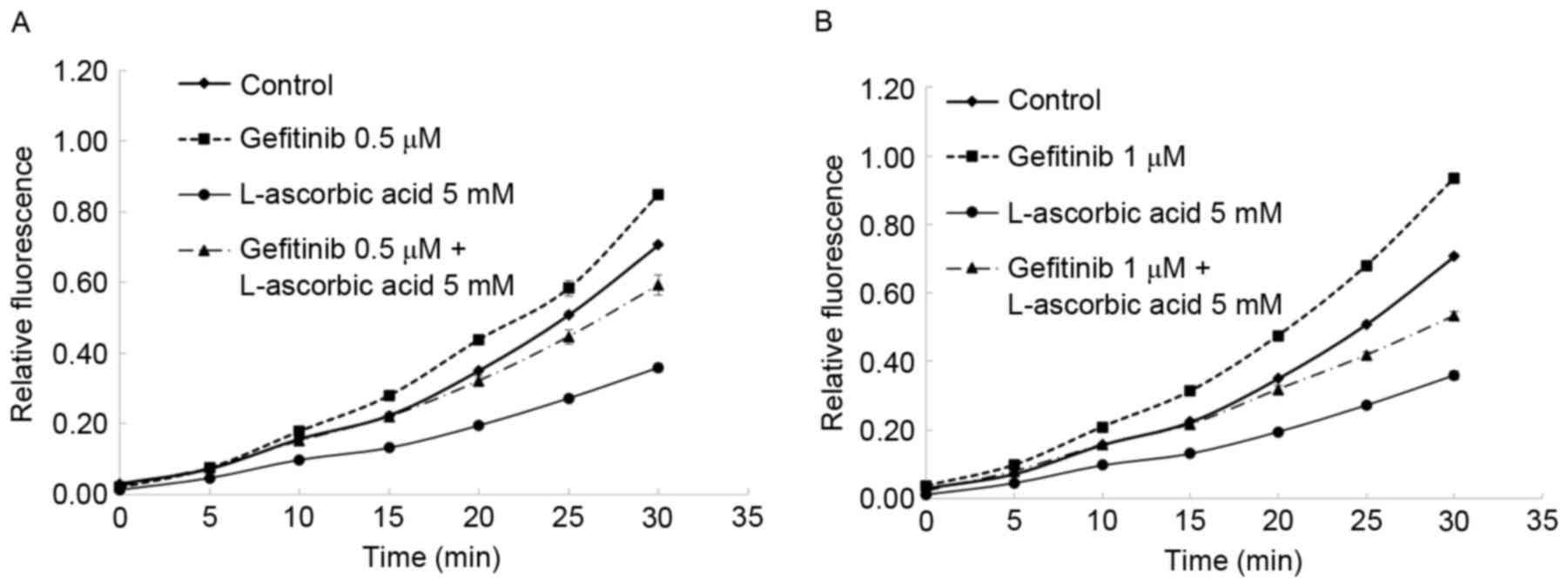
Changes in the cell cycle and ROS
production effected by gefitinib and L-ascorbic acid
The combination of L-ascorbic acid with gefitinib
demonstrated a synergistic reduction in the number of cells in the
S phase. In the Calu-3 cells, the percentage of cells in S phase of
the cell cycle was reduced from 35.8% in the control cells to 17.9%
in cells treated with gefitinib alone; this percentage was 23.1% in
cells treated with L-ascorbic acid alone and 11% in cells
co-treated with gefitinib and L-ascorbic acid. The highest observed
arrest in the G2 phase was with L-ascorbic acid alone,
and gefitinib did not induce G2 arrest. The effect of
L-ascorbic acid on G2 arrest was neutralized when it was
administered in combination with gefitinib. Also, the antioxidant
activity of L-ascorbic acid was observed: In Calu-3 cells, blocking
EGFR tyrosine kinase with gefitinib induced high intracellular
levels of ROS. This action was attenuated by the antioxidant
L-ascorbic acid, which reduced intracellular ROS levels in Calu-3
cells to approximately the level observed in the untreated cells
(Fig. 3).
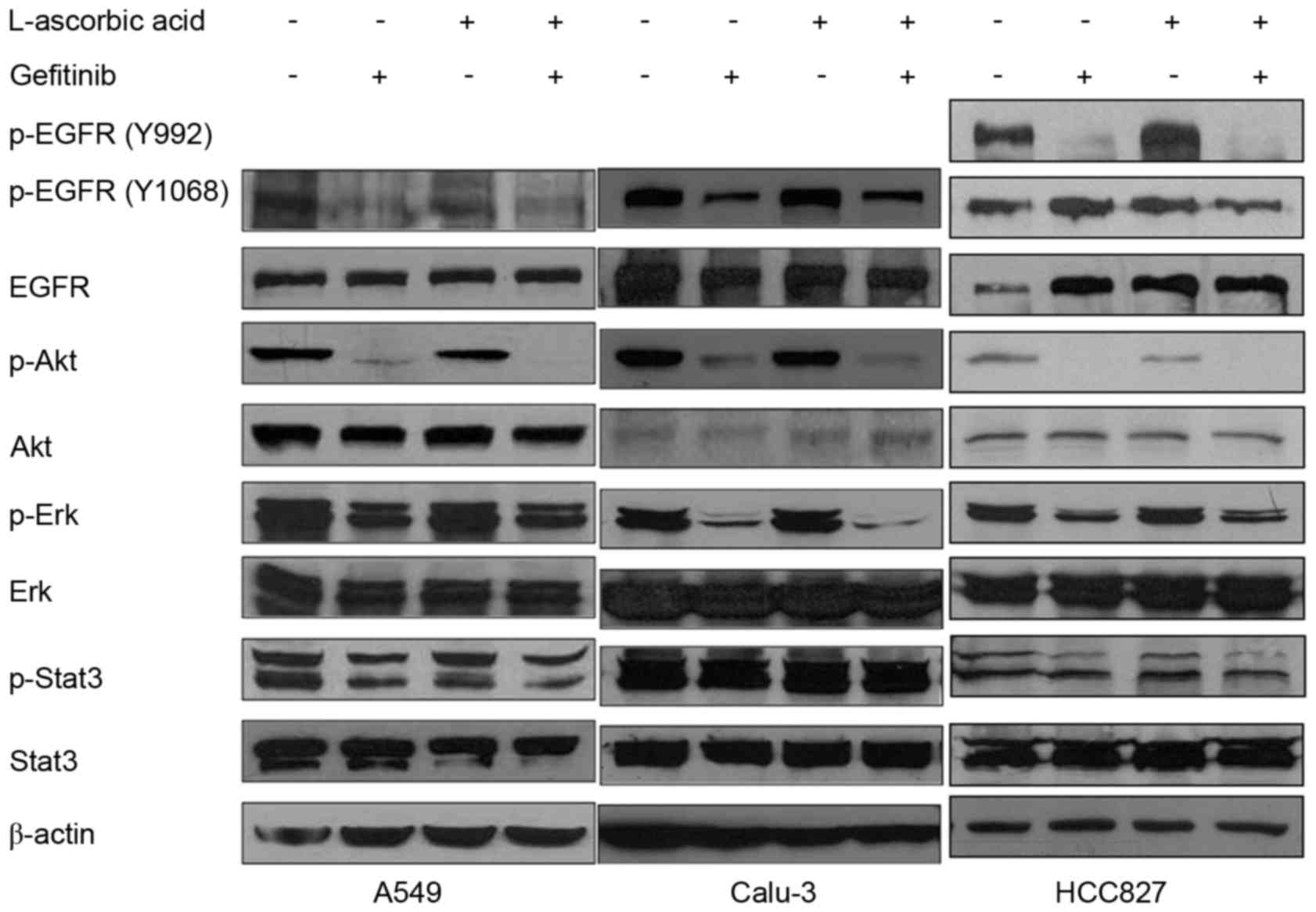
Changes in EGFR phosphorylation
following treatment with gefitinib and L-ascorbic acid
A total of three antibodies, Tyr992, Tyr1068 and
Tyr845, were used to identify different phosphorylation loci of the
EGFR. The addition of L-ascorbic acid to gefitinib did not
significantly modify the phosphorylation levels of Erk1/2, Stat3 or
EGFR in A549 cells, but slightly reduced the phosphorylation levels
of Akt (Fig. 4, left panel). In
Calu-3 cells, treatment with L-ascorbic acid alone was not observed
to have a significant effect on EGFR phosphorylation or the
associated downstream signaling pathways. By contrast, L-ascorbic
acid co-administered with gefitinib decreased the phosphorylation
levels of Akt and Erk1/2. The phosphorylation level of Stat3 was
not significantly altered following the co-administration of
L-ascorbic acid with gefitinib (Fig.
4, middle panel). In HCC827 cells, which exhibit an EGFR
mutation and are sensitive to gefitinib, L-ascorbic acid
administered in combination with gefitinib was not observed to
significantly affect the signaling cascades downstream of the EGFR
pathway. Monotherapy with gefitinib decreased the phosphorylation
levels of the downstream molecules of the EGFR pathway, including
Akt, in the HCC827 cell line (Fig. 4,
right panel), but no significance was observed. It was also
examined whether changes in ROS affected the signaling cascades
downstream of the EGFR signaling pathway.
Discussion
Numerous small molecules have been developed to
replace cytotoxic chemotherapy agents in cancer therapeutics;
however, the development of novel resistance to these drugs, along
with resistance mechanisms, remains problematic. Mutations in the
receptor kinase domains of cancer cells are the principal cause of
resistance to tyrosine kinase inhibitor therapy (12,17,29).
Imatinib mesylate, which is applied clinically in the treatment of
chronic myelogenous leukemia (CML), is the first small-molecule
drug for which resistance to tyrosine kinase inhibitors was
recognized; the mechanism for resistance was identified to
originate from a mutation in the kinase domain (7,13,31). The increase in intracellular ROS
production induced by imatinib mesylate in patients with CML causes
genetic instability and gene mutations, leading to mutation of the
kinase domain and subsequent drug resistance. Antioxidants may
decrease the levels of ROS present in cells and, therefore,
decrease the risk of gene mutations and associated drug resistance
(32).
Various previous in vitro and in vivo
studies have suggested that certain antioxidants may increase the
effects of cytotoxic therapy. However, clinical studies using
antioxidants as an adjuvant to chemotherapy have so far been
unsuccessful (10,19,25,28,33,34).
L-ascorbic acid may function as either a pro-oxidant or an
antioxidant at physiological micromolar concentrations (normal
range, <0.1 mM) in the presence of trace amounts of transition
metals including copper and iron (20,34–40). There
is controversy regarding the potential role and dose of L-ascorbic
acid that should be recommended for use in cancer therapy (40). Gefitinib is the first small-molecule
drug developed to inhibit tyrosine kinase in human NSCLC (41). Mutant forms of EGFR and increased EGFR
copy numbers are considered to be successful predictive biomarkers
for therapeutic responses.
Similar to imatinib mesylate, ROS may induce changes
in cell signaling, genetic instability and other various steps in
intracellular pathways. One of the functions of intracellular ROS
is to decrease phosphatase activity and to upregulate tyrosine
kinase once at a certain concentration; thus, ROS may serve a role
in carcinogenesis (31). At present,
there have been few studies on the effects of ROS on the
phosphorylation of EGFR (42).
The present study hypothesized that ROS affects EGFR
kinase activity and the sensitivity of cancer cells to EGFR
tyrosine kinase inhibitor therapy, and that L-ascorbic acid may
have an additive or synergistic effect when co-administered with an
EGFR tyrosine kinase inhibitor. As the EGFR mutation is an
important biomarker for the response to gefitinib, three human
NSCLC cell lines were used, including Calu-3 and A549 (wild-type
EGFR) and HCC827, which has a mutation in the EGFR tyrosine kinase
domain (E746-A750 deletion) and is known to respond to gefitinib.
The effects of L-ascorbic acid and gefitinib were synergistic in
all cell lines when administered at various dosages. In the present
study, gefitinib acted as a pro-oxidant and L-ascorbic acid
attenuated the elevation in ROS levels in lung cancer cell lines.
It was also revealed that L-ascorbic acid monotherapy reduced the
percentage of Calu-3 cells in the S phase, and that this effect was
augmented when administered in combination with gefitinib. Thomas
et al demonstrated that L-ascorbic acid induces transient
cell cycle arrest by delaying the accumulation and activation of
Cdc25C (42).
The antiproliferative and anticancer mechanisms of
L-ascorbic acid and gefitinib differ. L-ascorbic acid may kill or
inhibit the growth of numerous tumor cell lines and also increase
the potency of certain radiosensitizing drugs (43). Beyond its antioxidant role, the exact
mechanisms underlying the effects L-ascorbic acid have yet to be
elucidated. Although different concentrations of L-ascorbic acid
were chosen for each cell line, none of the selected concentrations
killed all the cells. In the present study, L-ascorbic acid
administered alone inhibited cancer cell growth through its
antioxidant function and by downregulating EGFR phosphorylation.
Gefitinib alone generated ROS, inhibited kinase activity and
downregulated Akt and Erk expression. In order to identify the
optimal redox environment for EGFR tyrosine kinase inhibitors to
obtain a maximum anticancer effect, the effect of ROS in EGFR
tyrosine kinase inhibitor therapy was examined using nonspecific
ROS blocking agents. It is not likely that ROS were the leading
cause of the antitumor effects in the present study. These results
indicate that L-ascorbic acid acts via its own antitumor mechanisms
that are separate to ROS activity, and that it is involved in the
inhibition of the EGFR signaling cascade.
In conclusion, the present study has demonstrated
that, when co-administered with gefitinib, L-ascorbic acid
functions as an antioxidant and is associated with the
downregulation of EGFR phosphorylation in three NSCLC cell lines.
However, several issues remain to be resolved, including the
practical delivery of a clinically relevant therapeutic dose of
L-ascorbic acid, the optimal redox status that exhibits an
antitumor effect and the in vivo responses. It is suggested
that L-ascorbic acid may overcome the wild-type EGFR tyrosine
kinase inhibitor resistance and induce a synergistic response when
co-administered with EGFR tyrosine kinase inhibitors in NSCLC.
These results also suggest that improved clinical outcomes in lung
cancer therapy may be obtained through L-ascorbic acid therapy
combined with an EGFR tyrosine kinase inhibitor therapy such as
gefitinib or erlotinib.
Acknowledgements
The present study was supported partly by Basic
Science Research Program through the National Research Foundation
of Korea (NRF) funded by the Ministry of Education (grant. no.
2013R1A2A2A01067836) and partly by intramural Research Promotion
Grants from Ewha Womans University, School of Medicine.
References
|
1
|
Ferlay J, Soerjomataram I, Ervik M,
Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and
Bray F: GLOBOCAN 2012 v1.0, Cancer Incidence, Mortality and
Prevalence Worldwide in 2012. IARC CancerBase No. 11 (Internet).
International Agency for Research on Cancer, Lyon, France.
2013.http://globocan.iarc.fr
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vincent TD, Steven AR and Theodore SL:
DeVita, Hellman and Rosenberg's Cancer: Principles & Practice
of Oncology. 9th. Wolters Kluwer Health/Lippincott Williams &
Wilkins; Philadelphia, PA: pp. 799–845. 2011
|
|
4
|
Lam KC and Mok TS: Targeted therapy: An
evolving world of lung cancer. Respirology. 16:13–21. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Braughler JM, Duncan LA and Chase RL: The
involvement of iron in lipid peroxidation. Importance of ferric to
ferrous ratios in initiation. J Biol Chem. 261:10282–10289.
1986.PubMed/NCBI
|
|
6
|
Cappuzzo F, Varella-Garcia M, Shigematsu
H, Domenichini I, Bartolini S, Ceresoli GL, Rossi E, Ludovini V,
Gregorc V, Toschi L, et al: Increased HER2 gene copy number is
associated with response to gefitinib therapy in epidermal growth
factor receptor-positive non-small-cell lung cancer patients. J
Clin Oncol. 23:5007–5018. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carter TA, Wodicka LM, Shah NP, Velasco
AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH III,
Edeen PT, et al: Inhibition of drug-resistant mutants of ABL KIT,
and EGF receptor kinases. Proc Natl Acad Sci USA. 102:pp.
11011–11016. 2005; View Article : Google Scholar : PubMed/NCBI
|
|
8
|
D'Incecco A and Cappuzzo F: Gefitinib for
non-small-cell lung cancer treatment. Expert Opin Drug Saf.
10:987–996. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang F, Wang LD, Li B and Sheng ZX:
Gefitinib compared with systemic chemotherapy as first-line
treatment for chemotherapy-naive patients with advanced non-small
cell lung cancer: A meta-analysis of randomised controlled trials.
Clin Oncol(R Coll Radiol). 24:396–401. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duarte TL and Lunec J: Review: When is an
antioxidant not an antioxidant? A review of novel actions and
reactions of vitamin C. Free Radic Res. 39:671–686. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Giordano CR, Mueller KL, Terlecky LJ,
Krentz KA, Bollig-Fischer A, Terlecky SR and Boerner JL: A targeted
enzyme approach to sensitization of tyrosine kinase
inhibitor-resistant breast cancer cells. Exp Cell Res.
318:2014–2021. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Koptyra M, Falinski R, Nowicki MO,
Stoklosa T, Majsterek I, Nieborowska-Skorska M, Blasiak J and
Skorski T: BCR/ABL kinase induces self-mutagenesis via reactive
oxygen species to encode imatinib resistance. Blood. 108:319–327.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tartarone A and Lerose R: Clinical
approaches to treat patients with non-small cell lung cancer and
epidermal growth factor receptor tyrosine kinase inhibitor acquired
resistance. Ther Adv Respir Dis. 9:242–250. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Russo A, Franchina T, Ricciardi GR, Picone
A, Ferraro G, Zanghì M, Toscano G, Giordano A and Adamo V: A decade
of EGFR inhibition in EGFR-mutated non small cell lung cancer
(NSCLC): Old successes and future perspectives. Oncotarget.
6:26814–26825. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang L and Fu L: Mechanisms of resistance
to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 5:390–401.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tan CS, Gilligan D and Pacey S: Treatment
approaches for EGFR-inhibitor-resistant patients with
non-small-cell lung cancer. Lancet Oncol. 16:e447–e459. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Meves A, Stock SN, Beyerle A, Pittelkow MR
and Peus D: H(2)O(2) mediates oxidative stress-induced epidermal
growth factor receptor phosphorylation. Toxicol Lett. 122:205–214.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miller DM, Buettner GR and Aust SD:
Transition metals as catalysts of ‘autoxidation’ reactions. Free
Radic Biol Med. 8:95–108. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Monteiro HP, Ivaschenko Y, Fischer R and
Stern A: Ascorbic acid inhibits protein tyrosine phosphatases in
NIH 3T3 cells expressing human epidermal growth factor receptors.
Int J Biochem. 25:1859–1864. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nicholson RI, Gee JM and Harper ME: EGFR
and cancer prognosis. Eur J Cancer. 37 Suppl 4:S9–S15. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ono M, Hirata A, Kometani T, Miyagawa M,
Ueda S, Kinoshita H, Fujii T and Kuwano M: Sensitivity to gefitinib
(Iressa, ZD1839) in non-small cell lung cancer cell lines
correlates with dependence on the epidermal growth factor (EGF)
receptor/extracellular signal-regulated kinase 1/2 and EGF
receptor/Akt pathway for proliferation. Mol Cancer Ther. 3:465–472.
2004.PubMed/NCBI
|
|
24
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wenzel U, Nickel A, Kuntz S and Daniel H:
Ascorbic acid suppresses drug-induced apoptosis in human colon
cancer cells by scavenging mitochondrial superoxide anions.
Carcinogenesis. 25:703–712. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pal SK and Pegram M: Epidermal growth
factor receptor and signal transduction: Potential targets for
anti-cancer therapy. Anticancer Drugs. 16:483–494. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Petrelli F, Borgonovo K, Cabiddu M and
Barni S: Efficacy of EGFR tyrosine kinase inhibitors in patients
with EGFR-mutated non-small-cell lung cancer: A meta-analysis of 13
randomized trials. Clin Lung Cancer. 13:107–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Prasad KN and Kumar R: Effect of
individual and multiple antioxidant vitamins on growth and
morphology of human nontumorigenic and tumorigenic parotid acinar
cells in culture. Nutr Cancer. 26:11–19. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin Y, Wang X and Jin H: EGFR-TKI
resistance in NSCLC patients: Mechanisms and strategies. Am J
Cancer Res. 4:411–435. 2014.PubMed/NCBI
|
|
30
|
Collins L and Franzblau SG: Microplate
alamar blue assay versus BACTEC 460 system for high-throughput
screening of compounds against Mycobacterium tuberculosis and
Mycobacterium avium. Antimicrob Agents Chemother. 41:1004–1009.
1997.PubMed/NCBI
|
|
31
|
Tarumoto T, Nagai T, Ohmine K, Miyoshi T,
Nakamura M, Kondo T, Mitsugi K, Nakano S, Muroi K, Komatsu N and
Ozawa K: Ascorbic acid restores sensitivity to imatinib via
suppression of Nrf2-dependent gene expression in the
imatinib-resistant cell line. Exp Hematol. 32:375–381. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Prasad KN, Sinha PK, Ramanujam M and
Sakamoto A: Sodium ascorbate potentiates the growth inhibitory
effect of certain agents on neuroblastoma cells in culture. Proc
Natl Acad Sci USA. 76:pp. 829–832. 1979; View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pathak AK, Bhutani M, Guleria R, Bal S,
Mohan A, Mohanti BK, Sharma A, Pathak R, Bhardwaj NK, Prasad KN and
Kochupillai V: Chemotherapy alone vs. chemotherapy plus high dose
multiple antioxidants in patients with advanced non small cell lung
cancer. J Am Coll Nutr. 24:16–21. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sane AT, Cantin AM, Paquette B and Wagner
JR: Ascorbate modulation of H(2)O(2) and camptothecin-induced cell
death in Jurkat cells. Cancer Chemother Pharmacol. 54:315–321.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rhee SG: Cell signaling. H2O2, a necessary
evil for cell signaling. Science. 312:1882–1883. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rosell R, Ichinose Y, Taron M, Sarries C,
Queralt C, Mendez P, Sanchez JM, Nishiyama K, Moran T, Cirauqui B,
et al: Mutations in the tyrosine kinase domain of the EGFR gene
associated with gefitinib response in non-small-cell lung cancer.
Lung Cancer. 50:25–33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sasazuki S, Hayashi T, Nakachi K, Sasaki
S, Tsubono Y, Okubo S, Hayashi M and Tsugane S: Protective effect
of vitamin C on oxidative stress: A randomized controlled trial.
Int J Vitam Nutr Res. 78:121–128. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chakraborthy A, Ramani P, Sherlin HJ,
Premkumar P and Natesan A: Antioxidant and pro-oxidant activity of
Vitamin C in oral environment. Indian J Dent Res. 25:499–504. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Buettner GR and Jurkiewicz BA: Catalytic
metals, ascorbate and free radicals: Combinations to avoid. Radiat
Res. 145:532–541. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cieslak JA and Cullen JJ: Treatment of
pancreatic cancer with pharmacological ascorbate. Curr Pharm
Biotechnol. 16:759–770. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Miller VA, Kris MG, Shah N, Patel J,
Azzoli C, Gomez J, Krug LM, Pao W, Rizvi N, Pizzo B, et al:
Bronchioloalveolar pathologic subtype and smoking history predict
sensitivity to gefitinib in advanced non-small-cell lung cancer. J
Clin Oncol. 22:1103–1109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Thomas CG, Vezyraki PE, Kalfakakou VP and
Evangelou AM: Vitamin C transiently arrests cancer cell cycle
progression inS phase and G2/M boundary by modulating the kinetics
of activation and the subcellular localization of Cdc25C
phosphatase. J Cell Physiol. 205:310–318. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Leekha A, Gurjar BS, Tyagi A, Rizvi MA and
Verma AK: Vitamin C in synergism with cisplatin induces cell death
in cervical cancer cells through altered redox cycling and p53
upregulation. J Cancer Res Clin Oncol. 142:2503–2514. 2016.
View Article : Google Scholar : PubMed/NCBI
|