Introduction
Estrogens act as important regulators of cell
proliferation, cell survival and differentiation in a variety of
organs and tissues, and have been implicated in the etiology of
various types of malignant and benign tumors (1). These compounds may induce de novo
breast cancer development by receptor-dependent or -independent
mechanisms, or actions mediated by the estrogen receptor (ER)
(2). A number of estrogen metabolites
have also been revealed to be more potent estrogenic compounds
compared with their precursor (3,4).
An endogenous estrogen of 17β-estradiol (E2),
2-methoxyestradiol (2-ME), is produced by sequential hydroxylation
of parental compounds followed by methylation by
catechol-o-methyltransferase, an enzyme present in numerous tissues
including the liver, kidney, brain, placenta, uterus and mammary
gland (5). Unlike the growth effects
of other estrogen metabolites, including 4-hydroxyestradiol and
16α-hydroxyestrone, 2-ME has been reported to elicit antitumor
effects on various types of cancer in vitro and in
vivo (6–10). A number of mechanisms underlying 2-ME
activity have been proposed, including effects on G2/M
cell-cycle arrest (11).
Additionally, this compound is able to induce mitochondrial
apoptotic signaling (8,12). In ER-positive MCF7 breast cancer
cells, 2-ME was demonstrated to increase p53 expression levels and
inhibit proliferation. These results have implications in
understanding the role of estrogen metabolite(s) in the regulation
of tumor progression (13). In a
previous study, apoptosis has received attention, as this process
is the primary mechanism underlying the anticancer drug-mediated
induction of tumor cell death (14).
However, autophagy has recently emerged as a key regulator of cell
death pathways, and may be involved in promoting cell death via
caspase-dependent and -independent mechanisms (15,16).
Furthermore, previous studies have revealed that apoptosis and
autophagy share certain common signaling pathways (16,17).
Among the ER-positive tumors, uterine leiomyosarcoma
(ULMS) is a relatively rare malignant smooth muscle cell tumor in
the uterus muscle layer and is divided into subtypes (18). ULMS accounts for 1% of all uterine
malignancies and ~30% of all uterine sarcomas including
carcinosarcomas, leiomyosarcomas and endometrial stromal sarcomas
(19). Although the pathogenesis and
molecular events that result in the development of leiomyosarcomas
remain largely unknown, this malignancy shares similar
morphological characteristics with leiomyoma that are considered to
promote transformation into ULMS and develop during reproductive
years with growth dependent on ovarian steroids (18,20). Local
therapy consisting of total hysterectomy and bilateral
salpingo-oophorectomy are generally recommended; however, these
surgical management strategies may not be appropriate for young
patients who desire to preserve their fertility potential (21). Therefore, there is a requirement to
develop effective therapeutics that are able to reverse ULMS
growth.
To the best of our knowledge, no previous studies
have evaluated the growth inhibitory effect of 2-ME on human ULMS.
In human clinical trials, 2-ME is well tolerated and was revealed
to exhibit low systematic toxicity (6). Therefore, the effects of 2-ME on
proliferation and programmed cell death in human ULMS cases were
determined in vitro using SK-LMS-1 cells. The present study
investigated whether 2-ME may be useful for treating human
ULMS.
Materials and methods
Chemicals
E2 was obtained from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany), Flavopiridol was supplied by Cayman Chemical
Company (Ann Arbor, MI, USA) and 2-ME was purchased from Selleck
Chemicals (Houston, TX, USA).
Cell culture
SK-LMS-1 cells were obtained from American Type
Culture Collection (Manassas, VA, USA). Cells were grown as
monolayer cultures in Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), supplemented
with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin (Gibco;
Thermo Fisher Scientific, Inc.) at 37°C in a humidified atmosphere
containing 95% O2 and 5% CO2. SK-LMS-1 cells
were plated on 6-well plastic tissue culture dishes (Nalge Nunc
International; Thermo Fisher Scientific, Inc.) at a density of
3×104 cells/well and grown for 24 h at 37°C. The medium
was replaced with phenol red-free Opti-MEM (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 5% charcoal dextran-stripped
FBS and 100 U/ml penicillin-streptomycin for 48 h to ensure the
depletion of steroid hormones in the cells at 37°C. Following 48 h,
the cells were exposed to various doses of 2-ME (10−7,
10−6 and 10−5 M) and Flavopiridol
(2×10−6 and 4×10−6 M). Each chemical was
dissolved in 100% dimethyl sulfoxide (DMSO) (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and added to phenol red-free
Opti-MEM containing 5% FBS and charcoal-dextran (starvation medium)
with a final DMSO concentration of 0.1%. Starvation medium
containing 2×10−6 or 4×10−6 M Flavopiridol
were used as positive controls (22)
and starvation medium containing 0.1% DMSO was used as a negative
control (vehicle). SK-LMS-1 cells were harvested 24 h after
treatment for evaluation of protein levels. All experiments were
performed in triplicate.
Cell viability assay
An MTT assay was performed to confirm the viability
of the endometrial cancer cells. SK-LMS-1 cells (1.0×103
cells/well) were seeded in 96-well plates for 24 h in an incubator
at 37°C. The following day, the medium was replaced with phenol
red-free Opti-MEM supplemented with 5% charcoal dextran-stripped
FBS and 100 U/ml penicillin-streptomycin in SK-LMS-1 cells for 48
h. After 48 h, the cells were exposed to E2 (10−9 M),
2-ME (10−7, 10−6 and 10−5 M) and
Flavopiridol (2×10−6 and 4×10−6 M) for 24 h.
Subsequently, an MTT assay was performed to determine cell
viability. A total of 5 mg/ml MTT stock solution (Sigma-Aldrich;
Merck KGaA) was diluted in PBS, then 20 µl MTT stock solution was
added to each well and the plates were incubated at 37°C for 4 h.
The yellow formazan crystals that formed were dissolved in 80 µl
DMSO. Following agitation, the absorbance was evaluated at 560 nm
using an ELISA plate reader. Each experiment was performed in
triplicate (n=6).
Terminal
deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL)
assay
The TUNEL assay was performed on chamber slides
(Thermo Fisher Scientific, Inc.) using the In Situ Cell Death
Detection kit, Fluorescein (Roche Diagnostics GmbH, Mannheim,
Germany). SK-LMS-1 cells were fixed in 4% paraformaldehyde. The
cells were then rinsed with PBS and incubated in blocking solution
(3% H2O2 in methanol) for 10 min at room
temperature and subsequently washed with PBS. Subsequently, cells
were permeabilized with 0.1% Triton X-100 (Amresco, LLC, Solon, OH,
USA) in 0.1% sodium citrate for 2 min on ice and then cells were
washed with PBS and the area around the sample was dried. The cells
were incubated with 50 µl TUNEL reaction mixture at 37°C for 60 min
in a dark humidified atmosphere. Using a BX51 fluorescence
microscope (Olympus Corporation, Tokyo Japan) the numbers of
TUNEL-positive cells were counted using fluorescence microscopy.
Positive control sections were treated with the same reagents and
pre-treated with 1,000 U/ml DNase I (Takara Bio Inc., Otsu, Japan)
for 10 min at room temperature prior to the TUNEL assay (data not
shown). The nuclei were stained with DAPI at 2 µg/ml
(Sigma-Aldrich; Merck KGaA).
Immunocytochemistry
The SK-LMS-1 cells (1.0×103 cells/well)
were cultured on 4-well plates (Nalge Nunc International; Thermo
Fisher Scientific, Inc.) for 24 h in an incubator at 37°. The
following day, the medium was replaced with phenol red-free
Opti-MEM supplemented with 5% charcoal dextran-stripped FBS and 100
U/ml penicillin-streptomycin in SK-LMS-1 cells for 48 h at 37°C.
Following 48 h, the cells were exposed to 2-ME (10−7,
10−6 and 10−5 M) and Flavopiridol
(2×10−6 and 4×10−6 M) for 24 h at 37°C. For
the staining procedure, the SK-LMS-1 cells were fixed in 4%
paraformaldehyde/PBS for 15 min at room temperature and in 100%
methanol (chilled at −20°C) at room temperature for 10 min and then
cells were immediately washed three times with PBS. The cells were
permeabilized with 0.2% Triton X-100 in PBS for 10 min at room
temperature and washed twice for 5 min with PBS. Subsequently, the
cells were incubated for 60 min in a blocking solution [PBS/5%
bovine serum albumin (BSA)] at room temperature. The primary
antibody against light chain 3 (LC3; #4108; Cell Signaling
Technology Inc., Danvers, MA, USA) was diluted 1:200 in PBS, 5% BSA
and 0.3% Triton X-100. Following incubation with the primary
antibodies overnight at 4°C, the cells were washed three times for
5 min with PBS and then incubated with DyLight 550 conjugated goat
anti-rabbit IgG (heavy and light chains; dilution, 1:250 in PBS,
#84541; Thermo Fisher Scientific, Inc.) for 1 h at room
temperature. Cells were then washed three times for 5 min with PBS
and incubated with 2 µg/ml DAPI to stain the nuclei for 5 min at
room temperature in the dark. Images of the cells were captured
using a BX51 fluorescence microscope.
Western blot analysis
Cellular protein was extracted using
radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 7.4, 150
mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM PMSF, 1 mM EDTA
and proteinase inhibitor cocktail (no. S8830; Sigma-Aldrich; Merck
KGaA) and was rapidly excised and washed in ice-cold sterile 0.9%
NaCl. Soluble proteins were extracted using Proprep (Intron
Biotechnology, Inc., Seongnam, Korea). Protein concentration was
determined using the bicinchoninic acid assay (Sigma-Aldrich; Merck
KGaA). A total of 40 µg protein per lane was separated using
SDS-PAGE (10–15% gel) and then transferred onto polyvinylidene
fluoride transfer membranes (Merck KGaA). The membranes were
blocked in Tris-buffered saline with 5% Tween-20 (TBS-T)
supplemented with 5% skimmed milk for 60 min at room temperature
and then incubated with primary antibodies against B-cell lymphoma
2-associated X protein (Bax; 1:1,000; no. sc-493; Santa Cruz
Biotechnology, Inc.), B-cell lymphoma-2 (Bcl-2; 1:1,000; no.
sc-509; Santa Cruz Biotechnology, Inc.), p53 (1:1,000; no.
sc-377567; Santa Cruz Biotechnology, Inc.), caspase-3 (1:1,000; no.
9662; Cell Signaling Technology Inc.), LC3 (1:1,000; no. 4108; Cell
Signaling Technology Inc.), protein kinase B (Akt; 1:1,000; no.
sc-11520 Santa Cruz Biotechnology, Inc.), phosphorylated (p)-Akt
(1:1,000; no. sc-33437; Santa Cruz Biotechnology, Inc.),
extracellular-signal-related kinase (ERK) 1/2 (1:1,000; no. 9102;
Cell Signaling Technology Inc.), p-ERK1/2 (1:1,000; no. 4370; Cell
Signaling Technology Inc.) and α-tubulin (1:1,000) at 4°C
overnight. Following washing in TBS-T, the membranes were incubated
with the appropriate horseradish peroxidase-conjugated secondary
antibodies (anti-rabbit; 1:3,000; no. sc-2004; Santa Cruz
Biotechnology, Inc.; or anti-mouse; 1:3,000; no. bs-0296G; BIOSS,
Beijing, China) for 2 h at room temperature. Following washing of
the membranes with TBS-T, antibody binding was detected using an
enhanced chemiluminescence reagent (Santa Cruz Biotechnology, Inc.)
and detected using ChemiDoc equipment GeneGnome 5 (Syngene,
Frederick, MD, USA). Optical density of the target band was
analyzed using ImageJ software (Version 1.50e; National Institutes
of Health, Bethesda, MD, USA).
Statistical analysis
The experiments were performed in triplicate.
Results are presented as the mean ± standard error of the mean.
P-values were determined using one-way analysis of variance,
followed by Tukey's test for multiple comparisons of columns.
P<0.05 was considered to indicate a statistically significant
difference.
Results
2-ME exerts an anti-proliferative
effect on ULMS cells
To determine whether 2-ME influenced the viability
of ULMS cells, SK-LMS-1 cells were incubated for 24 h with 2-ME
(10−7, 10−6 and 10−5 M).
Flavopiridol and E2 were used as the controls. Flavopiridol, a
synthetic derivative isolated initially from the stem bark of
Dysoxylum binectariferum, is a pan inhibitor of
cyclin-dependent kinases and is capable of inducing either cell
cycle arrest or apoptosis (22).
Concentrations of the chemicals were based on results from previous
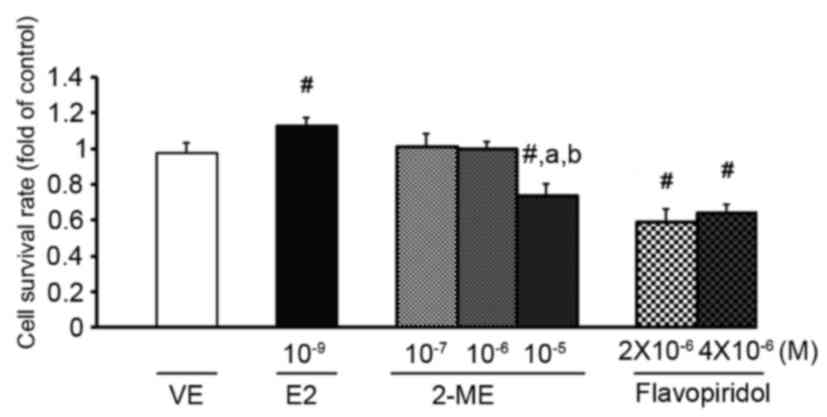
studies (9,22). As presented in Fig. 1, analysis of cell cytotoxicity was
determined using an MTT assay and indicated that 10−9 M
E2 significantly increased cell proliferation. In contrast,
treatment with 10−5 M 2-ME or flavopiridol resulted in
cell cytotoxicity (Fig. 1).
2-ME exerts a cytotoxic effect on ULMS
cells via apoptosis
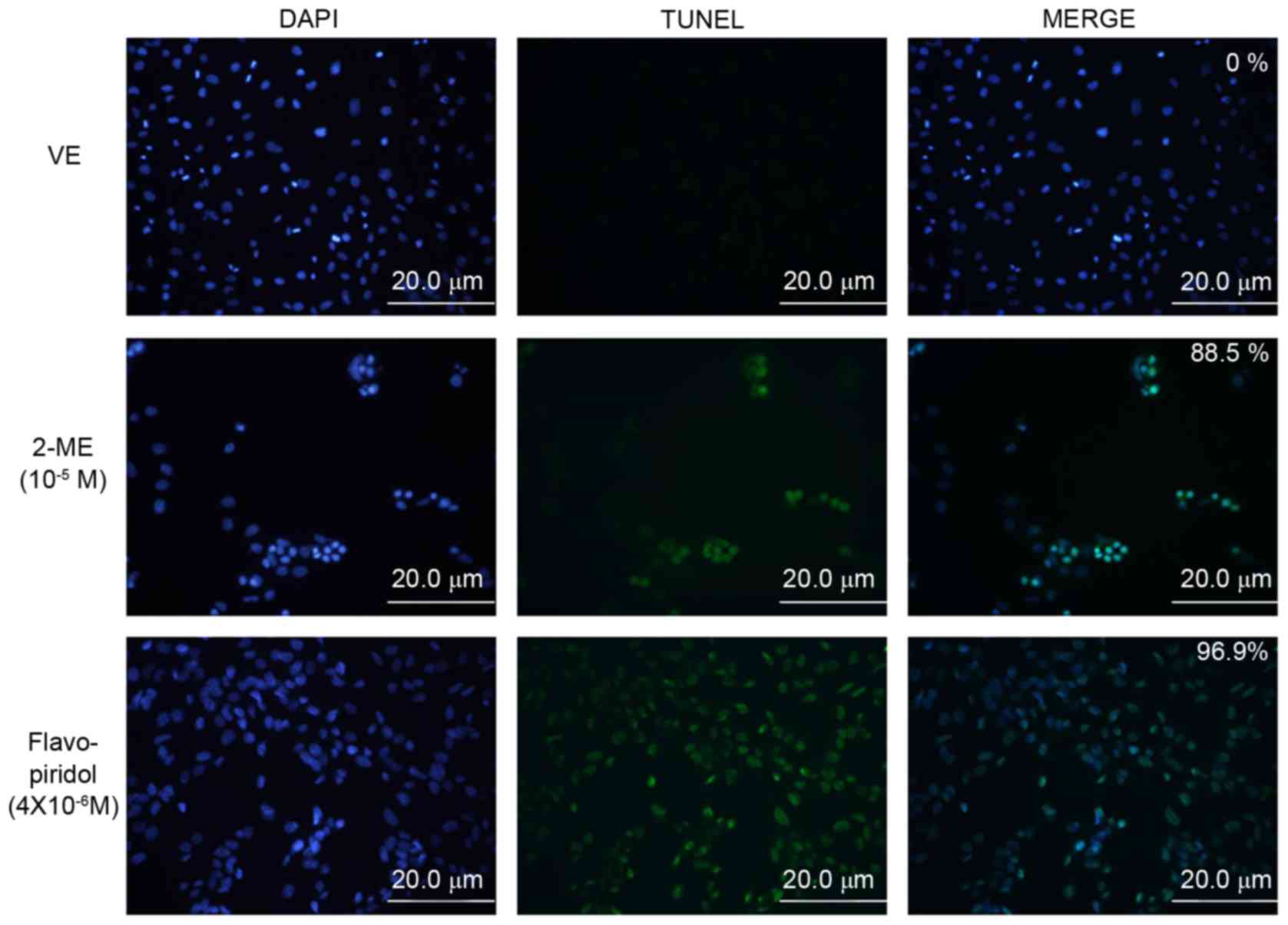
To examine apoptosis induced by 2-ME
(10−5 M), DNA fragmentation was visualized using a TUNEL
assay. As presented in Fig. 2,
SK-LMS-1 cells treated with 10−5 M 2-ME and
4×10−6 M flavopiridol exhibited increased levels of
green fluorescent signals compared with those treated with the
vehicle. TUNEL-positive cells were counted and compared with the
number of nuclei (blue signals) as morphological differences were
observed between cells treated with 2-ME or flavopiridol. The
results revealed that 88.5% of SK-LMS-1 cells incubated with 2-ME
(10−5 M) were TUNEL-positive and 96.9% of the
flavopiridol-treated cells were TUNEL-positive. Treatment with
10−5 M 2-ME was therefore revealed to increase the
number of apoptotic cells based on DNA fragmentation compared with
the untreated group (Fig. 2). To
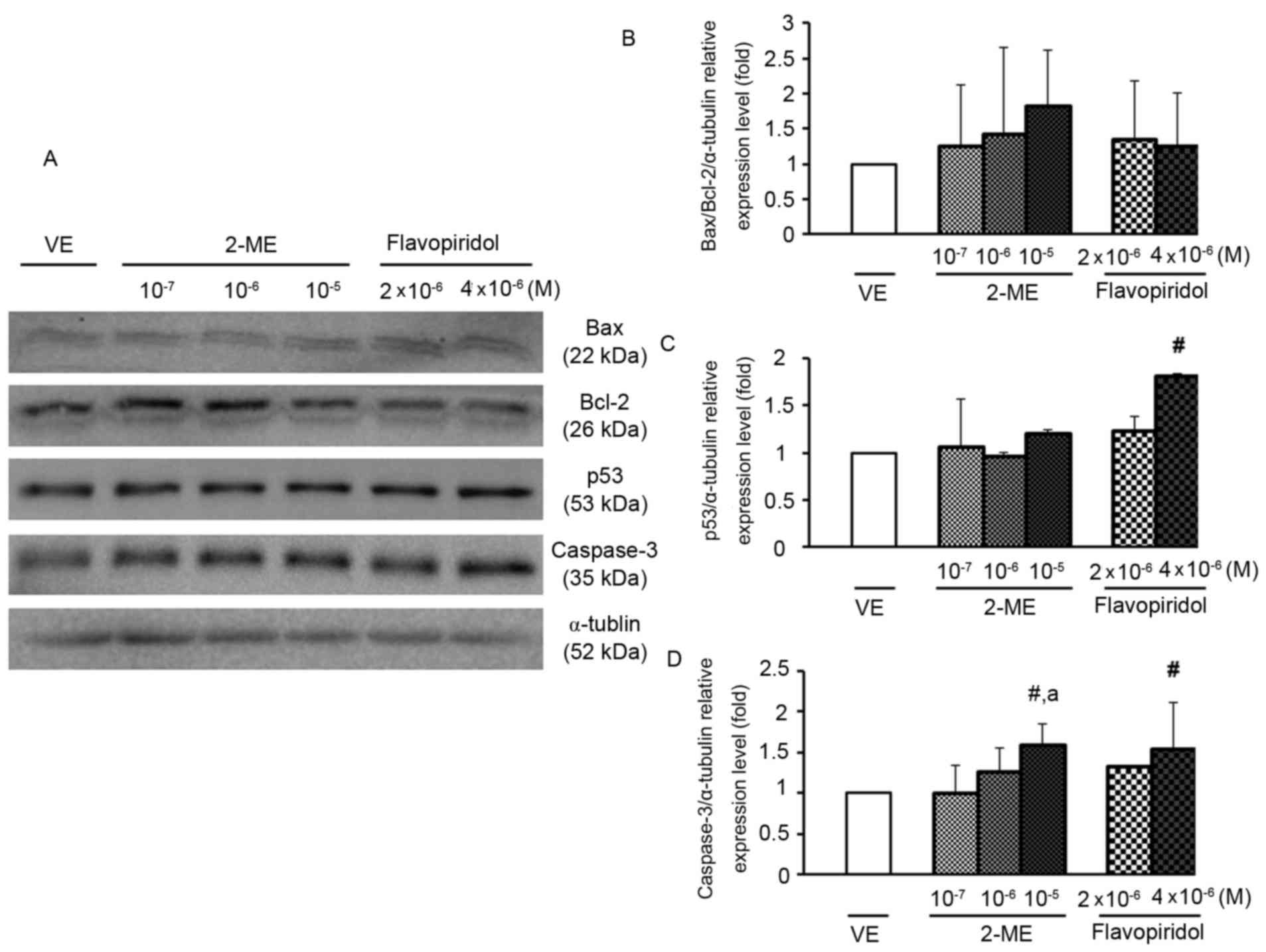
confirm that cell death induced by 2-ME was due to apoptosis, cell
lysates were analyzed for the expression of apoptosis markers Bax,
Bcl-2, caspase-3 and p53 by western blotting. Bax is a member of
the Bcl-2 family that promotes apoptosis and the ratio of Bax/Bcl-2
determines the sensitivity of a cell to apoptosis (23). Caspases, particularly caspase-3, are
known to act downstream of Bax/Bcl-2 and serve a key role in
apoptosis (24). p53 has the ability
to activate transcription of various pro-apoptotic genes, including
those encoding members of the Bcl-2 family, and may also trigger
apoptosis (25). As presented in
Fig. 3, caspase-3 protein expression
level was significantly increased following treatment with 2-ME
(10−5 M) or flavopiridol (4×10−6 M; Fig. 3). This result indicated that 2-ME at
10−5 M exerted an anti-proliferative effect on ULMS
cells via the induction of apoptosis.
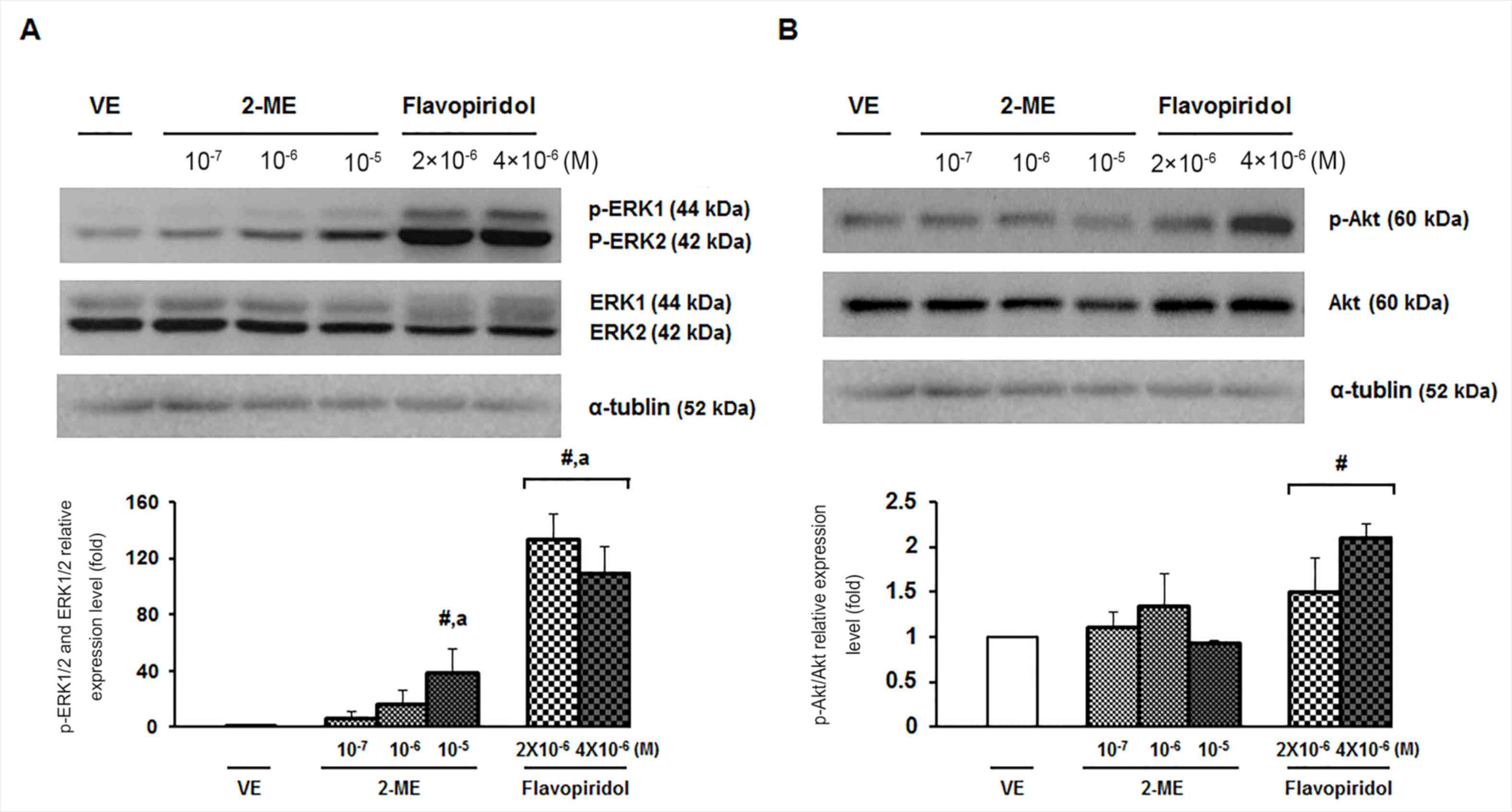
2-ME upregulates p-ERK1/2 signaling in
ULMS cells
It has been reported that ERK/mitogen-activated
protein kinase positively regulated the phosphorylation of Bcl-2 at
Ser70, inducing suppressed Bcl-2 expression (26). In order to determine whether 2-ME
influenced intrinsic apoptotic signaling via the ERK1/2 signaling
pathways, western blotting was performed, as presented in Fig. 4A. The expression levels of p-ERK1/2
and ERK1/2 were significantly upregulated in ULMS cells following
treatment with 10−5 M 2-ME and flavopiridol
(2×10−6 and 4×10−6 M) for 24 h (Fig. 4A). However, the p-Akt/Akt expression
level was decreased in the presence of 2-ME (10−5 M;
Fig. 4B). The
downregulation/inactivation of Akt has been reported to serve a key
role in mediating the cytotoxic effects of 2-ME in human leukemia
cells (27). However, the results of
the present study demonstrated that the p-Akt/Akt expression level
in ULMS cells was not significantly affected by 2-ME at any of the
doses investigated (10−7, 10−6 or
10−5 M) compared with the vehicle (Fig. 4B). Therefore, the results indicated
that 2-ME-induced apoptosis is mediated by the activation of the
(p)-ERK1/2 signaling pathway in ULMS cells.
2-ME enhances autophagy in ULMS
cells
Evaluation of apoptosis in tumor cells has been
identified to be an important factor for detecting the ability of
therapeutic drugs to prevent tumor growth. However, numerous
previous studies have demonstrated that autophagy is also a cell
death mechanism that may occur in the absence of detectable signs
of apoptosis or concomitantly with apoptosis (14). To determine whether 2-ME induces
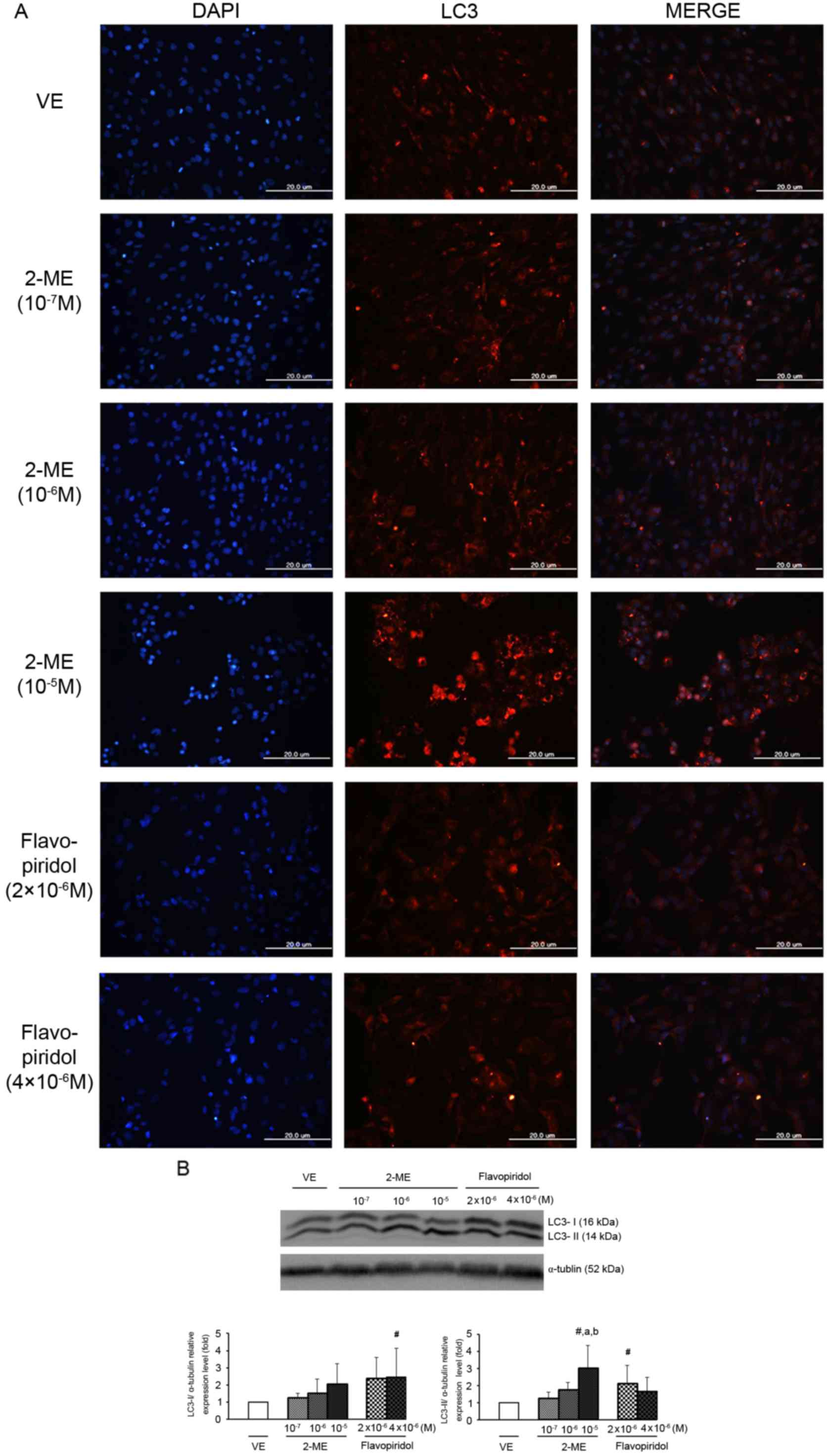
autophagy, the LC3 expression level was evaluated using
immunocytochemistry (Fig. 5A). In
cells treated with 10−5 M 2-ME, a marked LC3 expression
level was detected, indicating that 2-ME induced the formation of
autophagosomes in ULMS cells. To further confirm that 2-ME induced
autophagy, western blotting was also performed. As presented in
Fig. 5B, LC3 expression level
increased in the presence of 2-ME (10−7, 10−6
and 10−5 M). Flavopiridol also elevated LC3 expression
levels at a concentration of 4×10−6 M. Additionally, the
LC3-II expression level was significantly increased with
10−5 M 2-ME and 2×10−6 M flavopiridol
compared with the vehicle (Fig. 5B).
During the autophagy cascade, LC3-I is converted into LC-II via
lipidation by an ubiquitin-like system. LC3-II then remains with
autophagosomes until fusion with lysosomes is completed and it may
therefore be used as a marker of autophagy (28). Consequently, results from the present
study indicated that 2-ME induced autophagy in SK-LMS-1 cells.
Discussion
The estrogen metabolite 2-ME has been reported to
inhibit cell proliferation by apoptosis in a wide range of tumor
cells (7–9). In cases of uterine leiomyoma, 2-ME was
identified to exert an anti-proliferative effect by arrest in the
G2/M phase and inhibited collagen synthesis (29). Uterine leiomyomas, or fibroids, are
benign smooth muscle cell tumors and are the most common type of
pelvic tumors in females (30). These
lesions are estrogen/progesterone-dependent and occur primarily
during the reproductive years (29).
Ovarian steroid hormones, particularly estrogen,
mediate uterine leiomyoma development. Leiomyoma has been reported
to progress with estrogen stimulation (31). ULMS, a rare type of uterine tumor,
accounts for between 1 and 2% of uterine malignancies, and has
symptoms similar to those of common leiomyoma along with
preoperative distinction (19). It
was also reported that ULMS tissue expressed ERs and progesterone
receptors (32). ULMS poses a threat
to females of reproductive age, particularly since there are no
effective and safe medications for treating this disease. Surgical
procedures including hysterectomy or myomectomy are the primary
option for treatment (21).
Therefore, there is a requirement for novel drugs or compounds that
may be effective therapies for ULMS.
In the present study, 2-ME was demonstrated to
exhibit antitumor activity that resulted in autophagic and
apoptotic death of ULMS cells. The results revealed that the
anti-proliferative effects of 2-ME occur only at a concentration of
10−5 M in SK-LMS-1 cells. Consistent with previous
studies (8,9), 10−5 M 2-ME induced growth
inhibition and apoptosis. Similar to numerous other types of tumor,
homeostatic control of the growth of leiomyosarcoma is hypothesized
to be the result of the dynamic balance between cell proliferation
and cell death (29). In the present
study, 2-ME decreased the expression level of the anti-apoptotic
protein Bcl-2. This factor promotes cell survival and extends the
lifespan of certain cells (33).
Additionally, treatment with 2-ME and flavopiridol does not affect
the expression level of the pro-apoptotic protein Bax. Therefore,
treatment of ULMS cells with 2-ME increased the ratio of Bax/Bcl-2,
which reflected the induction of apoptosis. Similarly, Joubert
et al (34) reported that
treatment of esophageal carcinoma cells with 2-ME significantly
decreased Bcl-2 expression levels without affecting Bax expression
level (34). Furthermore, 2-ME was
reported to induce apoptosis as a result of c-Jun N-terminal
kinase, ERK and p38 kinase activation, in addition to activation of
the intrinsic apoptotic pathway via inactivation of Bcl-2 and
Bcl-extra large proteins. Upregulation of the extrinsic signaling
pathway by increased expression level of death receptor 5 induced
activation of caspase-8 has also been observed (35,36).
Caspases are crucial mediators of programmed cell
death, including apoptosis. Among these factors, caspase-3 is a
frequently activated death protease that induces DNA fragmentation
(37). Results of the present study
demonstrated that 2-ME induced the apoptotic death of SK-LMS-1
cells by increasing protein levels of caspase-3 and DNA
fragmentation (TUNEL-positive cells).
The tumor suppressor gene p53 is an important
regulator of apoptosis. Normally, this factor prevents the
replication of damaged DNA by inducing G1/S cell cycle
arrest or apoptosis. Increased p53 protein expression level induced
by 2-ME has also been observed in colorectal cancer cells (7). However, the expression level of p53 was
not significantly increased in the present study.
It has been well established that ERK1/2 activation
serves a role in proliferative signaling pathways (38); however, this activation was previously
reported to aid ERK1/2 phosphorylation involved in apoptotic events
(39). Tsai et al (40) demonstrated that upregulation of the
ERK signaling pathway was associated with the
mitochondria-dependent apoptotic signaling pathway in MDA-MB-468
human breast cancer cells and positively regulated the
phosphorylation of Bcl-2 at Ser70, thus inhibiting Bcl-2
expression (40). It has also been
revealed that ERK activation cooperatively acts with p53 to promote
apoptosis or cell cycle arrest in numerous cell lines (41). In the present study, increasing the
phosphorylation of ERK1/2 using 10−5 M 2-ME in SK-LMS-1
cells indicated that the intrinsic pathway was upregulated by death
signals to mitochondria-dependent associated apoptosis.
The phosphatidylinositol 3-kinase/Akt signal
transduction pathway serves an important role in cell survival
decisions (42). In human leukemia
cells, 2-ME-induced-Akt inactivation demonstrated a critical
functional role in mediating 2-ME lethality (27). However, phosphorylation of Akt was not
affected in SK-LMS-1 cells by 2-ME at any concentration, which
revealed that 2-ME-induced apoptosis occurs via an intrinsic
signaling pathway associated with the upregulation of ERK1/2
activity.
The role of the ERK signaling pathway in the
induction of cell death should not be restricted to apoptosis
(i.e., caspase-dependent cell death). Under certain conditions,
autophagy induces cell death associated with ERK activity.
ERK-dependent autophagic activity is associated with classical
markers of autophagy, including the induction of LC3 activity and
conversion of LC3-I into LC3-II (43,44).
Similarly, the results of the present study demonstrated that 2-ME
activated the ERK signaling pathway, which may mediate autophagic
type II programmed cell death by increasing LC3 expression level in
SK-LMS-1 cells.
The results of the present study indicated that
2-ME, a well-tolerated natural metabolite of E2, exerted
anti-proliferative effects on human ULMS cells at a concentration
of 10−5 M. This compound inhibited cell proliferation by
inducing apoptosis and autophagy. However, in a previous study,
treatment with 2-ME at concentrations lower than anticancer
therapeutic concentrations, revealed an estrogenic effect based on
uterine weight gain and increasing expression levels of estrogen
biomarkers in in vitro and in vivo models (45). Therefore, it may affect the estrogen
and/or progesterone receptor, and retaining 2-ME in the body may be
affect endocrine homeostasis and its function. Therefore, careful
selection of the optimal dose of 2-ME is required for treating
human ULMS.
Acknowledgements
The present study was supported by the National
Research Foundation of Korea grant of the Korean government (MEST;
grant no. 2017R1A2B2005031).
References
|
1
|
Spady TJ, McComb RD and Shull JD: Estrogen
action in the regulation of cell proliferation, cell survival, and
tumorigenesis in the rat anterior pituitary gland. Endocrine.
11:217–233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yue W, Yager JD, Wang JP, Jupe ER and
Santen RJ: Estrogen receptor-dependent and independent mechanisms
of breast cancer carcinogenesis. Steroids. 78:161–170. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nandi S, Guzman RC and Yang J: Hormones
and mammary carcinogenesis in mice, rats, and humans: A unifying
hypothesis. Proc Natl Acad Sci USA. 92:pp. 3650–3657. 1995;
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Seegers JC, Aveling ML, van Aswegen CH,
Cross M, Koch F and Joubert WS: The cytotoxic effects of
estradiol-17 beta, catecholestradiols and methoxyestradiols on
dividing MCF-7 and HeLa cells. J Steroid Biochem. 32:797–809. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu BT and Conney AH: Is
2-methoxyestradiol an endogenous estrogen metabolite that inhibits
mammary carcinogenesis? Cancer Res. 58:2269–2277. 1998.PubMed/NCBI
|
|
6
|
Lakhani NJ, Sarkar MA, Venitz J and Figg
WD: 2-Methoxyestradiol, a promising anticancer agent.
Pharmacotherapy. 23:165–172. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carothers AM, Hughes SA, Ortega D and
Bertagnolli MM: 2-Methoxyestradiol induces p53-associated apoptosis
of colorectal cancer cells. Cancer Lett. 187:77–86. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee YM, Ting CM, Cheng YK, Fan TP, Wong
RN, Lung ML and Mak NK: Mechanisms of 2-methoxyestradiol-induced
apoptosis and G2/M cell-cycle arrest of nasopharyngeal carcinoma
cells. Cancer Lett. 268:295–307. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li L, Bu S, Bäckström T, Landström M,
Ulmsten U and Fu X: Induction of apoptosis and G2/M arrest by
2-methoxyestradiol in human cervical cancer HeLaS3 cells.
Anticancer Res. 24:873–880. 2004.PubMed/NCBI
|
|
10
|
Seeger H, Wallwiener D, Kraemer E and
Mueck AO: Comparison of possible carcinogenic estradiol
metabolites: Effects on proliferation, apoptosis and metastasis of
human breast cancer cells. Maturitas. 54:72–77. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Minorics R, Bózsity N, Molnár J, Wölfling
J, Mernyák E, Schneider G, Ocsovszki I and Zupkó I: A molecular
understanding of D-homoestrone-induced G2/M cell cycle arrest in
HeLa human cervical carcinoma cells. J Cell Mol Med. 19:2365–2374.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chang I, Majid S, Saini S, Zaman MS,
Yamamura S, Chiyomaru T, Shahryari V, Fukuhara S, Deng G, Dahiya R
and Tanaka Y: Hrk mediates 2-methoxyestradiol-induced mitochondrial
apoptotic signaling in prostate cancer cells. Mol Cancer Ther.
12:1049–1059. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Siebert AE, Sanchez AL, Dinda S and
Moudgil VK: Effects of estrogen metabolite 2-methoxyestradiol on
tumor suppressor protein p53 and proliferation of breast cancer
cells. Syst Biol Reprod Med. 57:279–287. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Codogno P and Meijer AJ: Autophagy and
signaling: Their role in cell survival and cell death. Cell Death
Differ. 12 Suppl 2:S1509–S1518. 2005. View Article : Google Scholar
|
|
15
|
Debatin KM and Krammer PH: Death receptors
in chemotherapy and cancer. Oncogene. 23:2950–2966. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chuang TD, Ho M and Khorram O: The
regulatory function of miR-200c on inflammatory and cell-cycle
associated genes in SK-LMS-1, a leiomyosarcoma cell line. Reprod
Sci. 22:563–571. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
D'Angelo E and Prat J: Uterine sarcomas: A
review. Gynecol Oncol. 116:131–139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Segars JH, Parrott EC, Nagel JD, Guo XC,
Gao X, Birnbaum LS, Pinn VW and Dixon D: Proceedings from the third
national institutes of health international congress on advances in
uterine leiomyoma research: Comprehensive review, conference
summary and future recommendations. Hum Reprod Update. 20:309–333.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Reichardt P: The treatment of uterine
sarcomas. Ann Oncol. 23 Suppl 10:x151–x157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee HG, Baek JW, Shin SJ, Kwon SH, Cha SD,
Park WJ, Chung R, Choi ES, Lee GH and Cho CH: Antitumor effects of
flavopiridol on human uterine leiomyoma in vitro and in a xenograft
model. Reprod Sci. 21:1153–1160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rudel T: Caspase inhibitors in prevention
of apoptosis. Herz. 24:236–241. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Amaral JD, Xavier JM, Steer CJ and
Rodrigues CM: The role of p53 in apoptosis. Discov Med. 9:145–152.
2010.PubMed/NCBI
|
|
26
|
Deng X, Kornblau SM, Ruvolo PP and May WS
Jr: Regulation of Bcl2 phosphorylation and potential significance
for leukemic cell chemoresistance. J Natl Cancer Inst Monogr.
30–37. 2001.PubMed/NCBI
|
|
27
|
Gao N, Rahmani M, Dent P and Grant S:
2-Methoxyestradiol-induced apoptosis in human leukemia cells
proceeds through a reactive oxygen species and Akt-dependent
process. Oncogene. 24:3797–3809. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kondo Y and Kondo S: Autophagy and cancer
therapy. Autophagy. 2:85–90. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Salama SA, Nasr AB, Dubey RK and Al-Hendy
A: Estrogen metabolite 2-methoxyestradiol induces apoptosis and
inhibits cell proliferation and collagen production in rat and
human leiomyoma cells: A potential medicinal treatment for uterine
fibroids. J Soc Gynecol Investig. 13:542–550. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Robboy SJ, Bentley RC, Butnor K and
Anderson MC: Pathology and pathophysiology of uterine smooth-muscle
tumors. Environ Health Perspect. 108 Suppl 5:S779–S784. 2000.
View Article : Google Scholar
|
|
31
|
Walker CL and Stewart EA: Uterine
fibroids: The elephant in the room. Science. 308:1589–1592. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Akhan SE, Yavuz E, Tecer A, Iyibozkurt CA,
Topuz S, Tuzlali S, Bengisu E and Berkman S: The expression of
Ki-67, p53, estrogen and progesterone receptors affecting survival
in uterine leiomyosarcomas. A clinicopathologic study. Gynecol
Oncol. 99:36–42. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reed JC, Talwar HS, Cuddy M, Baffy G,
Williamson J, Rapp UR and Fisher GJ: Mitochondrial protein p26 BCL2
reduces growth factor requirements of NIH3T3 fibroblasts. Exp Cell
Res. 195:277–283. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Joubert A, Maritz C and Joubert F:
Bax/Bcl-2 expression levels of 2-methoxyestradiol-exposed
esophageal cancer cells. Biomed Res. 26:131–134. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fukui M and Zhu BT: Mechanism of
2-methoxyestradiol-induced apoptosis and growth arrest in human
breast cancer cells. Mol Carcinog. 48:66–78. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
LaVallee TM, Zhan XH, Johnson MS,
Herbstritt CJ, Swartz G, Williams MS, Hembrough WA, Green SJ and
Pribluda VS: 2-methoxyestradiol up-regulates death receptor 5 and
induces apoptosis through activation of the extrinsic pathway.
Cancer Res. 63:468–475. 2003.PubMed/NCBI
|
|
37
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Meloche S and Pouysségur J: The ERK1/2
mitogen-activated protein kinase pathway as a master regulator of
the G1- to S-phase transition. Oncogene. 26:3227–3239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen JR, Plotkin LI, Aguirre JI, Han L,
Jilka RL, Kousteni S, Bellido T and Manolagas SC: Transient versus
sustained phosphorylation and nuclear accumulation of ERKs underlie
anti-versus pro-apoptotic effects of estrogens. J Biol Chem.
280:4632–4638. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tsai SC, Huang WW, Huang WC, Lu CC, Chiang
JH, Peng SF, Chung JG, Lin YH, Hsu YM, Amagaya S and Yang JS:
ERK-modulated intrinsic signaling and G(2)/M phase arrest
contribute to the induction of apoptotic death by allyl
isothiocyanate in MDA-MB-468 human breast adenocarcinoma cells. Int
J Oncol. 41:2065–2072. 2012.PubMed/NCBI
|
|
41
|
Tang D, Wu D, Hirao A, Lahti JM, Liu L,
Mazza B, Kidd VJ, Mak TW and Ingram AJ: ERK activation mediates
cell cycle arrest and apoptosis after DNA damage independently of
p53. J Biol Chem. 277:12710–12717. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hemmings BA: Akt signaling: Linking
membrane events to life and death decisions. Science. 275:628–630.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cagnol S and Chambard JC: ERK and cell
death: Mechanisms of ERK-induced cell death-apoptosis, autophagy
and senescence. FEBS J. 277:2–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cheng Y, Qiu F, Tashiro S, Onodera S and
Ikejima T: ERK and JNK mediate TNFalpha-induced p53 activation in
apoptotic and autophagic L929 cell death. Biochem Biophys Res
Commun. 376:483–488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lee JS, Kim YK, Yang H, Kang HY, Ahn C and
Jeung EB: Two faces of the estrogen metabolite 2-methoxyestradiol
in vitro and in vivo. Mol Med Rep. 12:5375–5382. 2015.PubMed/NCBI
|