Introduction
The epithelial-to-mesenchymal transition (EMT)
serves important roles in embryonic development, cancer invasion,
metastasis and chemoresistance (1–3). EMT cells
demonstrate lower expression levels of epithelial markers,
including E-cadherin, and higher expression levels of mesenchymal
markers, including vimentin and fibronectin (1,2). These
expressional changes occur through the upregulation of
transcriptional factors, including the zinc finger proteins, zinc
finger E-box binding homeobox (ZEB)1 and 2, the basic helix-loop
helix protein Twist, and the Snail family, including Snail and Slug
(1,2).
Although the underlying mechanisms remain unclear, transforming
growth factor-β (TGF-β) and fibroblast growth factor-2 (FGF-2)
constitute the main EMT-inducing factors in numerous types of
cancer (1,4,5). Due to
its roles in cancer invasion, metastasis and chemoresistance, the
inhibition or reversion of the EMT has been regarded as a promising
strategy for treating cancer. Several agents, including a
mechanistic target of rapamycin inhibitor (6) and metformin (7), have been reported to suppress EMT in
lung cancer. However, effective approaches for inhibiting the EMT
have not been established.
Recently, the anti-fibrotic agents pirfenidone and
nintedanib have been approved in numerous countries for the
treatment of idiopathic pulmonary fibrosis (IPF). Randomized
controlled clinical trials have demonstrated that pirfenidone
suppresses the deterioration of the percentage forced vital
capacity (FVC), with manageable toxicities, in patients with IPF
(8,9).
Furthermore, pirfenidone has been revealed to suppress lung
fibrosis through the downregulation of TGF-β, platelet-derived
growth factor (PDGF) and collagen synthesis in a hamster model
(10–12). In addition, it was shown to suppress
liver fibrosis through the downregulation of TGF-β (13,14).
Nintedanib has been demonstrated to suppress the deterioration of
FVC and the incidence of acute exacerbation in patients with IPF
(15,16). Additionally, it has been revealed to
inhibit fibrosis through the downregulation of extracellular matrix
proteins in pulmonary fibroblast cells (17). The expression levels of the receptor
tyrosine kinases of PDGF, FGF and vascular endothelial growth
factor were observed to be inhibited following treatment with
nintedanib (17). Recently,
nintedanib was demonstrated to inhibit the early signaling of TGF-β
by suppressing the phosphorylation of the TGF-β type II receptor
(18).
Numerous studies have reported that the relative
risk of lung cancer among patients with IPF is 7–14 times higher
compared with that in patients without IPF (19–21). Under
such circumstances, these agents may soon be widely used for the
treatment of patients with lung cancer accompanied by IPF. Thus,
elucidating the potential effects of these agents on lung cancer
cells is required. Nintedanib, which is assumed to suppress TGF-β
and FGF, was recently reported to suppress the EMT in ovarian
cancer cells in vitro (22).
As pirfenidone also has the ability to suppress TGF-β, it may be
useful for suppressing or reverting the EMT. Therefore, the present
study aimed to evaluate the effect of pirfenidone on the EMT in
human lung cancer and compare its efficacy to the activity of
nintedanib.
Materials and methods
Cells and reagents
The human lung adenocarcinoma cell lines A-549,
HCC-827 (American Type Culture Collection, Manassas, VA, USA) and
PC-9 (Riken Cell Bank, Tsukuba, Japan) were used throughout the
present study. HCC-827 and PC-9 cells have a deletion in exon 19
(del E746-A750) of the epidermal growth factor receptor gene. Cells
were cultured as a monolayer in RPMI-1640 medium (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) supplemented with 10% fetal bovine
serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin (all
Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in a
humidified atmosphere containing 5% CO2. Pirfenidone
(Sigma-Aldrich; Merck KGaA) was dissolved in water at a
concentration of 1.0 M and stored at −20°C. Nintedanib (Selleck
Chemicals, Houston, TX, USA) was dissolved in DMSO (Sigma-Aldrich;
Merck KGaA) at a concentration of 10 mM and stored at −20°C. Prior
to use in the experiments, each agent was diluted in RPMI-1640
medium with 10% FBS, 100 U/ml penicillin and 100 mg/ml
streptomycin. Mouse monoclonal anti-fibronectin antibody (cat. no.
F3648) was purchased from Sigma-Aldrich (Merck KGaA). The anti-E
cadherin antibody was purchased from BD Biosciences (cat. no.
610181; Franklin Lakes, NJ, USA). Anti-mouse IgG Fab2 Alexa
Fluor® 488 molecular probes (cat. no. 4408S) and
anti-rabbit IgG Fab2 Alexa Fluor 555 molecular probes (cat. no.
4413S) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA).
In vitro induction and reversion of
EMT
EMT was induced by treatment with recombinant human
TGF-β (PeproTech, Inc., Rock Hill, NJ, USA) and FGF-2 (Cell
Signaling Technology, Inc.). Based on preliminary experiments (data
not shown), a combination of TGF-β (10 ng/ml) and FGF-2 (10 ng/ml)
was admixed into the complete medium for induction of EMT following
24 h of serum starvation. To evaluate the EMT phenotypes, cells
harvested at 48 h after the admixture of the agents were used.
Pirfenidone (0.2 and 2.0 mM) or nintedanib (0.1 and 1.0 µM) was
then added to cells in which the EMT had already been induced, with
TGF-β/FGF-2 being supplemented continuously. The phenotypic
alterations were evaluated after 72 h of culture with these
agents.
Evaluation of cell viability
Cell viability was determined using an MTT assay
according to the manufacturer's protocol (Promega Corp., Madison,
WI, USA). Cells were seeded into 96-well culture plates at a
density of 1×103 cells/well. Cells were allowed to
attach for 24 h prior to drug treatment. Cells were treated with
various concentrations of pirfenidone (0, 0.02, 0.2, 2.0 and 10 mM)
or nintedanib (0, 0.01, 0.1, 1.0 and 10 µM) for 48 h of culture at
a 37°C. The absorbance at 570 nm in the resulting solution was
measured using the Infinite® 200 PRO microplate reader
(Tecan Schweiz AG, Seestrasse, Switzerland).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Cells were cultured until 80% confluence was
achieved in 6-well culture plates, the total RNA was extracted
using the RNeasy® Mini kit (cat. no. 74104; Qiagen GmbH,
Hilden, Germany) and the cDNA was synthesized using the
SuperScript® First-Strand Synthesis kit for RT-PCR (cat.
no. 11904-018; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. The expression levels of various mRNAs
were quantified using RT-qPCR with the TaqMan® Gene
Expression Assay kit, TaqMan Fast Advanced Master mix (cat. no.
4444557) and StepOnePlus Real-Time PCR system (all Thermo Fisher
Scientific, Inc.). A total of 25 ng synthesized cDNA was used for
each qPCR reaction. Each sample was measured in triplicate. GAPDH
was used for normalization. Relative expression levels were
calculated as using the 2−∆∆Cq method
(23). TaqMan probes for GAPDH,
E-cadherin, vimentin, fibronectin, Slug and programmed death-ligand
1 (PD-L1) were purchased from Applied Biosystems (Thermo Fisher
Scientific, Inc.; assay identification nos. Hs02758991_g1,
Hs01023894_m1, Hs00185584_m1, Hs00365052_m1, Hs00950344_m1 and
Hs01125301_m1, respectively). The following thermocycling
conditions were maintained: 10 min at 95°C, followed by 40 cycles
of 15 sec at 95°C and 1 min at 60°C.
Fluorescent immunohistochemistry
For fluorescent immunohistochemical evaluation,
cells were seeded into a 4-well chamber slide (cat. no. 177399;
Thermo Fisher Scientific, Inc.) at density of 1×104
cells/well and cultured until 80% confluence was achieved. Cells
grown on the chamber slide were fixed with 4% paraformaldehyde
(Sigma-Aldrich; Merck KGaA) for 15 min at room temperature and with
acetone (both Sigma-Aldrich; Merck KGaA) for 10 min at −20°C.
Non-specific binding was blocked using 1% bovine serum albumin with
0.2% Triton X-100 (both Sigma-Aldrich; Merck KGaA) in PBS for 1 h
at room temperature. Then, the cells were incubated with primary
antibodies for 2 h at room temperature. Subsequently, the cells
were incubated with the corresponding secondary antibodies and
counterstained with DAPI (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan) at room temperature for 30 min prior to
observation using the EVOS FL Imaging system (Thermo Fisher
Scientific, Inc.).
Wound healing assay
Cell motility was assessed using a wound healing
assay. Confluent cells were scratched with micropipette tips
(Thermo Fisher Scientific, Inc.). Following washing with PBS, the
cells were incubated for a further 9 h at 37°C. Scratch areas were
viewed using an inverted microscope (magnification, ×10; Nikon
Corporation, Tokyo, Japan) and were quantified using ImageJ
software (version 1.46v; National Institutes of Health, Bethesda,
MD, USA). The wound closure rates were determined as a percentage
of the total repaired area per hour, and were normalized to the
control.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Statistical analysis was performed using Microsoft Excel
software (version 2010; Microsoft Corporation, Redmond, WA, USA).
Differences between groups were analyzed using the two-tailed
Student's t-test. P<0.05 was considered to indicate a
statistically significant difference. All experiments were repeated
≥3 times, in triplicate.
Results
Cytotoxicity of pirfenidone and
nintedanib
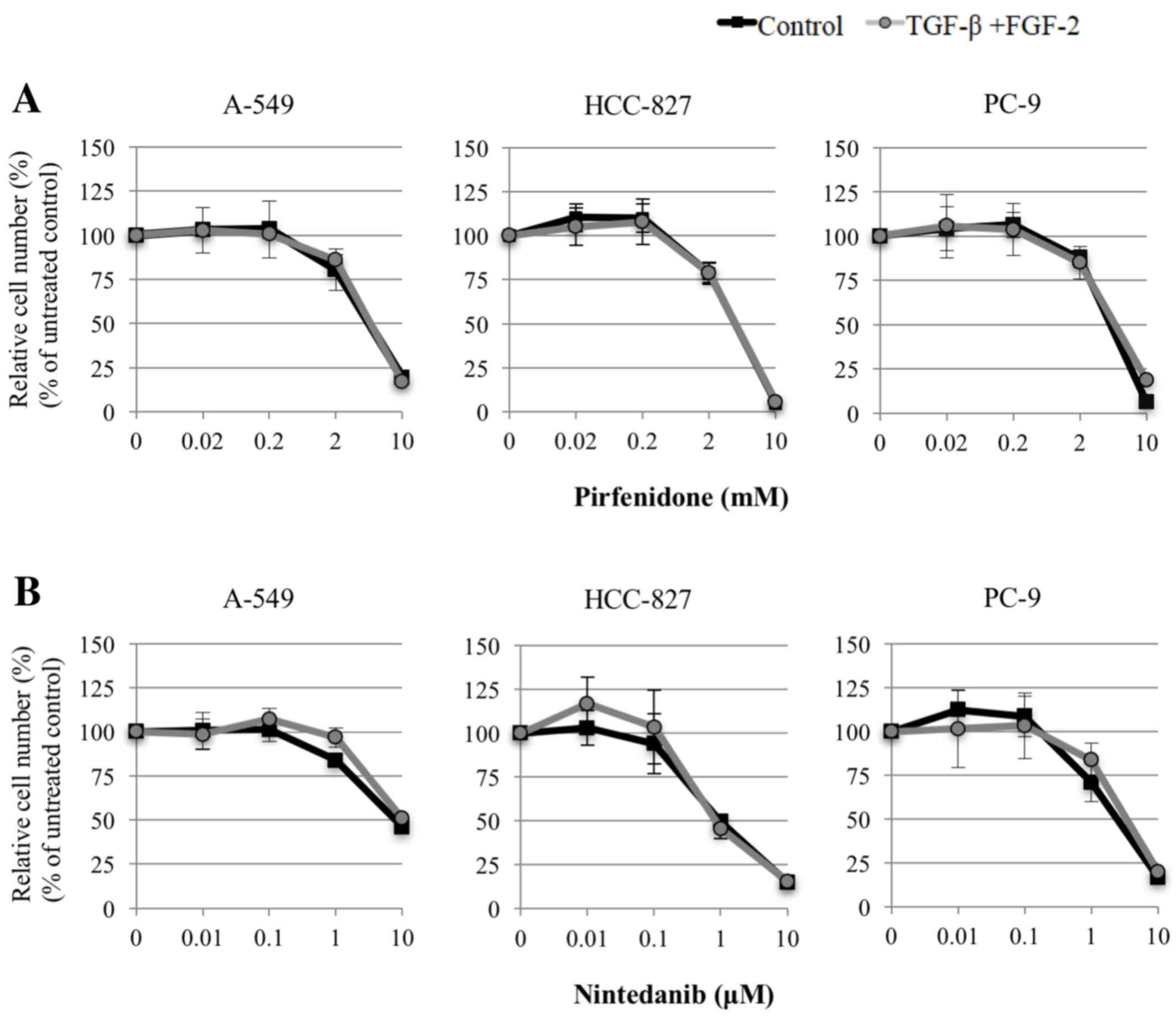
Fig. 1 shows the
cytotoxicity of the agents, as assessed using the MTT assay. Based
on these results, the borderline sublethal dose points, at which
cell viability began to decline, and the adjacent lower dose point
for each agent were selected for use in subsequent experiments (0.2
and 2.0 mM for pirfenidone; 0.1 and 1.0 µM for nintedanib).
In vitro induction of EMT
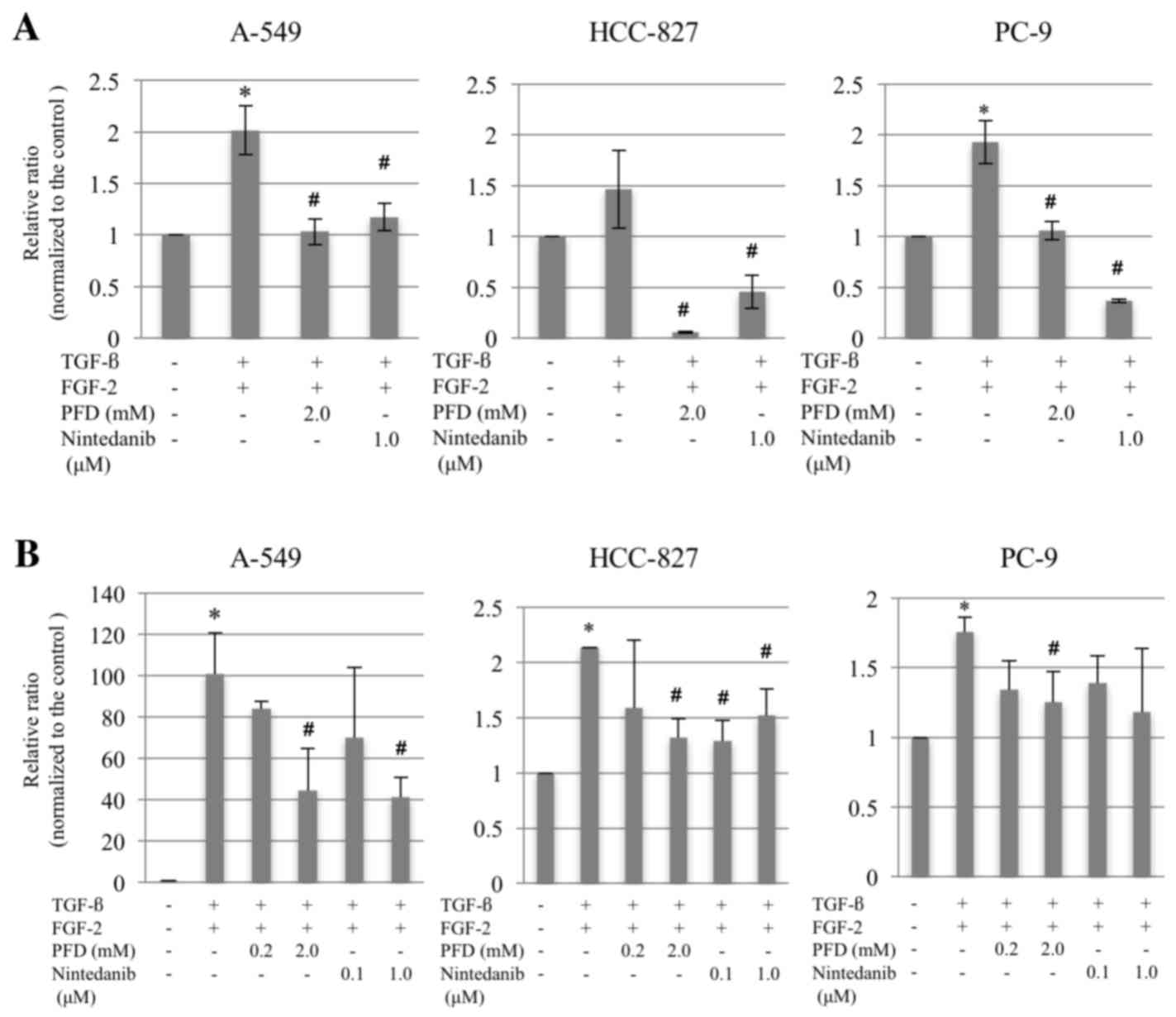
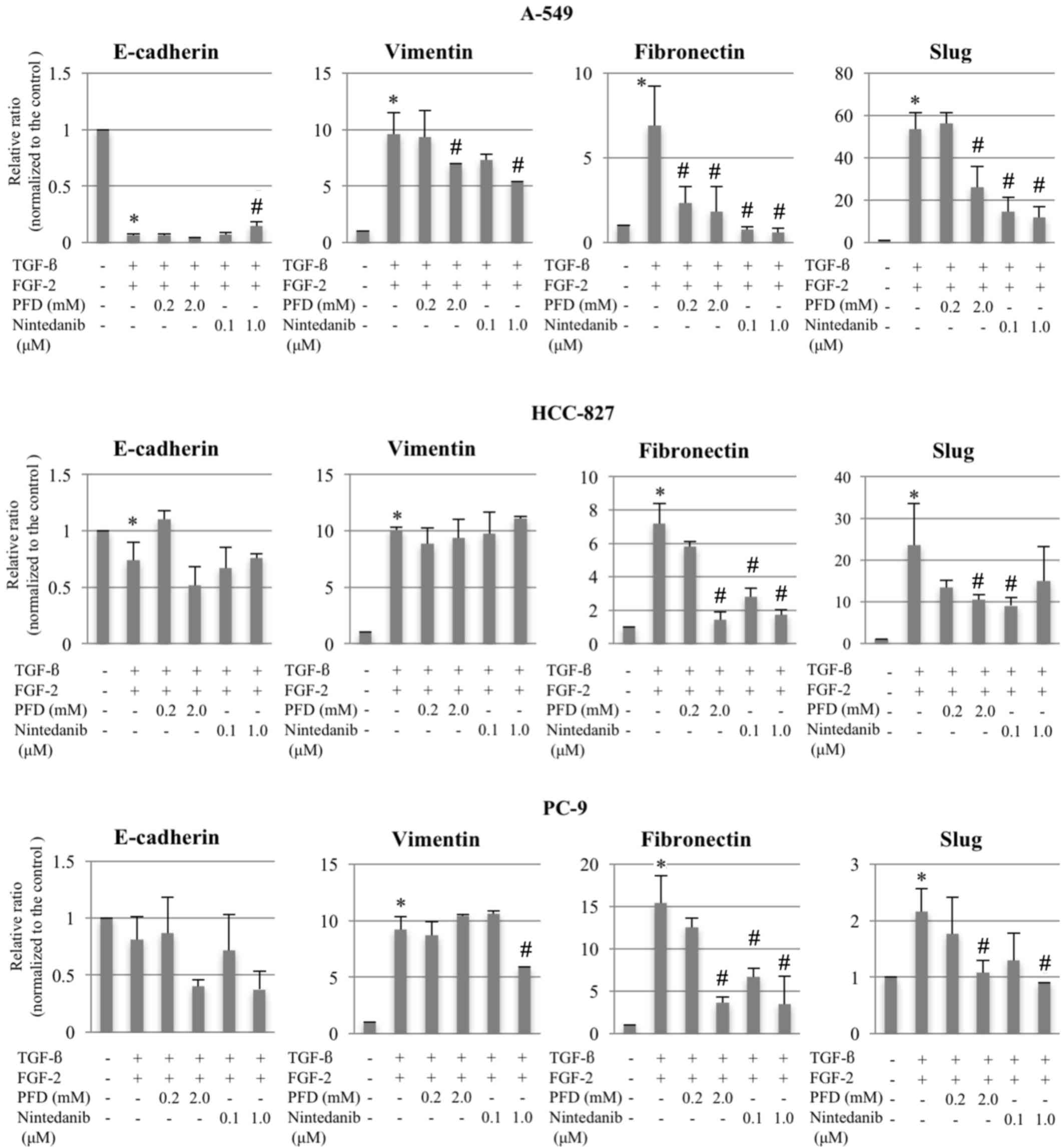
RT-qPCR in the three cell lines revealed that,
compared with the untreated control group, the combination of TGF-β
and FGF-2 significantly downregulated E-cadherin and upregulated
vimentin, fibronectin, and Slug, resulting in an EMT phenotype,
with the exception of E-cadherin expression in PC-9 cells where no
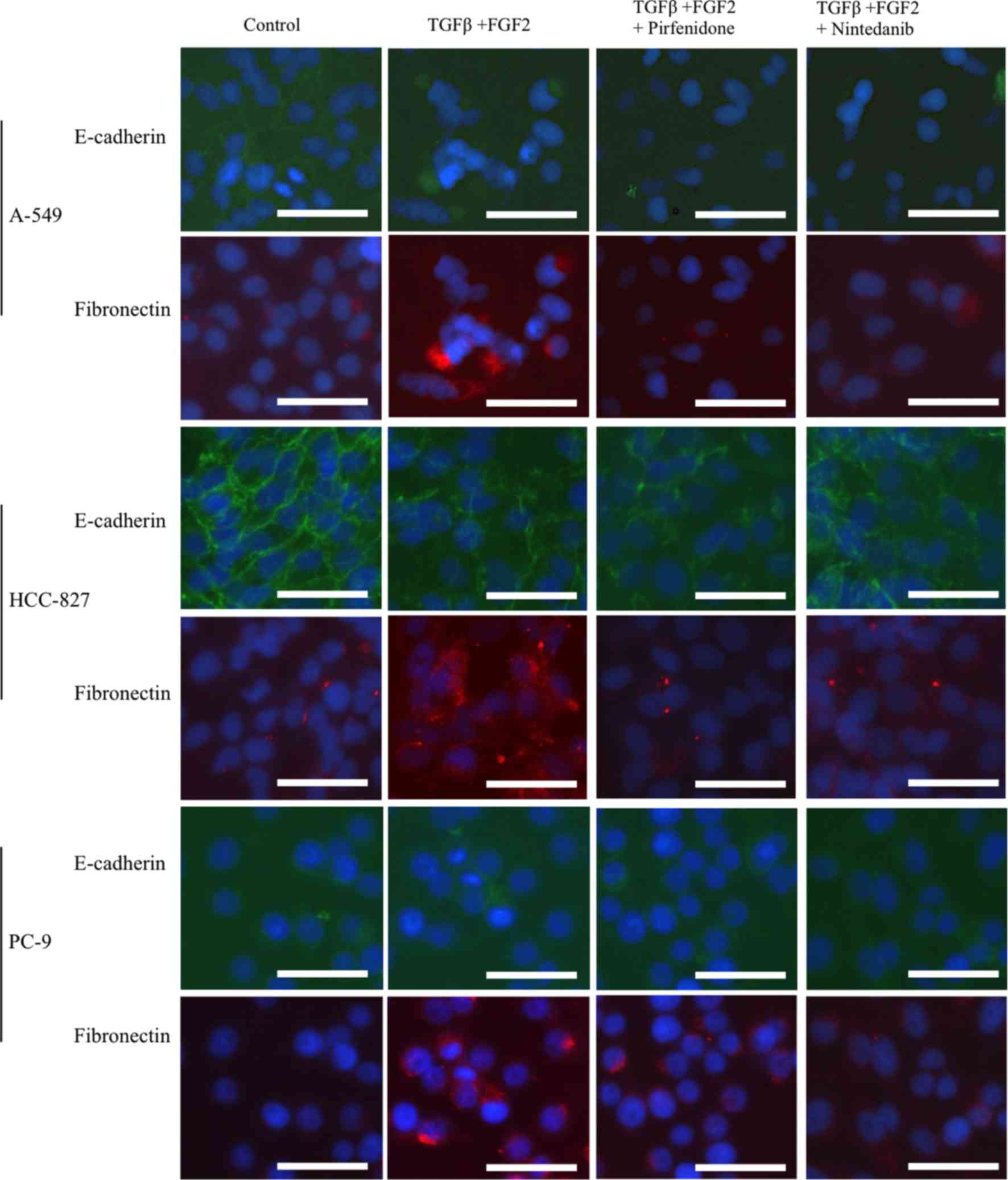
significant differences were observed (Fig. 2). Fluorescent immunohistochemical
analysis of E-cadherin at the cell junction and fibronectin in the
cytoplasm supported the results of the RT-qPCR analyses (Fig. 3). The wound-healing assay showed
significantly increased cell motility associated with the EMT
phenotype in A-549 and PC-9 cells (both P<0.05), and a degree of
increased cell motility in HCC-827 cells compared with the
untreated control groups (Fig. 4A).
The expression of PD-L1 assessed using RT-qPCR was significantly
increased following EMT induction in all cell lines compared with
the untreated cells (Fig. 4B). These
findings appear to support the successful induction of EMT in all
three cell lines.
 | Figure 2.Changes in mRNA expression levels of
E-cadherin, vimentin, fibronectin and Slug following EMT induction,
and treatment with pirfenidone or nintedanib in A-549, HCC-827 and
PC-9 cells. The relative ratios of mRNA in the treatment groups vs.
the controls were evaluated using reverse
transcription-quantitative polymerase chain reaction for cells
treated with TGF-β (10 ng/ml) and FGF-2 (10 ng/ml). Following EMT
induction, pirfenidone (0.2 or 2.0 mM) or nintedanib (0.1 or 1.0
µM) were added. Data are presented as the mean ± standard error of
the mean (n=6, in triplicate) of the relative ratios of expression
compared with the levels of the respective controls. GAPDH was used
for normalization. *P<0.05 compared with untreated control (no
TGF-β/FGF-2); #P<0.05 compared with EMT-phenotype
control (TGF-β/FGF-2-treated cells). PFD, pirfenidone; TGF-β,
transforming growth factor-β; FGF-2, fibroblast growth factor-2;
EMT, epithelial-to-mesenchymal transition. |
Effects of pirfenidone and nintedanib
on EMT-induced cells
Although no significant or consistent effects on the
expression levels of E-cadherin and vimentin were observed,
pirfenidone or nintedanib treatment at various doses significantly
suppressed fibronectin and Slug expression levels compared with the
EMT-phenotype control groups (Figs. 2
and 3). Furthermore, the wound
healing assay results demonstrated that both of the agents
significantly reverted the enhanced cell motility in all three cell
lines compared with the EMT-phenotype control groups (Fig. 4A). Furthermore, following treatment
with pirfenidone or nintedanib at various doses, PD-L1 expression
was significantly downregulated compared with the EMT-induced
cells, except in response to nintedanib in PC-9 cells (Fig. 4B).
Discussion
The present study demonstrated that a borderline
sublethal dose of pirfenidone or nintedanib could revert the EMT
phenotype that had been induced by a combination of TGF-β and FGF-2
in three different human lung adenocarcinoma cell lines. The
altered expression of mesenchymal markers and Slug, together with
the alteration to cell motility, support this observation. The
present results are consistent with a previous report indicating
that nintedanib suppressed the EMT in ovarian cancer cells
(22). The same authors also reported
that the promotion of E-cadherin expression in A-549 cells occurred
through ZEB1 downregulation (22).
Furthermore, nintedanib was demonstrated to inhibit TGF-β-induced
myofibroblast differentiation through the inhibition of early
events in TGF-β signaling and the activation of SMAD family member
3 (Smad3) in lung fibroblast cells (18). Therefore, the present study has
provided novel evidence that pirfenidone possesses a similar
ability to revert the EMT in human lung adenocarcinoma cells.
Previous studies have demonstrated that pirfenidone
is able to suppress the differentiation of several cell types
(24–28). Conte et al (24) reported that pirfenidone reduced
fibroblast proliferation, attenuated TGF-β-induced α-smooth muscle
actin and inhibited the TGF-β-induced phosphorylation of Smad3 in
human lung fibroblast cells. In addition, pirfenidone has been
reported to suppress the differentiation of nasal polyp-derived
fibroblasts (25), retinal pigment
epithelial cells (26,27) and cardiac fibroblasts (28). Kozono et al (29) reported that pirfenidone-treated
pancreatic stellate cells suppressed the invasiveness and migration
of pancreatic cancer cells. Pirfenidone's ability to induce EMT
reversion in cancer cells may be similar to the previously reported
findings. Furthermore, the present study confirmed the previously
reported observation that PD-L1 expression was enhanced and
suppressed according to the induction and reversion of EMT,
respectively (30), suggesting the
involvement of the EMT phenotype in the process of immune evasion
in cancer.
The limitations of the present study should be
considered when interpreting these results. Firstly, only the
combination of TGF-β and FGF-2 was used to induce EMT, and other
known EMT-inducing factors were not examined. Secondly, the
important signaling pathways underlying EMT induction and reversion
were not identified. Lastly, the study only included in
vitro experiments, and thus may not reflect in vivo
effects. Further studies associating the phenomena observed in the
present study and investigations into their clinical relevance are
warranted.
In conclusion, the results of the present study
demonstrated that pirfenidone and nintedanib could each revert EMT
induction and downregulate the expression of PD-L1 in human lung
adenocarcinoma cells. Pirfenidone, thereby, may modify tumor
progression and responsiveness to chemotherapy and/or immunotherapy
for cancer.
Acknowledgements
The present study was supported by the Ministry of
Education, Culture, Sports, Science and Technology in Japan
(Kiban-C; grant no. 26461182).
References
|
1
|
Gavert N and Ben-Ze'ev A:
Epithelial-mesenchymal transition and the invasive potential of
tumors. Trends Mol Med. 14:199–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sato M, Shames DS and Hasegawa Y: Emerging
evidence of epithelial-to-mesenchymal transition in lung
carcinogenesis. Respirology. 17:1048–1059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shirakihara T, Horiguchi K, Miyazawa K,
Ehata S, Shibata T, Morita I, Miyazono K and Saitoh M: TGF-β
regulates isoform switching of FGF receptors and
epithelial-mesenchymal transition. EMBO J. 30:783–795. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kurimoto R, Iwasawa S, Ebata T, Ishiwata
T, Sekine I, Tada Y, Tatsumi K, Koide S, Iwama A and Takiguchi Y:
Drug resistance originating from a TGF-β/FGF-2-driven
epithelial-to-mesenchymal transition and its reversion in human
lung adenocarcinoma cell lines harboring an EGFR mutation. Int J
Oncol. 48:1825–1836. 2016.PubMed/NCBI
|
|
6
|
Lamouille S and Derynck R: Cell size and
invasion in TGF-beta-induced epithelial to mesenchymal transition
is regulated by activation of the mTOR pathway. J Cell Biol.
178:437–451. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li L, Han R, Xiao H, Lin C, Wang Y, Liu H,
Li K, Chen H, Sun F, Yang Z, et al: Metformin sensitizes
EGFR-TKI-resistant human lung cancer cells in vitro and in vivo
through inhibition of IL-6 signaling and EMT reversal. Clin Cancer
Res. 20:2714–2726. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Noble PW, Albera C, Bradford WZ, Costabel
U, Glassberg MK, Kardatzke D, King TE Jr, Lancaster L, Sahn SA,
Szwarcberg J, et al: Pirfenidone in patients with idiopathic
pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet.
377:1760–1769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Taniguchi H, Ebina M, Kondoh Y, Ogura T,
Azuma A, Suga M, Taguchi Y, Takahashi H, Nakata K, Sato A, et al:
Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J.
35:821–829. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Iyer SN, Gurujeyalakshmi G and Giri SN:
Effects of pirfenidone on procollagen gene expression at the
transcriptional level in bleomycin hamster model of lung fibrosis.
J Pharmacol Exp Ther. 289:211–218. 1999.PubMed/NCBI
|
|
11
|
Iyer SN, Gurujeyalakshmi G and Giri SN:
Effects of pirfenidone on transforming growth factor-beta gene
expression at the transcriptional level in bleomycin hamster model
of lung fibrosis. J Pharmacol Exp Ther. 291:367–373.
1999.PubMed/NCBI
|
|
12
|
Gurujeyalakshmi G, Hollinger MA and Giri
SN: Pirfenidone inhibits PDGF isoforms in bleomycin hamster model
of lung fibrosis at the translational level. Am J Physiol.
276:L311–L318. 1999.PubMed/NCBI
|
|
13
|
Di Sario A, Bendia E, Baroni G Svegliati,
Ridolfi F, Casini A, Ceni E, Saccomanno S, Marzioni M, Trozzi L,
Sterpetti P, et al: Effect of pirfenidone on rat hepatic stellate
cell proliferation and collagen production. J Hepatol. 37:584–591.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Garcia L, Hernández I, Sandoval A, Salazar
A, Garcia J, Vera J, Grijalva G, Muriel P, Margolin S and
Armendariz-Borunda J: Pirfenidone effectively reverses experimental
liver fibrosis. J Hepatol. 37:797–805. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Richeldi L, Costabel U, Selman M, Kim DS,
Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G,
et al: Efficacy of a tyrosine kinase inhibitor in idiopathic
pulmonary fibrosis. N Engl J Med. 365:1079–1087. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Richeldi L, du Bois RM, Raghu G, Azuma A,
Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y,
et al: Efficacy and safety of nintedanib in idiopathic pulmonary
fibrosis. N Engl J Med. 370:2071–2082. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wollin L, Maillet I, Quesniaux V, Holweg A
and Ryffel B: Antifibrotic and anti-inflammatory activity of the
tyrosine kinase inhibitor nintedanib in experimental models of lung
fibrosis. J Pharmacol Exp Ther. 349:209–220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rangarajan S, Kurundkar A, Kurundkar D,
Bernard K, Sanders YY, Ding Q, Antony VB, Zhang J, Zmijewski J and
Thannickal VJ: Novel mechanisms for the Antifibrotic action of
Nintedanib. Am J Respir Cell Mol Biol. 54:51–59. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Turner-Warwick M, Lebowitz M, Burrows B
and Johnson A: Cryptogenic fibrosing alveolitis and lung cancer.
Thorax. 35:496–499. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hubbard R, Venn A, Lewis S and Britton J:
Lung cancer and cryptogenic fibrosing alveolitis. A
population-based cohort study. Am J Respir Crit Care Med. 161:5–8.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matsushita H, Tanaka S, Saiki Y, Hara M,
Nakata K, Tanimura S and Banba J: Lung cancer associated with usual
interstitial pneumonia. Pathol Int. 45:925–932. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang RY, Kuay KT, Tan TZ, Asad M, Tang
HM, Ng AH, Ye J, Chung VY and Thiery JP: Functional relevance of a
six mesenchymal gene signature in epithelial-mesenchymal transition
(EMT) reversal by the triple angiokinase inhibitor, nintedanib
(BIBF1120). Oncotarget. 6:22098–22113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Conte E, Gili E, Fagone E, Fruciano M,
Iemmolo M and Vancheri C: Effect of pirfenidone on proliferation,
TGF-β-induced myofibroblast differentiation and fibrogenic activity
of primary human lung fibroblasts. Eur J Pharm Sci. 58:13–19. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shin JM, Park JH, Park IH and Lee HM:
Pirfenidone inhibits transforming growth factor β1-induced
extracellular matrix production in nasal polyp-derived fibroblasts.
Am J Rhinol Allergy. 29:408–413. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Choi K, Lee K, Ryu SW, Im M, Kook KH and
Choi C: Pirfenidone inhibits transforming growth factor-β1-induced
fibrogenesis by blocking nuclear translocation of Smads in human
retinal pigment epithelial cell line ARPE-19. Mol Vis.
18:1010–1020. 2012.PubMed/NCBI
|
|
27
|
Wang J, Yang Y, Xu J, Lin X, Wu K and Yu
M: Pirfenidone inhibits migration, differentiation, and
proliferation of human retinal pigment epithelial cells in vitro.
Mol Vis. 19:2626–2635. 2013.PubMed/NCBI
|
|
28
|
Shi Q, Liu X, Bai Y, Cui C, Li J, Li Y, Hu
S and Wei Y: In vitro effects of pirfenidone on cardiac
fibroblasts: Proliferation, myofibroblast differentiation,
migration and cytokine secretion. PLoS One. 6:e281342011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kozono S, Ohuchida K, Eguchi D, Ikenaga N,
Fujiwara K, Cui L, Mizumoto K and Tanaka M: Pirfenidone inhibits
pancreatic cancer desmoplasia by regulating stellate cells. Cancer
Res. 73:2345–2356. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen L, Gibbons DL, Goswami S, Cortez MA,
Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, et al: Metastasis
is regulated via microRNA-200/ZEB1 axis control of tumour cell
PD-L1 expression and intratumoral immunosuppression. Nat Commun.
5:52412014. View Article : Google Scholar : PubMed/NCBI
|