Introduction
Hepatocellular carcinoma (HCC) is the most common
type of liver cancer and is rapidly becoming one of the most
prevalent types of cancer, being the sixth most common type of
neoplasm worldwide and the third leading cause of cancer-associated
mortality, responsible for more than 650,000 mortalities per year
globally (1). The most efficient
treatments for this disease include complete surgical resection of
the tumor and liver transplantation. The outcome of HCC is
typically poor since only 10–20% of HCCs may be completely removed
by surgery and the lack of complete removal leads to the relapse of
the disease (2).
HCC is a tumor characterized by being highly
resistant to systemic chemotherapy. The use of the kinase inhibitor
sorafenib has previously increased the overall survival of patients
suffering from this disease (3–6). This
agent, which may be orally administered, has antiproliferative and
antiangiogenic activity and delays tumor progression, being at
present the standard of care in patients diagnosed with an
advanced-stage disease (7). However,
the active search for other treatments that may be used to treat
this malignancy is underway. Thus, during previous years, several
phase III trials, including patients with intermediate and
advanced-stages, have been carried out to test the effects of
various drugs; however, in general, they have failed to present
survival benefits (5), driving the
search for the identification of novel oncogenic drivers and
molecular therapeutics that may be used to treat HCC. In the
previous decade, molecular analyses of patients with HCC have
resulted in the identification of several genomic subclasses
(5), although, thus far, there is no
clear consensus with respect to the association between molecular
subclass and patient outcome.
Several precedents indicate that natural products
may be useful anticancer supplements. Numerous studies have
indicated that green tea-derived polyphenol
epigallocatechin-3-gallate exerts chemopreventive and
hepatoprotective effects against HCC in preclinical models
(8–10). The mechanism of action of this product
has not been fully elucidated, but may include the reduction of
prostaglandin biosynthesis by HCC cells (11), which may then induce apoptotic cell
death in HCC. In addition, epigallocatechin-3-gallate may reduce
the metastatic ability of HCC cells by decreasing the production of
osteopontin (12).
Ocoxin® oral solution (OOS) is an oral nutritional
supplement that contains several compounds with anticancer
activities, including epigallocatechin-3 gallate (13). In addition, OOS is comprised of a
variety of other components, including vitamins B6 and C and
cinnamic acid, which exhibit anticancer properties (14–16). In
addition, OOS contains glycyrrhizinic acid, which exhibits
anti-inflammatory and immunomodulatory effects (17). As OOS contains several compounds with
an antitumor action, it is currently being investigated in clinical
trials as part of the treatment of several types of cancer, in
which an improvement in the quality of life of the patients has
been reported (18,19). Additionally, several recent reports
have demonstrated that OOS exhibits in vitro and in
vivo antitumoral action in various tumor models, including HER2
positive breast cancer (13) and
acute myeloid leukemia (20). These
two tumor types are dissimilar at the molecular and cellular
levels; however, the clear antitumoral action of OOS has been
reported in the two. In addition, the antitumor action appeared to
have been mediated by the slowing of cell cycle progression induced
by an increase in expression of the cell cycle inhibitor p27.
Furthermore, a recent clinical study revealed preliminary data that
suggested that OOS may be useful in patients with end-stage HCC
(18).
On the basis of these precedents, the present study
aimed to explore the potential antitumoral effect of OOS in
preclinical models of HCC. The present study demonstrated that OOS
exhibits antitumoral effects in various HCC cellular models in
vitro. In addition, when tested in vivo, an antitumoral
effect was also demonstrated in terms of tumor growth reduction.
Combinational studies with sorafenib, the standard of care for the
treatment of HCC, indicated increased antitumoral activity in the
combination, providing the rationale to additionally explore the
clinical value of this therapy in HCC.
Materials and methods
Reagents and antibodies
The Dulbecco's modified Eagle's medium (DMEM), MTT,
hematoxylin and eosin were purchased from Sigma-Aldrich (Merck
Millipore, Darmstadt, Germany). The fetal bovine serum (FBS) and
antibiotics were purchased from Thermo Fisher Scientific, Inc.
(Waltham, MA, USA) and Immobilon P polyvinylidene fluoride
membranes from Merck Millipore. OOS was provided by Catalysis, S.L.
(Madrid, Spain). The other generic chemicals were purchased from
Sigma-Aldrich, Roche Applied Science (Mannheim, Germany) or Merck
Millipore.
The origins of the various antibodies used in the
Western blot analyses are as follows: Anti-GAPDH (cat. no.
sc-166574; 1:10,000) and anti-cyclin B (cat. no. sc-245; 1:5,000)
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA) and the anti-phospho-histone H3 (cat. no. 06-570;
1:3,500) from Merck Millipore. The horseradish
peroxidase-conjugated secondary antibodies (goat anti-rabbit; cat.
no. 170-6515; 1:20,000; and goat anti-mouse; cat. no. 170-6516;
1:10,000), were obtained from Bio-Rad Laboratories Inc. (Hercules,
CA, USA).
Cell culture, cell proliferation
measurement, protein purification and western blot analysis
The HepG2 and Huh7 HCC cell lines were donated by Dr
Mollinedo (University of Salamanca, Salamanca, Spain) and grown in
DMEM supplemented with 10% FBS and antibiotics. The cells were
cultured at 37°C in a humidified atmosphere in the presence of 5%
CO2-95% air.
The proliferation of HCC cells was examined using a
modified MTT metabolization assay (13). The cells were plated in triplicate and
treated the next day as indicated, with various concentrations of
OOS alone or in combination with other drugs: Sorafenib (LC
Laboratories, Woburn, MA, USA) 0.1, 0.3, 1 or 3 µM; docetaxel
(Hospira UK Ltd., Leamington Spa, UK) 1 or 3 µM; and cisplatin
(Pharmacia Grupo Pfizer, Madrid, Spain) 2 and 20 µM. On the day of
the experiment, MTT was added to the wells at 0.5 mg/ml and
incubated at 37°C for 1 h. The MTT-formazan crystals were dissolved
in DMSO and the absorbance of the samples was recorded at 570 nm
using a Tecan spectrophotometer with X-Fluor 4 (version 4.50; Tecan
Trading AG, Männedorf, Switzerland) software. A minimum of 3 wells
were analyzed for each condition, and the results are presented as
the mean ± standard deviation (SD) of a representative experiment
repeated at least twice. In order to prepare the cells for protein
analyses, they were washed in PBS and lysed in an ice-cold lysis
buffer (140 mM NaCl, 10 mM EDTA, 10% glycerol, 11% Nonidet P-40, 20
mM Tris, pH 8.0, 1 mM pepstatin, 1 µg/ml aprotinin, 1 µg/ml
leupeptin, 1 mM PMSF and 1 mM sodium orthovanadate). Protein
quantification, SDS-PAGE and western blot analysis were performed
as previously described (21). For
the western blot analysis, a total of 50 µg protein were probed
with the indicated antibodies.
Transfection and fluorimetric
quantification
The HepG2 and Huh7 cells were transfected with a
plasmid coding for the luciferase gene (pCDNA3.1-Luc) using the
jetPEI® reagent (Polyplus-transfection®, Illkirch, France)
according to the protocol of the manufacturer. The plasmid coding
for the luciferase gene (pCDNA3.1-Luc) was donated by Dr J.
Massagué (Memorial Sloan-Kettering Cancer Center, New York, NY,
USA). Positive transfected cells were obtained subsequent to
selection with the appropriate antibiotic, 450 µg/ml hygromycin.
Once the clones had been isolated they were independently harvested
by cloning rings, grown in multiwell plates and expanded.
Hygromycin positive clones were tested for the presence of the
luciferase gene by adding 150 µg/ml luciferin to the culture media,
and then reading the luminescence in triplicate wells using a
Synergy4 multi-mode microplate reader with Gen5 1.05 software
(BioTek Instruments, Inc., Winooski, VT, USA). Each clone was
analyzed at least twice and one of them, HepG2-Luc#22, was chosen
for further experiments. In addition, a previously published
MCF7-lucifesase expressing clone was established as previously
described (22) and analyzed in
parallel as a positive control. The correct behavior of these
clones in terms of general aspect, proliferation (measured as MTT
metabolization) and cell cycle profile was determined before
further proceeding.
Cell death, cell cycle and cell
synchronization and release experiments
To analyze apoptotic cell death, the cells were
treated for 48 h with the indicated treatments (OOS; dilutions,
1:25, 1:50 or 1:100 in culture media), resuspended in binding
buffer (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulphonic
acid/NaOH; 140 mM NaCl; 2,5 nM CaCl2; pH=7.4) containing
5 µl Annexin V-fluorescein isothiocyanate (BD Biosciences) and 5 µl
50 µg/ml propidium iodide (PI), and stained at room temperature for
15 min. A total of 50,000 cells were acquired using a BD Accuri C6
flow cytometer and C6 software (version 1.0.264.21; BD
Biosciences).
For cell cycle analysis by flow cytometry, the
ethanol-fixed cells were stained with 5 µg/ml PI and 250 µg
DNase-free RNase. A total of 50,000 cells were acquired as
previously described. For the cell cycle synchronization
experiments, the cells were arrested at various phases of the cell
cycle as previously described (21).
Thus, for G2-M synchronization, nocodazole treatment was
used, whereas for G1-S arrest a double thymidine block
was carried out. Subsequent to synchronization, the cells were
washed twice in PBS and released in normal medium, or in medium
supplemented with OOS (dilution, 1:25), and the cell samples were
harvested after 1, 2, 3, 6, 9, 12 or 24 h for additional flow
cytometry or western blot analysis.
In vivo experiments
For the animal studies, 12 7-week-old female athymic
mice (BALB/C nu/nu) weighing between 18 and 20 g, were purchased
from Charles River Laboratories (Wilmington, MA, USA) and kept in
pathogen-free housing at Universidad de Salamanca Animal Care
Facility (Salamanca, Spain). All animal experiments were performed
according to the institutional guidelines and protocol approved by
the Ethics Committee of Universidad de Salamanca (Salamanca,
Spain). A total of 1 week subsequent to the purchase of the mice,
they were injected with the appropriate cells to generate tumors.
For the xenograft model, 1×106 HepG2 cells resuspended
in 50 µl DMEM and 50 µl Matrigel were subcutaneously injected into
the right caudal flank of each mouse. When the tumors became
palpable, the mice were randomly assigned into two groups (six per
group), which received either vehicle alone, the control group, or
100 µl OOS per 20 g weight of mouse. The treatments were
administered with a once daily schedule (Monday to Friday) by oral
gavage until the humane endpoint (tumor volume, 2.000
mm3) was achieved by the control group and they were
subsequently sacrificed by isofluorane euthanasia. The mice were
weighed and the tumors were measured twice a week with a digital
caliper (Proinsa, Vitoria, Spain). Tumor volumes were calculated
using the formula: V= (L/2) × (W/2)2 × 4/3 × π, where V
= volume (cubic mm), L= length (mm) and W= width (mm). Subsequent
to the sacrifice of the mice, the tumor tissue was resected and
immediately frozen at −80°C.
For the disseminated model, HepG2-Luc cells were
directly injected into the liver of 14 BALB/C nu/nu mice who were
anesthetized with ketamine and xylazine, and a 5-mm incision was
carried out in the upper left ventral region of the abdomen,
through the skin and the muscle layer to access the abdominal
cavity. A total of 0.5×106 HepG2-Luc cells resuspended
in 25 µl DMEM and 25 µl Matrigel were injected in the liver, which
was observed through the incision. The muscle and the skin layers
were independently sutured, the region was cleaned and the mice
were treated with buprenorfine, used as an analgesic. The correct
localization of the cells was analyzed in vivo by
bioluminescent imaging, performed with the IVIS 50 imaging system
(Xenogen Corporation, Alameda, CA, USA), and the results were
analyzed using Living Image software (version 4.1; Perking Elmer,
Boston, MA, USA). The mice were intraperitoneally injected with 150
mg/kg aqueous solution of D-luciferin (Perking Elmer, Boston, MA,
USA) and anesthetized by isoflurane inhalation, and images were
captured. The status of the mice, including general status and
weight, and tumor growth were determined twice a week.
Histological and immunohistochemical
(IHC) analyses
Representative tumor areas were fixed in formalin,
embedded in paraffin, cut into 2–3-µm sections and either stained
with hematoxylin and eosin or prepared for IHC, which was performed
as previously described (23). Thus,
two cell conditioning periods of 8 min at 95°C and 4 min at 100°C
on a hot plate using Tris-EDTA, pH=8, buffer were performed on
previously dewaxed formalin-fixed paraffin-embedded tissue
sections. Sections were incubated for 42 min at 37°C with a 1:50
dilution anti-Ki67 antibody (Master Diagnóstica, Granada, Spain;
cat. no. MAD-000310QD), and the staining was performed with the IHC
3,3′-diaminobenzidine system (Ventana Medical Systems, Tuscon, AZ,
USA). The results were evaluated by pathologists blinded to the
clinicopathological and molecular data. The extension of the
necrotic area present in each tissue sample was assessed by
measuring the total tumor and necrotic areas. The tumors were
scanned and the percentage of the necrotic region was normalized to
the total area using the dotSlide system and dotSlide 2.1 software
(Olympus Corporation, Tokyo, Japan). These procedures were
performed by independent personnel of the pathology unit of the
University of Salamanca (Salamanca, Spain).
Statistical analysis
Each condition was analyzed in triplicate or
quadruplicate and data are presented as the mean ± SD of ≥3
independent experiments. Comparisons of continuous variables
between two groups were performed using a two-sided Student's
t test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effect of OOS on the proliferation of
HCC cell lines
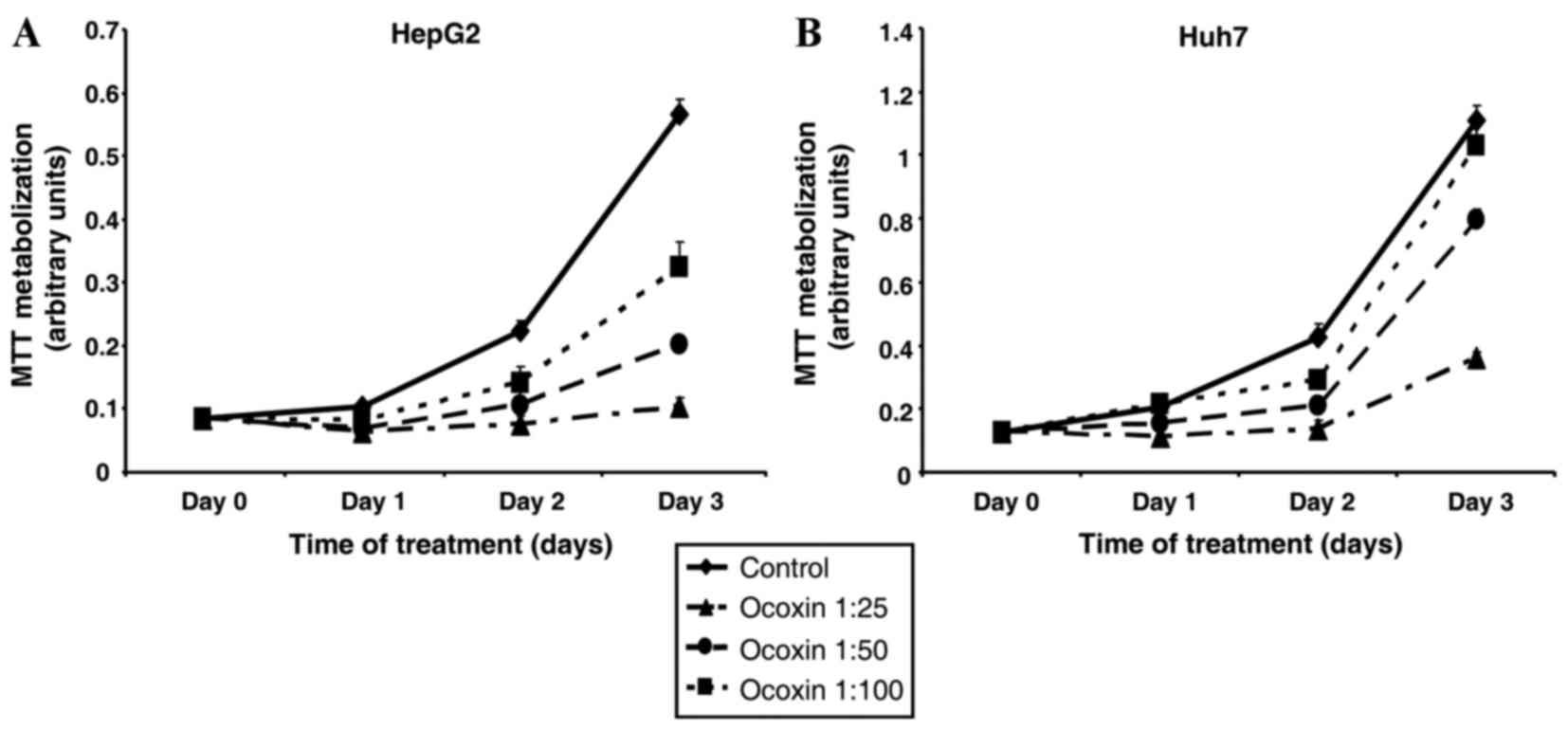
To study the potential effect of OOS on HCC, HepG2
and Huh7 HCC cell lines were treated for various days with
increasing amounts of OOS, and the cellular response in terms of
cellular proliferation was assessed. In those conditions, OOS
reduced MTT metabolization, used as an indicator of cell
proliferation, in the two cell lines (Fig. 1). The effect was time- and
dose-dependent. The HepG2 cell line was more sensitive to the
antiproliferative effect of OOS, compared with the Huh7 cell line.
Treatment of HepG2 cells with a 1:100 dilution of OOS inhibited
cell proliferation by 50% subsequent to three days of treatment
(P=0.00082; Fig. 1A), whereas an
equal dose of OOS did not substantially affect the proliferation of
Huh7 cells (P=0.21; Fig. 1B). Due to
the higher sensitivity of the HepG2 cell line to OOS, the present
study preferentially used the aforementioned cell line as the
principal model to explore the mechanism of action of OOS in
HCC.
Effect of OOS in combination with
other anti-HCC treatments
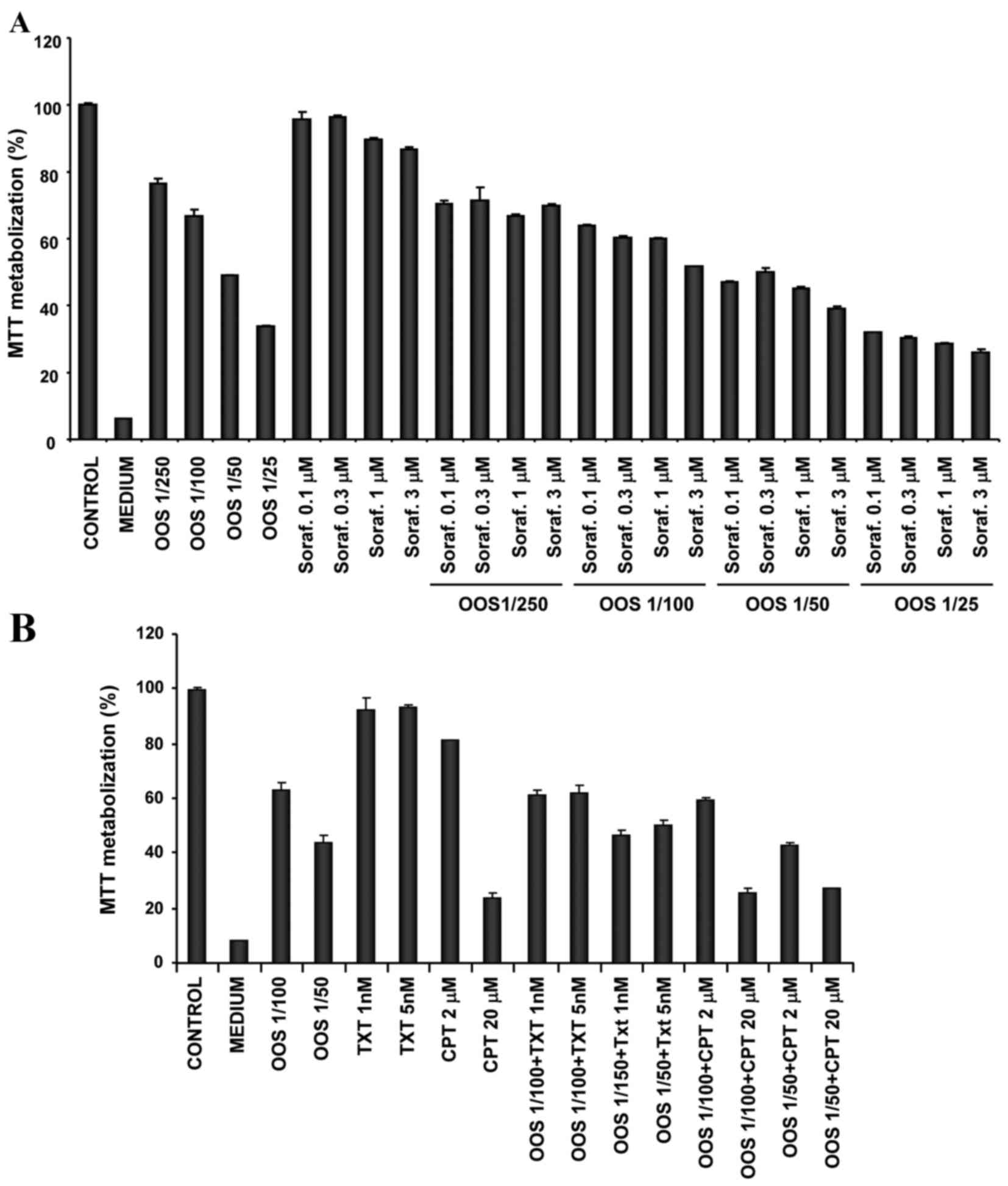
In the majority of the cases the success of
antitumoral treatments is based on the combination of various
agents. For that reason, the present study investigated if the
antitumoral effect of OOS on the HepG2 cell line was increased by
other drugs commonly used in the treatment of cancer. Since
sorafenib is the recommended systemic treatment for patients
diagnosed with advanced HCC, the present study tested the effect of
OOS in combination with sorafenib. The simultaneous use of the
agents was more efficient with respect to inhibiting the cell
growth, compared with any of the individual treatments (Fig. 2A). OOS was also evaluated in
combination with other drugs that had exhibited efficacy in the
treatment of patients with HCC, including the antitumoral agents
cisplatin (24) and docetaxel
(25,26). However, treatment with OOS in
combination with these agents did not substantially improve the
effect of the individual treatments (Fig.
2B).
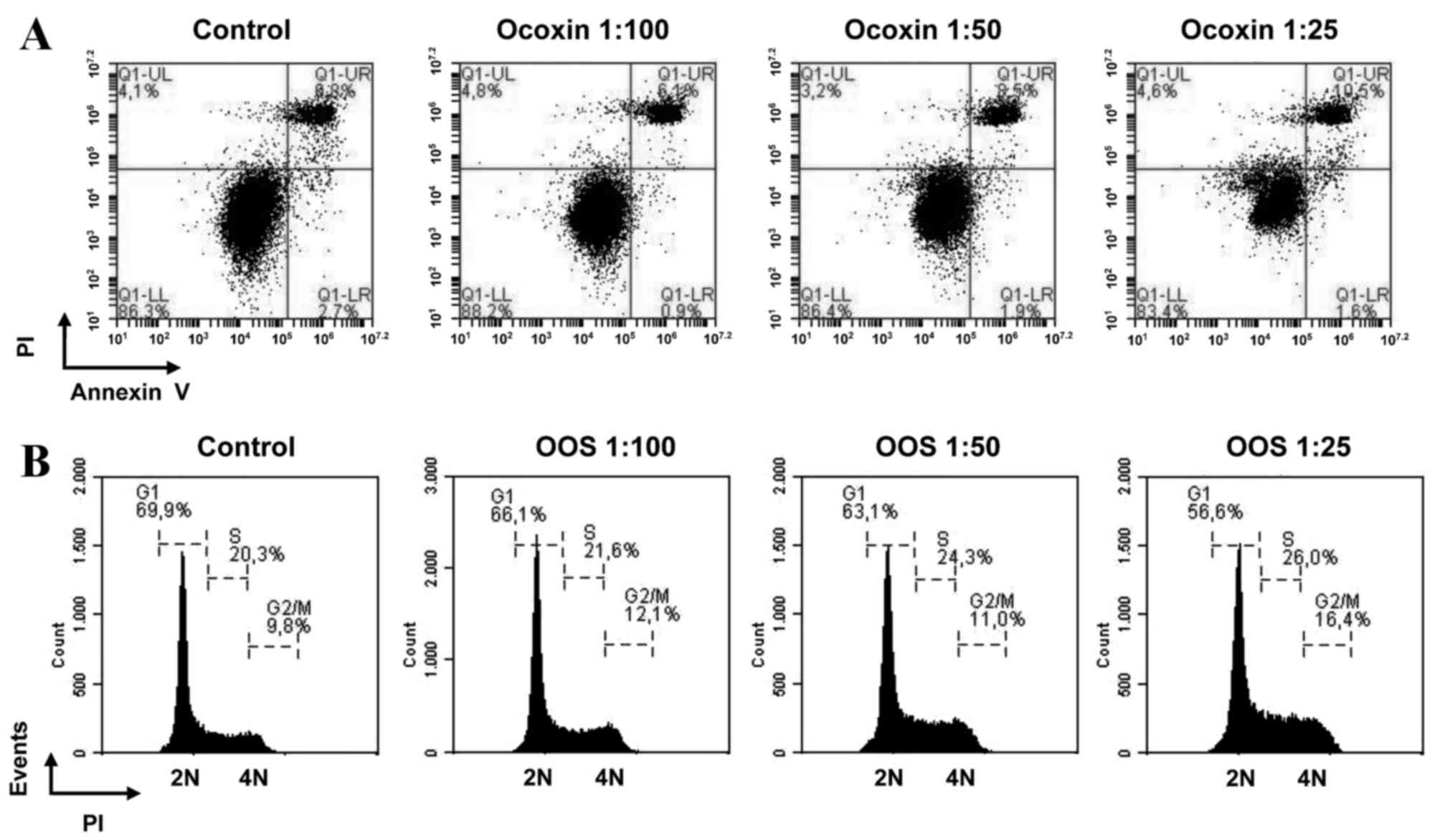
Effect of OSS on apoptotic cell death
and cell cycle progression
A decrease in MTT metabolization may be caused by an
increase in cell death, cell cycle arrest, or a combination of the
two. To additionally investigate the mechanism of action of OOS in
HCC cells, apoptotic cell death was initially assessed. The HepG2
cells were treated with increasing concentrations of OOS and
apoptotic cell death was determined by double staining with Annexin
V and PI. No major changes in the various populations were observed
in any of the concentrations tested, indicating that there was no
apoptotic cell death induced by OOS observed under the experimental
conditions of the present study (Fig.
3A).
The current study then investigated if OOS induced
cell cycle arrest in HCC cells. The cells treated for 48 h with
increasing concentrations of OOS were fixed and the cell cycle
profile was analyzed by PI staining of the DNA. A decrease in the
number of cells in the G1 phase of the cell cycle was
observed. In addition, treatment with OOS resulted in an increase
in the number of cells present in the G2-M section of
the histogram (Fig. 3B). These
results indicated that the antitumoral action of OOS was due to an
effect on cell cycle progression, without an apparent effect of OOS
on cell viability.
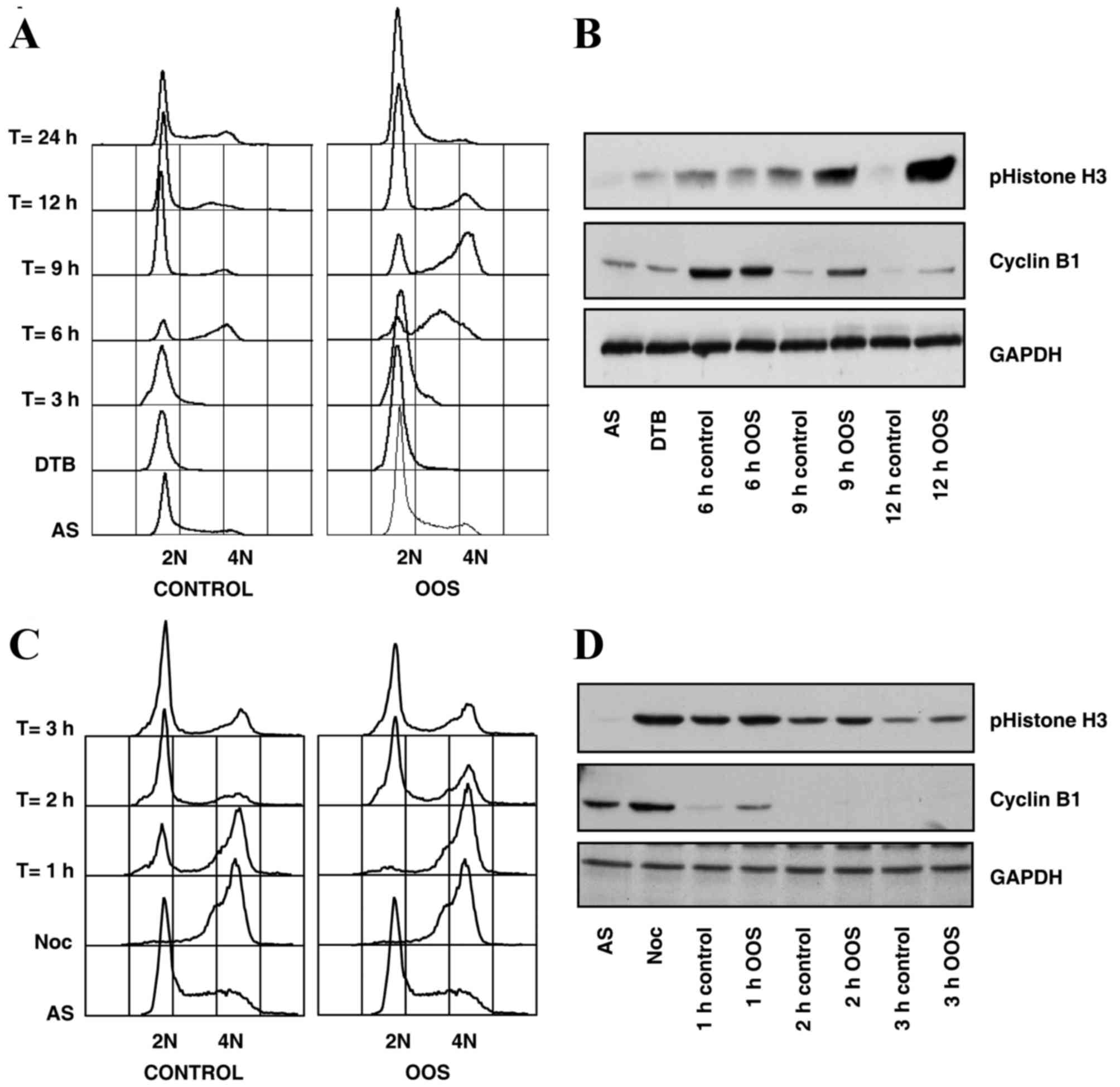
To more precisely map the effect of OOS on cell
cycle progression, synchronization and release experiments were
carried out. Two types of synchronization procedures were used to
arrest cells in various cell cycle phases. Firstly, HepG2 cells
were synchronized in the G1 phase of the cell cycle by
double thymidine treatment (21), and
then released from the double thymidine block by incubation in
normal medium, or in medium supplemented with OOS. A section of the
sample was fixed to analyze cell cycle progression by flow
cytometry, and the other section was lysed in order to explore the
biochemical markers of each cell cycle phase using western
blotting. Cells were observed to progress through the cell cycle
more slowly subsequent to OOS treatment (Fig. 4A and B). For example, when the cell
cycle distribution was analyzed using flow cytometry 9 h subsequent
to releasing the block, the majority of the cells had completed the
cell cycle in the control condition, as the majority of the cells
were already in the G1 phase of the cell cycle, with 2N
DNA content. At that time, the majority of the OOS-treated cells
were in the G2-M phase, with 4N DNA content (Fig. 4A). This observation was confirmed when
the expression levels of key cell cycle markers were determined by
western blot analysis. Thus, at the aforementioned time point, the
levels of the G2-M markers cyclin B and phospho-histone
H3 were higher in the OOS-treated cells, compared with the control
population, and demonstrated a larger number of cells in the
G2-M phase (Fig. 4B).
The delay of the cell cycle progression induced by
OOS was additionally confirmed by nocodazole synchronization
experiments. This is a reversible microtubule polymerization
inhibitor that arrests the cell cycle in the M phase, since cells
are unable to form the mitotic spindle in the presence of
nocodazole and the spindle checkpoint becomes active (27). When HepG2 cells were synchronized
using nocodazole and released into OOS-containing media, a delay in
mitotic exit was observed by flow cytometry and by western blot
analysis of key mitotic markers (Fig. 4C
and D). A total of 1 h subsequent to the release into the
media, ~1/2 of the cells were in G1 in the control
condition, whereas none of the cells had reached G1 in
the OOS-treated group as observed by flow cytometry cell cycle
analysis (Fig. 4C). In addition, a
delay in the degradation of the G2-M markers cyclin B1
and phospho-histone H3 by western blot analysis was observed
subsequent to OOS treatment, confirming that the cell cycle was
slowed under that condition (Fig.
4D).
Effect of OOS treatment on xenograft
tumor models
To determine if OOS exerted an effect on HCC growth
in vivo, two distinct mouse models were developed. In the
first one, HepG2 cells were engrafted in the flanks of BALB/c nu/nu
athymic mice. Subsequent to three days, when the tumors became
palpable, the mice were randomly assigned into two groups, one
receiving daily oral treatment with 100 µl OOS/20 g body weight and
the other, considered the control group, administered 100 µl
water/20 g body weight. The tumors in the mice receiving OOS
treatment grew at a slower rate, as compared with the tumors in the
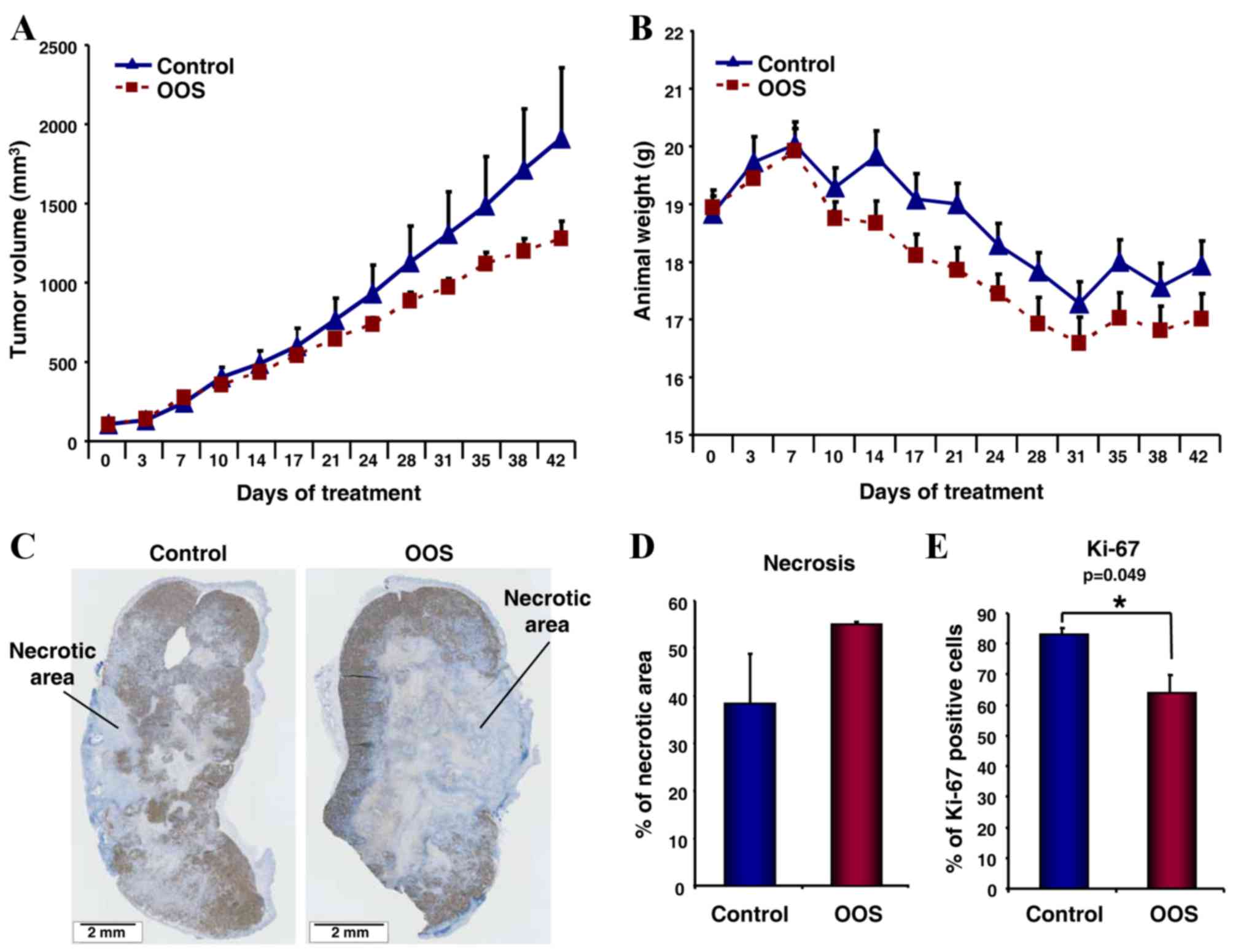
untreated control mice (Fig. 5A).
Notably, and due to the aggressive characteristics of the HCC cells
in vivo, all mice lost weight throughout the experiment.
However, that effect was independent of OOS administration as there
were no differences observed in the general behavior of the two
groups (Fig. 5B).
To investigate the reason leading to the diminished
tumor growth observed in OOS-treated mice, several tumors were
resected and fixed subsequent to the sacrifice of the mice, and
stained with the proliferation marker Ki-67. When the whole tumor
section was observed, a difference in proliferative vs. necrotic
regions was identified (Fig. 5C).
Thus, when the total necrotic area was measured and normalized to
total tumor area, an increase in necrosis was identified in the
tumors from OOS-treated mice (Fig.
5D). In addition, OOS induced an evident decrease in the
percentage of Ki-67 positive cells present in the proliferative
region of the tumor; this was a statistically significant reduction
compared with the control untreated tumors (P=0.049; Fig. 5E).
A second mouse model was used to follow the growth
and dissemination properties of HepG2 cells in vivo. For
this model, HepG2 cells were transfected with a plasmid coding for
the luciferase gene, so that cells carrying the gene would emit
light in the presence of luciferin. HepG2 and Huh7 cells positively
expressing the luciferase gene, HepG2-Luc and Huh7-Luc,
respectively, were isolated and luciferase expression was assessed.
One of the clones expressing high levels of the transgene,
HepG2-Luc#22 (Fig. 6A), was selected
for additional investigation. This clone, although expressing the
luciferase gene, exhibited normal proliferation and cell cycle
status compared with the parental cells (data not presented).
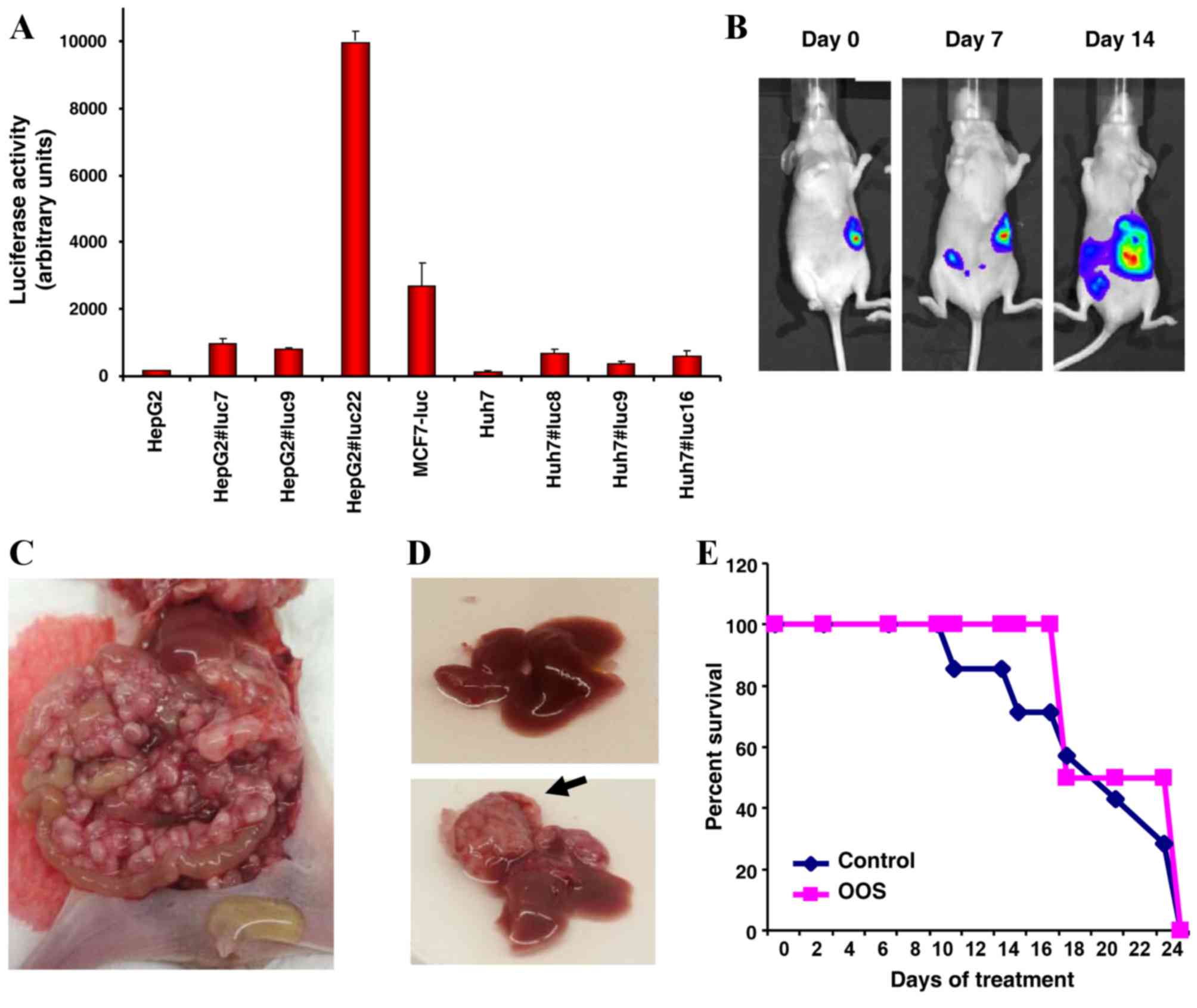 | Figure 6.OOS effect on a disseminated HCC tumor
model. (A) Generation of HepG2-luc cells. HepG2 and Huh7 cells were
transfected with the pCDNA3.1-Luc plasmid and positive clones
selected with 450 µg/ml hygromycin, individually harvested and
expanded. The expression of the luciferase gene was verified using
a Synergy4 multi-mode microplate reader and Gen5 1.05 software. An
MCF7-luc clone was analyzed in parallel and used as a positive
control. (B) HepG2-Luc cells were implanted directly in the liver
of female BALB/C nu/nu athymic mice by surgery. Subsequent to one
day, the correct localization of the cells was verified by
luciferin detection with the bioluminescence reader Xenogen IVIS 50
using Living Image software. Tumor progression and dissemination
were measured twice per week, and representative images of the mice
were obtained once per week. (C) Macroscopic image of a
representative mouse at the time of sacrifice, exhibiting a large
number of small tumoral masses extended throughout the abdominal
cavity, and ascitic fluid. (D) Liver status of the mice at the time
of sacrifice. The liver of the majority of the mice appeared to be
healthy (upper image). Only one of the mice exhibited a liver tumor
(black arrow, lower image). (E) Kaplan-Meier curves of the mice. A
total of 14 mice were intrahepatically injected with HepG2-Luc
cells, and subsequent to five days, randomly assigned into two
groups that were orally treated five days per week with 200 µl
OOS/20 g body weight, or with vehicle alone as the control. The
graph depicts the time at which the mice required sacrifice due to
their general status and abdominal enlargement with ascites. OOS,
Ocoxin® oral solution; Luc, luciferase; HCC, hepatocellular
carcinoma. |
HepG2-Luc cells were orthotopically placed in the
liver of BALB/c nu/nu athymic mice by a small incision in the skin
and the direct injection of HepG2 cells into the liver. The correct
location of the cells was verified in vivo by bioluminescent
imaging (Fig. 6B). The progression of
the tumors was followed by a luciferin injection twice a week.
Although the tumors were intrahepatically injected, they rapidly
disseminated throughout the abdomen, as demonstrated by imaging
(Fig. 6B) and symptoms including an
enlarged abdomen due to the accumulation of ascitic fluid (data not
presented). In addition, an abdominal inspection of the mice
revealed the infiltration of the peritoneal cavity by tumoral
masses (Fig. 6C). In general, the
liver was normal (Fig. 6D, top image)
and at the time of sacrifice only 1/14 of the mice that were
analyzed exhibited a mayor hepatic lesion (Fig. 6D, lower image, black arrow).
A total of five days subsequent to injection the
mice were randomly assigned into two groups that received water or
OOS (200 µl/20 g body weight) by daily oral gavage. The control and
the OOS-treated tumors were aggressive; however, the OOS-treated
tumors appeared to be less aggressive, as the control mice required
sacrifice prior to the OOS-treated mice (Fig. 6E).
Discussion
Even subsequent to curative resection of HCC in
early-stage disease, ~70% of patients exhibit recurrence after five
years (2), increasing the requirement
for the development of novel drugs for use in an adjuvant setting.
No effective neoadjuvant or adjuvant treatment options that reduce
the risk of recurrence are currently available. Additionally, in
previous years adoptive immunotherapy and retinoids have been used
with some success in HCC (28,29),
although the results have been difficult to reproduce.
The present study was conducted in order to explore
the potential antitumoral action of OOS in HCC, following several
precedents. Firstly, our previous studies demonstrated the
antitumoral value of OOS in highly proliferating cells, including
breast cancer and acute myeloid leukemia (13,20).
Secondly, numerous reports have demonstrated the effects of
epigallocatechin-3-gallate on HCC models inducing cell death
(30), suppressing hepatocellular
carcinoma growth or dissemination. The present study, using two HCC
cell lines, observed that OOS induced a decrease in MTT
metabolization, indicative of reduced proliferation in the
HCC-derived cell lines tested. In addition, combinatory studies
using OOS and standard of care drugs used in the therapy of HCC
indicated that OOS potentiated the effect of sorafenib, one of the
principal drugs used to treat advanced-stage HCC.
Mechanistically, the decrease in MTT metabolization
caused by OOS appeared to be cell death-independent, since no major
apoptotic population was detected in those conditions. This was a
noteworthy result as one of the product components is
epigallocatechin-3-gallate, which has formerly been reported to
induce apoptosis in HCC cells (30).
The reason for this discrepancy remains unknown, but may be
associated with the distinct experimental conditions used in these
studies, including the doses of the products employed. While OOS
did not induce a noticeable effect on apoptosis, it exhibited a
clear effect on cell cycle progression and that is a possible
mechanism underlying the antitumoral action of OOS (30). The effect of OOS on cell cycle
progression was more evident when analyzing the action of OOS on
synchronized cells. In cells synchronized in G1 by a
double thymidine block protocol, OOS delayed G1-S
progression. In addition, the nocodazole block experiments also
indicated that OOS delayed the progression from the M to
G1 phase. The observation that OOS affected progression
along various phases of the cell cycle is notable, and suggests
that OOS as a companion product may be used with other agents
acting on more specific cell cycle phases. Therefore, the action of
OOS in HCC appears to involve a delay in cell cycle progression
rather than cell cycle arrest, which is in line with the
conclusions observed when analyzing other tumor types (13,20).
The data presented in the current study indicates
that OOS may be beneficial for patients with HCC, particularly in
combination with standard of care drugs. This conclusion is
supported by the in vitro data and by the observation that
OOS also exerted an antitumoral action when administered to mice
carrying xenografted HepG2 cells. The effect of OOS on this in
vivo model resulted in a decrease in the rate of tumor growth,
suggesting that OOS exerted a cytostatic effect, in line with the
in vitro results on the mechanism of action of OOS. In
addition, the larger areas of necrotic tumor tissue observed in the
tissue samples from OOS-treated mice suggests that this compound
may provoke damage to the tumoral tissue, either by acting directly
on the tumoral cells or their supporting stroma. These findings are
relevant as, together with recent data indicating that patients
with terminal HCC exhibited a longer survival when administered OOS
(18), it opens the possibility of
using OOS to delay tumor progression when used in combination with
standard of care therapies. In addition, the preventive effect on
HCC previously reported for epigallocatechin-3-gallate (8), raises the possibility of using OOS in
patients with cirrhosis or patients previously affected with
hepatitis B in order to combat the progression of these diseases
into HCC. Future studies may assess this hypothesis using models of
the aforementioned diseases at pre-tumoral stages.
Acknowledgements
The plasmid coding for the luciferase gene
(pCDNA3.1-Luc) was donated by Dr J. Massagué (Memorial
Sloan-Kettering Cancer Center, New York, NY, USA). The present
study was supported by Catalysis S.L. and the Ministry of Economy
and Competitiveness of Spain (grant no., BFU2012-39151). The
present study was supported by a contract from the Spanish Cancer
Network (grant no. AECC12/GC02). The Institute of Molecular and
Cellular Cancer Biology (CSIC-University of Salamanca) laboratory
received support from the European Community through the regional
development funding program and from the Fundación Ramón
Areces.
References
|
1
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Llovet JM, Schwartz M and Mazzaferro V:
Resection and liver transplantation for hepatocellular carcinoma.
Semin Liver Dis. 25:181–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bruix J, Raoul JL, Sherman M, Mazzaferro
V, Bolondi L, Craxi A, Galle PR, Santoro A, Beaugrand M,
Sangiovanni A, et al: Efficacy and safety of sorafenib in patients
with advanced hepatocellular carcinoma: Subanalyses of a phase III
trial. J Hepatol. 57:821–829. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gish RG, Finn RS and Marrero JA: Extending
survival with the use of targeted therapy in the treatment of
hepatocellular carcinoma. Gastroenterol Hepatol (N Y). 9 4 Suppl
2:S1–S24. 2013.
|
|
5
|
Llovet JM, Villanueva A, Lachenmayer A and
Finn RS: Advances in targeted therapies for hepatocellular
carcinoma in the genomic era. Nat Rev Clin Oncol. 12:408–424. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mancuso A, Mazzarelli C, Perricone G and
Zavaglia C: Sorafenib efficacy for treatment of HCC recurrence
after liver transplantation is an open issue. J Hepatol.
60:6812014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. N Engl J
Med. 359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Darweish MM, Abbas A, Ebrahim MA and
Al-Gayyar MM: Chemopreventive and hepatoprotective effects of
Epigallocatechin-gallate against hepatocellular carcinoma: Role of
heparan sulfate proteoglycans pathway. J Pharm Pharmacol.
66:1032–1045. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee SI, Kim HJ and Boo YC: Effect of green
tea and (−)-epigallocatechin gallate on ethanol-induced toxicity in
HepG2 cells. Phytother Res. 22:669–674. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shimizu M, Shirakami Y, Sakai H, Tatebe H,
Nakagawa T, Hara Y, Weinstein IB and Moriwaki H: EGCG inhibits
activation of the insulin-like growth factor (IGF)/IGF-1 receptor
axis in human hepatocellular carcinoma cells. Cancer Lett.
262:10–18. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin J, Chang Y, Wei W, He YF, Hu SS, Wang
D and Wu YJ: Prostanoid EP1 receptor as the target of
(−)-epigallocatechin-3-gallate in suppressing hepatocellular
carcinoma cells in vitro. Acta Pharmacol Sin. 33:701–709. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zapf MA, Kothari AN, Weber CE, Arffa ML,
Wai PY, Driver J, Gupta GN, Kuo PC and Mi Z: Green tea component
epigallocatechin-3-gallate decreases expression of osteopontin via
a decrease in mRNA half-life in cell lines of metastatic
hepatocellular carcinoma. Surgery. 158:1039–1048. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hernaádez-García S, González V, Sanz E and
Pandiella A: Effect of oncoxin oral solution in her2-overexpressing
breast cancer. Nutr Cancer. 67:1159–1169. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hollman PC, Feskens EJ and Katan MB: Tea
flavonols in cardiovascular disease and cancer epidemiology. Proc
Soc Exp Biol Med. 220:pp. 198–202. 1999; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu L, Hudgins WR, Shack S, Yin MQ and
Samid D: Cinnamic acid: A natural product with potential use in
cancer intervention. Int J Cancer. 62:345–350. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang CS, Chung JY, Yang G, Chhabra SK and
Lee MJ: Tea and tea polyphenols in cancer prevention. J Nutr. 130
2S Suppl:S472–S478. 2000.
|
|
17
|
Gomez EV, Perez YM, Sanchez HV, Forment
GR, Soler EA, Bertot LC, Garcia AY, del Rosario Abreu Vazquez M and
Fabian LG: Antioxidant and immunomodulatory effects of Viusid in
patients with chronic hepatitis C. World J Gastroenterol.
16:2638–2647. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Al-Mahtab M, Akbar S, Khan M and Rahman S:
Increased survival of patients with end-stage hepatocellular
carcinoma due to intake of ONCOXIN®, a dietary supplement. Indian J
Cancer. 52:443–446. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Uddin MAI Dayem, Mahmood I, Ghosha AK and
Khatuns RA: Findings of the 3-Month supportive treatment with
ocoxin solution beside the standard modalities of patients with
different neoplastic diseases. TAJ. 22:172–175. 2009.
|
|
20
|
Diaz-Rodríguez E, Hernández-García S, Sanz
E and Pandiella A: Antitumoral effect of Ocoxin on acute myeloid
leukemia. Oncotarget. 7:6231–6242. 2016.PubMed/NCBI
|
|
21
|
Díaz-Rodríguez E and Pandiella A:
Multisite phosphorylation of Erk5 in mitosis. J Cell Sci.
123:3146–3156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seoane S, Montero JC, Ocaña A and
Pandiella A: Breast cancer dissemination promoted by a
neuregulin-collagenase 3 signalling node. Oncogene. 35:2756–2765.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
de Alava E, Ocaña A, Abad M, Montero JC,
Esparís-Ogando A, Rodríguez CA, Otero AP, Hernández T, Cruz JJ and
Pandiella A: Neuregulin expression modulates clinical response to
trastuzumab in patients with metastatic breast cancer. J Clin
Oncol. 25:2656–2663. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yoshikawa M, Ono N, Yodono H, Ichida T and
Nakamura H: Phase II study of hepatic arterial infusion of a
fine-powder formulation of cisplatin for advanced hepatocellular
carcinoma. Hepatol Res. 38:474–483. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alberts SR, Reid JM, Morlan BW, Farr GH
Jr, Camoriano JK, Johnson DB, Enger JR, Seay TE and Kim GP:
Gemcitabine and docetaxel for hepatocellular carcinoma: A phase II
north central cancer treatment group clinical trial. Am J Clin
Oncol. 35:418–423. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yata Y, Xue F, Takahara T, Kudo H, Hirano
K, Yasumura S, Minemura M, Scanga AE and Sugiyama T: Docetaxel
inhibits progression of human hepatoma cell line in vitro and is
effective in advanced hepatocellular carcinoma. Hepatol Res.
40:304–310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jordan MA, Thrower D and Wilson L: Effects
of vinblastine, podophyllotoxin and nocodazole on mitotic spindles.
Implications for the role of microtubule dynamics in mitosis. J
Cell Sci. 102:401–416. 1992.PubMed/NCBI
|
|
28
|
Muto Y, Moriwaki H, Ninomiya M, Adachi S,
Saito A, Takasaki KT, Tanaka T, Tsurumi K, Okuno M, Tomita E, et
al: Prevention of second primary tumors by an acyclic retinoid,
polyprenoic acid, in patients with hepatocellular carcinoma.
Hepatoma Prevention Study Group. N Engl J Med. 334:1561–1567. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takayama T, Sekine T, Makuuchi M, Yamasaki
S, Kosuge T, Yamamoto J, Shimada K, Sakamoto M, Hirohashi S, Ohashi
Y and Kakizoe T: Adoptive immunotherapy to lower postsurgical
recurrence rates of hepatocellular carcinoma: A randomised trial.
Lancet. 356:802–807. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Y, Duan W, Owusu L, Wu D and Xin Y:
Epigallocatechin-3-gallate induces the apoptosis of hepatocellular
carcinoma LM6 cells but not non-cancerous liver cells. Int J Mol
Med. 35:117–124. 2015.PubMed/NCBI
|