Introduction
A decade ago, bortezomib became the first member of
the novel chemotherapeutic class of proteasome inhibitors to
receive clinical approval (1).
Originally developed for the study of proteasome physiology,
proteasome inhibitors soon demonstrated significant antineoplastic
activity (2) that, starting with
bortezomib, was successfully applied in the treatment of multiple
myeloma (3). Subsequently, bortezomib
has become an almost indispensable part of the gold standard
therapy regimen, significantly improving the treatment outcomes of
affected patients (4). However, its
clinical applicability is considerably impeded by dose-limiting
toxicity (5) and by primary or
secondary drug-resistance (6,7). As acquired drug resistance can be
mediated by enhanced efflux transporter expression (8,9), it is
essential to determine whether anti-myeloma drugs are transported
by certain proteins. It has previously been demonstrated that
bortezomib is a substrate of the well-known ATP-binding cassette
(ABC) transporter P-glycoprotein (P-gp); however, the clinical
relevance of this finding remains unclear (10). Furthermore, antineoplastic drugs may
induce the expression of similar transporter genes in respective
target tissues or cells eventually leading to iatrogenic drug
resistance (8,9). Consequently, investigations into the
inducing properties of proteasome inhibitors are warranted.
In 2012, the epoxyketone carfilzomib became the
second proteasome inhibitor to be approved by the Food and Drug
Administration for the treatment of relapsed and refractory
multiple myeloma (11). Inducing
irreversible proteasome inhibition, carfilzomib not only
demonstrates greater preclinical antitumor activity (12), but it is also effective in cell lines
already resistant to bortezomib (13). The boronic acid ixazomib is another
reversible second-generation proteasome inhibitor that appears to
exhibit sufficient activity in bortezomib-resistant myeloma cells,
despite structural similarities (14). It has become the first orally
bioavailable proteasome inhibitor to be approved for the treatment
of recurrent multiple myeloma in the USA (15). In addition to the aspect of drug
resistance at a cellular level, it is important to understand the
merits and limitations of certain proteasome inhibitors in a given
clinical setting characterized by combination chemotherapy or the
co-administration of drugs against co-morbidities. Extensive
research, therefore, concentrates on the potential differences of
proteasome inhibitors in pharmacodynamics, pharmacokinetics and
vulnerability to drug-drug interactions (16). Such drug-drug interactions may
modulate the systemic availability/exposure of other
chemotherapeutics that are typically part of the complex therapy
regimen for patients with myeloma (17,18), or
that may be used in combination with proteasome inhibitors in the
future due to proven synergistic effects (19,20).
Transporter-based pharmacokinetic drug-drug interactions typically
comprise direct inhibition of drug transporters or alterations of
their gene expression. In the present study, the inhibitory and
induction potentials of bortezomib, carfilzomib and ixazomib on
various drug transporters known to relevantly affect systemic
pharmacokinetics, and thus the efficacy and safety of
pharmacotherapies, were evaluated (21). Furthermore, the effect of these
compounds on the expression levels of crucial drug transporters was
investigated (1) in a cell model used
for assessing inducing properties (LS180 cells) assessing possible
drug-drug interactions and (2) in
myeloma cell lines, where induction could contribute to iatrogenic
treatment failure. The findings indicate that proteasome inhibitors
neither relevantly inhibit nor induce drug transporters, suggesting
that bortezomib, carfilzomib and ixazomib do not provoke
transporter-mediated pharmacokinetic drug-drug interactions.
Furthermore, transporter expression remains unchanged in myeloma
cell lines upon exposure to proteasome inhibitors, indicating
cellular adaptation mechanisms to be of minor relevance. Together,
the ‘negative results’ presented in this study suggest that
proteasome inhibitors do not affect drug transporter expression at
certain physiological barriers, including the intestine; therefore,
proteasome inhibitors are devoid of transporter-based drug-drug
interactions.
Materials and methods
Materials
Bortezomib was purchased from Absource Diagnostics
GmbH (Munich, Germany); carfilzomib and ixazomib were purchased
from Sequoia Research Products, Ltd. (Pangbourne, UK). The
GenElute™ Mammalian Total RNA Miniprep kit, fumitremorgin C (FTC),
doxorubicin, rifampicin, verapamil hydrochloride, all cell culture
media [RPMI-1640, Iscove's modified Dulbecco's media (IMDM),
Dulbecco's modified Eagle's medium (DMEM)] and supplements were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Fetal calf serum (FCS) was purchased from Biochrom, Ltd.
(Cambridge, UK) and geneticin (G418) was supplied by PAA
Laboratories (GE Healthcare Life Sciences, Chalfont, UK). Calcein
acetoxymethylester (calcein-AM) was obtained from Invitrogen
(Thermo Fisher Scientific, Inc., Waltham, MA, USA), and
8-fluorescein-cAMP (8-FcA) from BIOLOG Life Science Institute
(Bremen, Germany) and pheophorbide A (PhA) from Frontier
Scientific, Ltd. (Carnforth, UK). The RevertAid™ H Minus First
Strand cDNA Synthesis kit from Fermentas (Thermo Fisher Scientific,
Inc., Pittsburgh, PA, USA) and the Absolute QPCR SYBR®
Green mix was obtained from ABgene (Thermo Fisher Scientific,
Inc.). The Cytotoxicity Detection kit was from Roche Applied
Science (Mannheim, Germany). Primers were synthesized by Eurofins
MWG Operon (Ebersberg, Germany).
Cell lines
The myeloma cell lines used (Karpas-620, L363,
OPM-2, EJM, KMM-1, LP-1, RPMI-8226 and U266) were purchased from
the German Collection of Microorganisms and Cell Cultures GmbH
(Braunschweig, Germany). With the exception of EJM and LP-1 cells,
which were cultured in IMDM, all myeloma cell lines were cultured
in RPMI-1640 medium. The media were each supplemented with 10% FCS,
2 mM glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin
sulfate. Cells were cultured at 37°C, 5% CO2 and 100%
humidity.
The LS180 human colon adenocarcinoma cell line
(American Type Culture Collection, Manassas, USA) is one model
frequently used for investigating pregnane-X-receptor (PXR) and
aryl hydrocarbon receptor mediated induction (22–24), and
was used as an induction model in the present study. LS180 cells
were cultured under standard cell culture conditions at 37°C, 5%
CO2 and 100% humidity in DMEM supplemented with 10% FCS,
2 mM glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin sulfate
and 0.1 mM nonessential amino acids.
Possible inhibitory effects on the human organic
anion transporting polypeptides [OATPs, also termed solute carriers
of organic anions (SLCOs)] were studied in HEK-293 cells
overexpressing SLCO1B1 (HEK-OATP1B1) or SLCO1B3
(HEK-OATP1B3) (25,26). The cell line transfected with the
empty control vector was used as a control (HEK-293-VC G418). These
cell lines were provided by Dr D. Keppler (German Cancer Research
Centre, Heidelberg, Germany) and cultured in DMEM supplemented with
10% FCS, 2 mM glutamine, 100 U/ml penicillin, 100 µg/ml
streptomycin sulfate, and 800 µg/ml G418 at 37°C, 5% CO2
and 100% humidity.
The ability of the proteasome inhibitors to inhibit
breast cancer resistance protein (BCRP) was investigated in
MDCKII-BCRP (overexpressing BCRP/ABCG2) cells (27) in comparison with the parental MDCKII
cell line. These cells lines were provided by Dr A. H. Schinkel
(The Netherlands Cancer Institute, Amsterdam, Netherlands) and
cultured in DMEM supplemented with 10% FCS, 2 mM glutamine, 100
U/ml penicillin and 100 µg/ml streptomycin sulfate. Cells were
cultured at 37°C, 5% CO2 and 100% humidity.
The P-gp inhibition assay was performed using the
P388 murine monocytic leukemia cell line and the corresponding
doxorubicin-resistant P388/dx cells overexpressing mdr1a/1b, which
are an ideal model for testing P-gp inhibition (28). These two cell lines were provided by
Dr D. Ballinari (Pharmacia & Upjohn, Milano, Italy). The
RPMI-1640 culture medium for these cells was supplemented with 10%
FCS, 2 mM glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin
sulfate and 100 µM 2-mercaptoethanol. Additionally, doxorubicin
(0.43 µM) was added to the medium of the P388/dx cells in order to
maintain P-gp overexpression; this was discontinued one day prior
to each assay. Cells were cultured at 37°C, 5% CO2 and
100% humidity.
Growth inhibition assays
To exclude any profound antiproliferative effects of
the proteasome inhibitors on LS180 and myeloma cells during the
induction assay, growth inhibition assays were conducted to define
the maximum concentration ensuring ~80% cell survival
(IC20). For the adherent LS180 cells, growth inhibition
was quantified following 48 h of incubation at standard cell
culture conditions via crystal violet staining of the surviving
cells, as previously described (29).
For the myeloma cells, an MTT assay was used to assess growth
inhibition following 48 h of incubation at standard cell culture
conditions, as described previously (29). Each experiment was performed in
quadruplicate with n=8 wells for each concentration (0.005, 0.01,
0.05, 0.1, 0.5, 1, 5, 10, 50 and 100 µM). Sigmoid
concentration-response curves and IC20 values were
calculated using GraphPad Prism software (version 6.02; GraphPad
Software Inc., La Jolla, CA, USA).
Induction assay
For the induction assays, the cells were treated
with bortezomib, carfilzomib, ixazomib, 20 µM rifampicin (positive
control) or culture medium only (negative control) for 4 days at
standard cell culture conditions. All incubation solutions were
adjusted to 0.02% dimethyl sulfoxide. Myeloma cells were exposed to
proteasome inhibitor concentrations representing the
IC20 value in the corresponding cell line (bortezomib,
1–5 nM; carfilzomib, 1–20 nM; ixazomib, 2–70 nM). LS180 cells were
treated with four distinct concentrations (bortezomib, 0.1–5 nM;
carfilzomib, 0.5–10 nM; ixazomib, 1–50 nM), whereas the maximum
concentration applied corresponded to the IC20 of
proliferation inhibition in this cell line (bortezomib, 7.6 nM;
carfilzomib, 13 nM; ixazomib, 62 nM). All incubations were
conducted at least in quadruplicate. Following harvesting by
centrifugation (1,000 × g, 5 min, 4°C), the cell pellets were
subjected to RNA extraction using the GenElute™ Mammalian Total RNA
Miniprep kit.
Quantification of mRNA expression
using reverse transcription-quantitative polymerase chain reaction
(RT-qPCR)
The purity and concentration of the isolated RNA
were determined spectrophotometrically. Using the RevertAid™ H
Minus First Strand cDNA Synthesis kit, cDNA was synthesized
according to the manufacturer's protocol. The mRNA expression of
ABCB1 (coding for P-gp), ABCC1 [coding for multidrug
resistance-associated protein 1 (MRP1)], ABCC2 (coding for
MRP2) and ABCG2 (coding for BCRP) was quantified using qPCR
with the LightCycler® 480 (Roche Applied Science). From
each biological sample, one technical PCR duplicate was prepared;
prior to PCR, amplification was performed in 20 µl total volume
containing 5 µl 1:10 diluted cDNA, 0.15 µM of each primer and 1X
Absolute QPCR SYBR® Green mix for 40 cycles. Primer
sequences and thermocycling conditions are listed in Table I.
 | Table I.Primer sequences and thermocycler
conditions used in reverse transcription-quantitative polymerase
chain reaction. |
Table I.
Primer sequences and thermocycler
conditions used in reverse transcription-quantitative polymerase
chain reaction.
| Gene | Primers | Thermocycler
conditions |
|---|
| β 2-mg |
| F |
5′CCAGCAGAGAATGGAAAGTC3′ | 15 s, 95°C; 30 s,
61°C; 30 s, 72°C |
| R |
5′CATGTCTCGATCCCACTTAAC3′ |
|
| GU |
| F |
5′TTCAACAGGATCCACCTCTG3′ | 15 s, 95°C; 30 s,
61°C; 30 s, 72°C |
| R |
5′AGCACTCTCGTCGGTGACTG3′ |
|
| G6PDH |
| F |
5′ATCGACCACTACCTGGGCAA3′ | 15 s, 95°C; 30 s,
61°C; 30 s, 72°C |
| R |
5′TTCTGCATCACGTCCCGGA3′ |
|
| HPRT |
| F |
5′CTGGCGTCGTGATTAGTG3′ | 15 s, 95°C; 30 s,
61°C; 30 s, 72°C |
| R |
5′CACACAGAGGGCTACAATG3′ |
|
| HUPO |
| F |
5′AGCTCTGGAGAAACTGCTG3′ | 15 s, 95°C; 30 s,
61°C; 30 s, 72°C |
| R |
5′CAGCAGCTGGCACCTTATTG3′ |
|
| RPL13 |
| F |
5′GCTCATGAGGCTACGGAAAC3′ | 15 s, 95°C; 30 s,
61°C; 30 s, 72°C |
| R |
5′TATTGGGCTCAGACCAGGAG3′ |
|
| ABCB1 |
| F |
5′CCCATCATTGCAATAGCAGG3′ | 15 s, 95°C; 30 s,
60°C; 50 s, 72°C |
| R |
5′TGTTCAAACTTCTGCTCCTGA3′ |
|
| ABCC1 |
| F |
5′ATGTCACGTGGAATACCAGC3′ | 15 s, 95°C; 30 s,
60°C; 50 s, 72°C |
| R |
5′GAAGACTGAACTCCCTTCCT3′ |
|
| ABCC2 |
| F |
5′ACAGAGGCTGGTGGCAACC3′ | 15 s, 95°C; 30 s,
63°C; 30 s, 72°C |
| R |
5′ACCATTACCTTGTCACTGTCCATGA3′ |
|
| ABCG2 |
| F |
5′AGATGGGTTTCCAAGCGTTCAT3′ | 15 s, 95°C; 30 s,
57°C; 30 s, 72°C |
| R |
5′CCAGTCCCAGTACGACTGTGACA3′ |
|
The most stable housekeeping genes for the treatment
of each cell line were identified using geNorm (version 3.4; Centre
for Medical Genetics, Ghent, Belgium) (30) and used for normalization. GAPDH was
the most stable in LS180, KMM-1, RPMI-8226 and U266 cells under
bortezomib treatment, in LS180 cells and LP-1 cells under
carfilzomib treatment, and in LP1 cells under ixazomib treatment.
β2-microglobulin was most stable in EJM cells and OPM-2 cells under
bortezomib treatment, in L363 cells under carfilzomib treatment and
in Karpas-620, L363, and OPM-2 cells under ixazomib treatment.
Hypoxanthine-phosphoribosyl transferase 1 was most stable in
Karpas-620, L363 and LP-1 cells under bortezomib treatment, in
OPM-2 cells under carfilzomib treatment and in RPMI-8226 cells
under ixazomib treatment. The 60S human acidic ribosomal protein P1
was most stable in EJM cells under carfilzomib treatment. Ribosomal
protein L13 was most stable in Karpas-620 cells, KMM-1 cells and
RPMI-8266 cells under carfilzomib treatment. Glucuronidase-β was
most stable in U266 cells under carfilzomib treatment, and in LS180
cells, EJM cells, KMM-1 cells and U266 cells under ixazomib
treatment.
Data were evaluated using calibrator-normalized
relative quantification with efficiency correction using
LightCycler® 480 software (version 1.5; Roche Applied
Science), which calculates the relative amount of the target gene
and the reference gene based on the crossing points (Cp) and the
underlying calibration curve. The results are expressed as the
target/reference ratio divided by the target/reference ratio of the
calibrator and are, therefore, corrected for sample inhomogeneities
and variance caused by detection. Whenever mRNA expression was
below the detection limit (Cp value >35), the respective cell
line was excluded from further analysis. The degree of
induction/repression was then calculated by the mean mRNA
expression ratio between the incubated samples and the respective
medium control. A threshold of a 1.5-fold change in mRNA expression
normalized to the respective negative control was defined, i.e.
normalized mRNA levels >150% or <67% compared with the
control, as induction or repression, respectively. Statistical
analysis was performed for these values only.
Drug transporter inhibition
assays
The potential inhibitory effects of bortezomib,
carfilzomib and ixazomib on the activity of various drug
transporters were evaluated and compared between model cell lines
overexpressing ABCB1, ABCG2, SLCO1B1 or
SLCO1B3 in relation to the respective parental cell line.
With the use of fluorescent substrates, three drug transporter
inhibition assays were performed, as previously described and
validated (22,31,32). All
experiments were conducted at least in triplicate.
In brief, for the P-gp inhibition assay, P388 and
P388/dx cells were pre-incubated with the proteasome inhibitors
(≤10 µM; each concentration was evaluated in octuplicate) for 15
min at 37°C in Hepes buffered Hank's balanced salt solution
(HHBSS). Following pre-incubation, calcein-AM was added at a final
concentration of 1 µM and the cells were further incubated for 30
min at 37°C. Following washing twice with ice-cold HHBSS, the cells
were lysed in 1% Triton X-100 for 15 min at 37°C and calcein
fluorescence was measured using a Fluoroskan Ascent™ fluorometer
(Thermo Fisher Scientific, Inc.) with 485 nm excitation and 535 nm
emission filters. The effect in the parental cell line P388 was
used to determine whether the effects observed could be attributed
to P-gp inhibition. The P-gp inhibitor verapamil served as the
positive control.
For the BCRP inhibition assay, MDCKII and
MDCKII-BCRP cells were incubated for 30 min at 37°C in RPMI-1640
with 2% FCS containing 1 µM pheophorbide A. Following washing,
cells were incubated for 60 min at 37°C with medium containing the
proteasome inhibitors at 0.05, 0.1, 0.5, 1, 5, 10, 50 or 100 µM
Following washing, intracellular fluorescence was analyzed using a
BD™ LSR II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA)
with a 633 nm helium/neon laser and a 660 nm bandpass filter. In
each sample, 30,000 cells were counted. To quantify the effects of
the proteasome inhibitors, the ratio between the median
fluorescence with the inhibitor and without the inhibitor during
the efflux period was calculated and normalized to the effect
observed in the parental cell line. The selective BCRP inhibitor
FTC served as the positive control.
For the OATP inhibition assays, HEK-OATP1B1,
HEK-OATP1B3 and HEK-293-VC G418 cells were incubated in PBS with 2%
FCS containing 2.5 µM 8-FcA with or without proteasome inhibitor
(≤100 µM) at 37°C for 10 min. Following washing, intracellular
fluorescence was analyzed using a BD™ LSR II flow cytometer with a
solid state coherent sapphire blue laser and a 530 nm bandpass
filter for 8-FcA. In each sample, 30,000 cells were counted. The
ratio between the median fluorescence of intracellular 8-FcA with
and without the inhibitor was calculated in order to evaluate the
inhibitor effects. The effect in the cell line HEK-293-VC G418 was
used to determine whether the effects observed could be attributed
to OATP inhibition. The potent OATP inhibitor rifampicin served as
the positive control.
To exclude confounding variables, including leakage
of the fluorescent agents due to membrane lesions, non-toxic
concentrations of each of the proteasome inhibitors were determined
prior to each assay using the Cytotoxicity Detection kit, according
to the manufacturer's protocol. As bortezomib demonstrated ≥20%
cytotoxicity in HEK-OATP1B3 cells at higher incubation
concentrations, this compound was only examined at a concentration
of ≤5 µM in the OATP1B3 inhibition assay.
Statistical analysis
Statistical analysis was conducted using GraphPad
Prism version 6.02 (GraphPad Software, Inc., La Jolla, CA, USA).
Differences >1.5-fold threshold in mRNA-expression provoked by
the proteasome inhibitors, compared with the medium control, in
myeloma cells were evaluated using the Student's two-tailed t-test.
Differences >1.5-fold threshold in mRNA-expression provoked by
the proteasome inhibitors or rifampicin, compared with the medium
control, in LS180 cells were analyzed using one way analysis of
variance with Dunnett's post hoc test (compared with the medium
control). P≤0.05 was considered to indicate a statistically
significant difference.
Results
Induction of drug transporter
expression
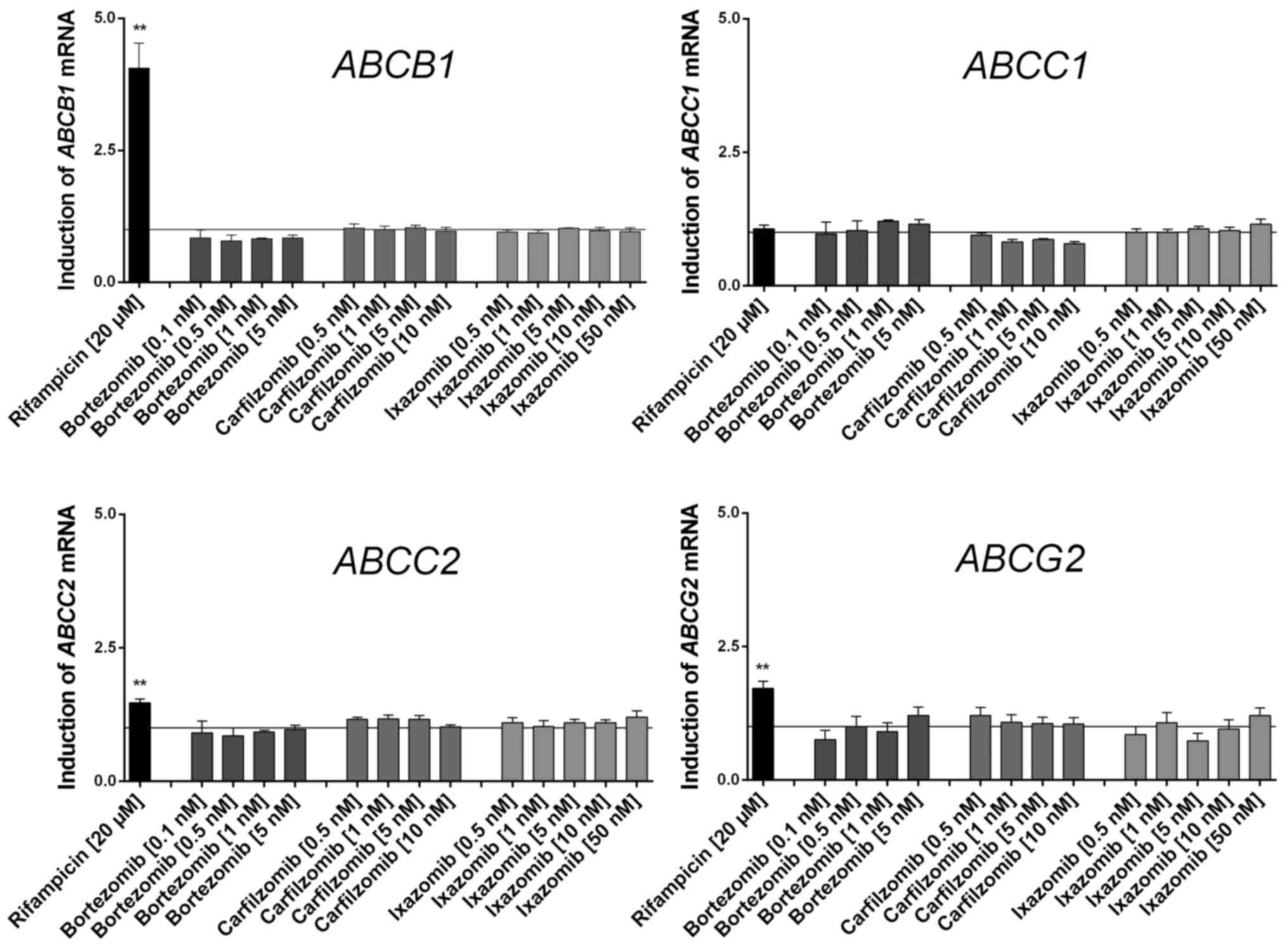
None of the proteasome inhibitors tested
significantly induced or repressed any of the drug transporter
genes investigated in LS180 cells (Fig.
1). As predicted, the positive control rifampicin induced the
expression of typical nuclear PXR-regulated genes, including
ABCB1.
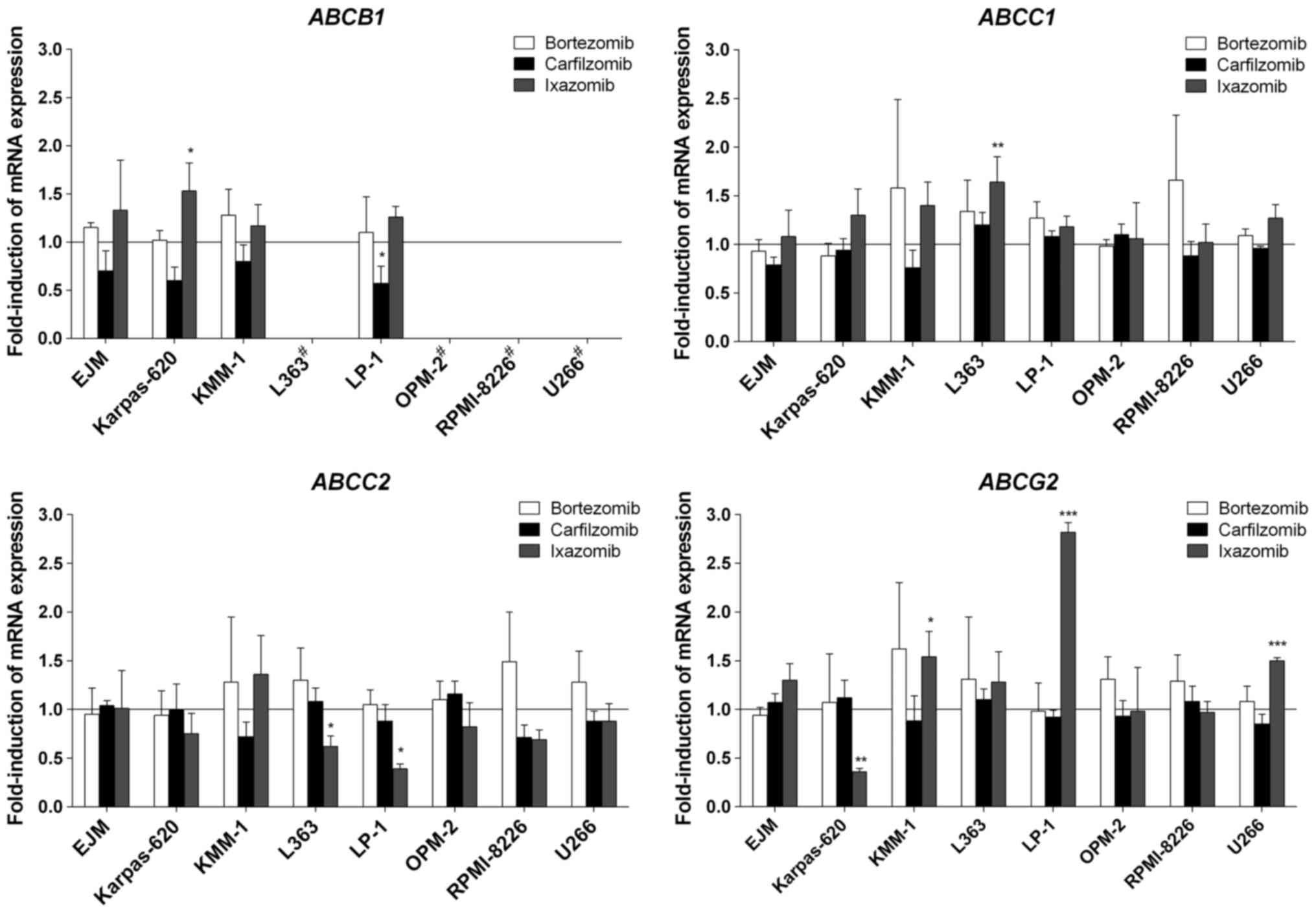
In the myeloma cells, only minor changes to gene
expression occurred. Specifically, bortezomib did not induce the
expression of any gene investigated in the present study (Fig. 2). Similarly, carfilzomib did not
induce gene expression in myeloma cells, but suppressed the mRNA
expression of ABCB1 in LP-1 cells (Fig. 2). Ixazomib provoked certain changes in
transporter gene expression, but only in a modest number of myeloma
cell lines. The most pronounced effect of ixazomib was observed for
ABCG2, for which mRNA expression was significantly increased
by 2.8-fold in LP-1 cells, and 1.5-fold in KMM-1 and U266 cells,
whereas it was significantly suppressed in Karpas-620 cells
(Fig. 2). In contrast to the
experiments with LS180 cells, the prototypical inducer rifampicin
exhibited no significant effects on mRNA expression levels in the
myeloma cells; even its typical target genes, including
ABCB1, were not altered upon exposure to rifampicin (data
not presented).
Inhibition of drug transporter
activity
Compared with carfilzomib, bortezomib and ixazomib
demonstrated weak or no inhibition of the examined drug
transporters. Bortezomib and ixazomib only inhibited OATP1B1, but
at higher concentrations compared with carfilzomib (Table II). Apart from OATP1B1, carfilzomib
also inhibited OATP1B3 with comparable potency compared with its
effect on OATP1B1. Furthermore, BCRP efflux activity was inhibited
with however 3-fold lower potency compared with its effect on
OATP1B1. All proteasome inhibitors studied did not significantly
increase intracellular calcein fluorescence at concentrations of
≥10 µM, indicating a lack of P-gp-mediated calcein-AM
transport.
| Table II.Comparison of the inhibitory
potential (IC50) of bortezomib, carfilzomib and ixazomib
on the activity of various drug transporters. |
Table II.
Comparison of the inhibitory
potential (IC50) of bortezomib, carfilzomib and ixazomib
on the activity of various drug transporters.
|
| IC50,
µM |
|---|
|
|
|
|---|
| Drug
transporter | Bortezomib | Carfilzomib | Ixazomib |
|---|
| BCRP | N/A | 12.4±2.0 | N/A |
| OATP1B1 | 140±9.5 | 3.6±0.5 | >100 |
| OATP1B3 | >10a | 4.7±1.0 | N/A |
| P-gp | N/A | N/A | N/A |
Discussion
Following its clinical approval in 2003, bortezomib
has unquestionably revolutionized the treatment of multiple myeloma
(31). Proteasome inhibitors in
particular have become a primary interest of myeloma-associated
research due to the significant improvements observed in the
outcomes of affected patients (4).
The novel proteasome inhibitors carfilzomib and ixazomib have been
clinically approved and have the potential to overcome previous
limitations associated with bortezomib treatment (6). To further optimize their clinical
application, a review on several studies investigated the potential
differences in their respective pharmacodynamic or pharmacokinetic
profiles (16). However, proteasome
inhibitors may also differ in their interactions with
co-administered drugs (17). This may
considerably alter certain factors, including the bioavailability
of other chemotherapeutics that are usually part of the complex
multi-drug therapy regimen for patients with myeloma (Palumbio,
Mai). Therefore, the present study aimed to identify and compare
such transporter-based systemic pharmacokinetic drug-drug
interactions possibly mediated by bortezomib, carfilzomib or
ixazomib. For experiments investigating inhibition, the focus was
on the most important drug transporters for drug-drug interactions
(21), whereas for experiments
investigating induction, the effects on ABCC1 (MRP1) and
ABCC2 (MRP2) were also evaluated.
None of the tested proteasome inhibitors relevantly
modified the mRNA expression of the investigated drug transporters
in the LS180 induction model cell line following four days of
constant exposure. This indicates that the proteasome inhibitors
investigated are not activators of PXR, which usually mediates the
induction of genes, including ABCB1, ABCC2 and
ABCG2 (32–34). This is concordant with previous in
vitro studies that reported no significant changes in the
expression of certain drug transporter genes (ABCB1,
ABCC1 and ABCG2), even following long-term exposure
to bortezomib (35) or an increase in
ABCB1 expression following six months of treatment with
increasing concentrations of carfilzomib only (36). As the data from the current study
demonstrated that induction via PXR could be excluded, an
iatrogenic increase in drug transporters, such as ABCB1, may
be attributed to a selection process rather than targeted
transcriptional induction. Such Darwinian selection processes
leading to drug resistance have previously been described for
kinase inhibitors, including imatinib (37), or for classical cytotoxic compounds
like docetaxel (38).
In contrast to experiments using the intestinal cell
model (LS180), certain statistically significant differences in
mRNA expression patterns in myeloma cells were observed following
treatment with ixazomib or carfilzomib. However, the majority of
the effects were statistically insignificant, and thus of debatable
clinical relevance. Indeed, ixazomib significantly induced the
expression of ABCB1 in Karpas-620 cells, of ABCC1 in
L363 cells and of ABCG2 in KMM-1, LP-1 and U266 cells.
However, targeted induction mediated by PXR appears to improbable
for the following reasons: i) These genes were not induced by
ixazomib in LS180 cells (the gold-standard for PXR-mediated gene
regulation); ii) the prototypical PXR ligand rifampicin had no
observable significant effects in the myeloma cell lines. Thus, the
mechanisms underlying the few differences in drug transporter mRNA
expression observed remain uncertain. The concurrent decrease in
the mRNA expression of certain genes mediated by carfilzomib and
ixazomib is challenging to elucidate, but increases and decreases
may have resulted from changes in mRNA stability, and not from
specific transcriptional regulation.
Another principal mechanism of drug-drug
interactions can be provoked by the inhibition of transporter
activity (39). Bortezomib and
ixazomib did not relevantly inhibit drug transporter activity. The
weak inhibition of OATP1B1 effected by bortezomib only occurred at
high incubation concentrations that exceeded frequently recorded
plasma levels (40). Possibly, this
observation results from competitive inhibition due to the weak
uptake of bortezomib via OATP1B1, as previously described (10). Potentially due to structural
similarities with bortezomib, the results obtained for ixazomib
were predominantly comparable. In contrast to bortezomib, for which
only incomplete data concerning drug transporter inhibition is
available, the data for ixazomib matches the Declaration in the
Summary of Product Characteristics of Ninlaro (41), indicating that ixazomib does not
inhibit P-gp, BCRP, OATP1B1 and OATP1B3. By contrast with the two
proteasome inhibitors containing boronic acids, carfilzomib
inhibited OATP1B1 and OATP1B3 at relatively low concentrations. The
data for OATP1B1 are concordant with those indicated in the Summary
of Product Characteristics of Kyprolis published by the European
Medicines Agency (42). Although
inhibition principally occurred within the range of the maximum
plasma concentrations (Cmax) (41), its relevance for clinical drug-drug
interactions remains limited as carfilzomib is rapidly eliminated
from the systemic circulation, leading to a rapid decline in plasma
concentrations (42,43). Consequently, the observed BCRP
inhibition following the treatment of cells with carfilzomib
appears to be less relevant. Thus far, significant drug-drug
interactions have not been reported for carfilzomib in clinical
trials (42). The question arises
whether the effects observed in vitro are also relevant
clinically. The Cmax of ixazomib are between 64–213 nM
following an oral application of 4 mg (44), distinctly exceeding the applied
concentrations of 2–70 nM in the present induction study.
Therefore, the in vitro conditions utilized in this
experimental set up are realistic in terms of in vivo plasma
exposure. For carfilzomib, the induction concentrations in the
present study were low, compared with the observable plasma peak
concentrations measured in a previous study (43). However, due to high systemic
clearance, carfilzomib is eliminated so rapidly from the
circulation that concentrations around the Cmax are only
maintained for a short period of time (45). Thus, it remains open whether changes
in mRNA expression may also occur in vivo; in either case
they will most likely be irrelevant to pharmacokinetic drug-drug
interactions, but may contribute to the resistance of myeloma cells
towards the proteasome inhibitors and other antineoplastic drugs.
Together, these data indicate that no transporter-mediated systemic
drug-drug interactions are to be reasonably expected in
vivo.
In conclusion, the transporter-mediated systemic
pharmacokinetic drug-drug interaction potential of the proteasome
inhibitors bortezomib, carfilzomib and ixazomib as perpetrator
drugs appears to be low. As these proteasome inhibitors did not
affect the transcription of drug transporter genes in the induction
model using LS180 cells, drug resistance through the iatrogenic
transcriptional induction of respective transporter genes is
unlikely. As proteasome inhibitors are substrates of drug
transporters, Darwinian selection of pre-existing transporter
overexpressing myeloma subclones may still lead to enhanced
transporter expression in a given myeloma cell population.
Acknowledgements
The present study was supported by the Deutsche
Forschungsgemeinschaft (grant no. SFB/TRR79) and the EU 7th
Framework Program (grant no. ‘OverMyR’). The authors would like to
thank J. Kocher, C. Mueller, S. Rosenzweig and A. Fautsch for their
technical assistance.
References
|
1
|
Adams J and Kauffman M: Development of the
proteasome inhibitor Velcade (Bortezomib). Cancer Invest.
22:304–311. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kisselev AF and Goldberg AL: Proteasome
inhibitors: From research tools to drug candidates. Chem Biol.
8:739–758. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Richardson PG, Barlogie B, Berenson J,
Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina
M, Alexanian R, et al: A phase 2 study of bortezomib in relapsed,
refractory myeloma. N Engl J Med. 348:2609–2617. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ludwig H, Sonneveld P, Davies F, Bladé J,
Boccadoro M, Cavo M, Morgan G, de la Rubia J, Delforge M,
Dimopoulos M, et al: European perspective on multiple myeloma
treatment strategies in 2014. Oncologist. 19:829–844. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Argyriou AA, Iconomou G and Kalofonos HP:
Bortezomib-induced peripheral neuropathy in multiple myeloma: A
comprehensive review of the literature. Blood. 112:1593–1599. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dispenzieri A, Jacobus S, Vesole DH,
Callandar N, Fonseca R and Greipp PR: Primary therapy with single
agent bortezomib as induction, maintenance and re-induction in
patients with high-risk myeloma: Results of the ECOG E2A02 trial.
Leukemia. 24:1406–1411. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Richardson PG, Xie W, Mitsiades C,
Chanan-Khan AA, Lonial S, Hassoun H, Avigan DE, Oaklander AL, Kuter
DJ, Wen PY, et al: Single-agent bortezomib in previously untreated
multiple myeloma: Efficacy, characterization of peripheral
neuropathy, and molecular correlations with response and
neuropathy. J Clin Oncol. 27:3518–3525. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen Z, Shi T, Zhang L, Zhu P, Deng M,
Huang C, Hu T, Jiang L and Li J: Mammalian drug efflux transporters
of the ATP binding cassette (ABC) family in multidrug resistance: A
review of the past decade. Cancer Lett. 370:153–164. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Szakács G, Paterson JK, Ludwig JA,
Booth-Genthe C and Gottesman MM: Targeting multidrug resistance in
cancer. Nat Rev Drug Discov. 5:219–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Clemens J, Seckinger A, Hose D, Theile D,
Longo M, Haefeli WE, Burhenne J and Weiss J: Cellular uptake
kinetics of bortezomib in relation to efficacy in myeloma cells and
the influence of drug transporters. Cancer Chemother Pharmacol.
75:281–291. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Siegel DS, Martin T, Wang M, Vij R,
Jakubowiak AJ, Lonial S, Trudel S, Kukreti V, Bahlis N, Alsina M,
et al: A phase 2 study of single-agent carfilzomib (PX-171-003-A1)
in patients with relapsed and refractory multiple myeloma. Blood.
120:2817–2825. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Demo SD, Kirk CJ, Aujay MA, Buchholz TJ,
Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, et al:
Antitumor activity of PR-171, a novel irreversible inhibitor of the
proteasome. Cancer Res. 67:6383–6391. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kuhn DJ, Chen Q, Voorhees PM, Strader JS,
Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan
AA, et al: Potent activity of carfilzomib, a novel, irreversible
inhibitor of the ubiquitin-proteasome pathway, against preclinical
models of multiple myeloma. Blood. 110:3281–3290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gentile M, Offidani M, Vigna E, Corvatta
L, Recchia AG, Morabito L, Morabito F and Gentilli S: Ixazomib for
the treatment of multiple myeloma. Expert Opin Investig Drugs.
24:1287–1298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ocio EM, Mitsiades CS, Orlowski RZ and
Anderson KC: Future agents and treatment directions in multiple
myeloma. Expert Rev Hematol. 7:127–141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Teicher BA and Tomaszewski JE: Proteasome
inhibitors. Biochem Pharmacol. 96:1–9. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Palumbo A, Bringhen S, Rossi D, Cavalli M,
Larocca A, Ria R, Offidani M, Patriarca F, Nozzoli C, Guglielmelli
T, et al: Bortezomib-melphalan-prednisone-thalidomide followed by
maintenance with bortezomib-thalidomide compared with
bortezomib-melphalan-prednisone for initial treatment of multiple
myeloma: A randomized controlled trial. J Clin Oncol. 28:5101–5109.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mai EK, Bertsch U, Dürig J, Kunz C, Haenel
M, Blau IW, Munder M, Jauch A, Schurich B, Hielscher T, et al:
Phase III trial of bortezomib, cyclophosphamide and dexamethasone
(VCD) versus bortezomib, doxorubicin and dexamethasone (PAd) in
newly diagnosed myeloma. Leukemia. 29:1721–1729. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aras B and Yerlikaya A: Bortezomib and
etoposide combinations exert synergistic effects on the human
prostate cancer cell line PC-3. Oncol Lett. 11:3179–3184.
2016.PubMed/NCBI
|
|
20
|
Yerlikaya A, Erdoğan E, Okur E, Yerlikaya
Ş and Savran B: A novel combination treatment for breast cancer
cells involving BAPTA-AM and proteasome inhibitor bortezomib. Oncol
Lett. 12:323–330. 2016.PubMed/NCBI
|
|
21
|
Müller F and Fromm MF:
Transporter-mediated drug-drug interactions. Pharmacogenomics.
12:1017–1037. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Weiss J, Theile D, Spalwisz A, Burhenne J,
Riedel KD and Haefeli WE: Influence of sildenafil and tadalafil on
the enzyme- and transporter-inducing effects of bosentan and
ambrisentan in LS180 cells. Biochem Pharmacol. 85:265–273. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gupta A, Mugundu GM, Desai PB, Thummel KE
and Unadkat JD: Intestinal human colon adenocarcinoma cell line
LS180 is an excellent model to study pregnane X receptor; but not
constitutive androstane receptor; mediated CYP3A4 and multidrug
resistance transporter 1 induction: Studies with anti-human
immunodeficiency virus protease inhibitors. Drug Metab Dispos.
36:1172–1180. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Harper PA, Prokipcak RD, Bush LE, Golas CL
and Okey AB: Detection and characterization of the Ah receptor for
2;3;7;8-tetrachlorodibenzo-p-dioxin in the human colon
adenocarcinoma cell line LS180. Arch Biochem Biophys. 290:27–36.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
König J, Cui Y, Nies AT and Keppler D:
Localization and genomic organization of a new hepatocellular
organic anion transporting polypeptide. J Biol Chem.
275:23161–23168. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
König J, Cui Y, Nies AT and Keppler D: A
novel human organic anion transporting polypeptide localized to the
basolateral hepatocyte membrane. Am J Physiol Gastrointest Liver
Physiol. 278:G156–G164. 2000.PubMed/NCBI
|
|
27
|
Pavek P, Merino G, Wagenaar E, Bolscher E,
Novotna M, Jonker JW and Schinkel AH: Human breast cancer
resistance protein: Interactions with steroid drugs, hormones, the
dietary carcinogen 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine,
and transport of cimetidine. J Pharmacol Exp Ther. 312:144–152.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boesch D, Gavériaux C, Jachez B,
Pourtier-Manzanedo A, Bollinger P and Loor F: In vivo circumvention
of P-glycoprotein-mediated multidrug resistance of tumor cells with
SDZ PSC 833. Cancer Res. 51:4226–4233. 1991.PubMed/NCBI
|
|
29
|
Peters T, Lindenmaier H, Haefeli WE and
Weiss J: Interaction of the mitotic kinesin Eg5 inhibitor monastrol
with P-glycoprotein. Naunyn Schmiedebergs Arch Pharmacol.
372:291–299. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vandesompele J, De Preter K, Pattyn F,
Poppe B, Van Roy N, De Paepe A and Speleman F: Accurate
normalization of real-time quantitative RT-PCR data by geometric
averaging of multiple internal control genes. Genome Biol.
3(RESEARCH0034)2002.PubMed/NCBI
|
|
31
|
Fröhlich M, Albermann N, Sauer A,
Walter-Sack I, Haefeli WE and Weiss J: In vitro and ex vivo
evidence for modulation of P-glycoprotein activity by progestins.
Biochem Pharmacol. 68:2409–2416. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weiss J, Rose J, Storch CH,
Ketabi-Kiyanvash N, Sauer A, Haefeli WE and Efferth T: Modulation
of human BCRP (ABCG2) activity by anti-HIV drugs. J Antimicrob
Chemother. 59:238–245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moreau P, Richardson PG, Cavo M, Orlowski
RZ, San Miguel JF, Palumbo A and Harousseau JL: Proteasome
inhibitors in multiple myeloma: 10 years later. Blood. 120:947–959.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Geick A, Eichelbaum M and Burk O: Nuclear
receptor response elements mediate induction of intestinal MDR1 by
rifampin. J Biol Chem. 276:14581–14587. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oerlemans R, Franke NE, Assaraf YG, Cloos
J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova
K, Lemos C, et al: Molecular basis of bortezomib resistance:
Proteasome subunit beta5 (PSMB5) gene mutation and overexpression
of PSMB5 protein. Blood. 112:2489–2499. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ao L, Wu Y, Kim D, Jang ER, Kim K, Lee DM,
Kim KB and Lee W: Development of peptide-based reversing agents for
p-glycoprotein-mediated resistance to carfilzomib. Mol Pharm.
9:2197–2205. 2012.PubMed/NCBI
|
|
37
|
Blagosklonny MV: STI-571 must select for
drug-resistant cells but ‘no cell breathes fire out of its nostrils
like a dragon’. Leukemia. 16:570–572. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
De Souza R, Zahedi P, Badame RM, Allen C
and Piquette-Miller M: Chemotherapy dosing schedule influences drug
resistance development in ovarian cancer. Mol Cancer Ther.
10:1289–1299. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu DK: The contribution of P-glycoprotein
to pharmacokinetic drug-drug interactions. J Clin Pharmacol.
39:1203–1211. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Moreau P, Karamanesht II, Domnikova N,
Kyselyova MY, Vilchevska KV, Doronin VA, Schmidt A, Hulin C, Leleu
X, Esseltine DL, et al: Pharmacokinetic, pharmacodynamic, and
covariate analysis of subcutaneous versus intravenous
administration of bortezomib in patients with relapsed multiple
myeloma. Clin Pharmacokinet. 51:823–829. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Summary of product characteristics for
Ninlaro. https://www.accessdata.fda.gov/drugsatfda_docs/label/…/208462lbl.pdf
|
|
42
|
Summary of product characteristics for
Kyprolis. simpleec.europa.eu/health/documents/…register/…/anx_133351_en.pdf
|
|
43
|
Wang Z, Yang J, Kirk C, Fang Y, Alsina M,
Badros A, Papadopoulos K, Wong A, Woo T, Bomba D, et al: Clinical
pharmacokinetics, metabolism, and drug-drug interaction of
carfilzomib. Drug Metab Dispos. 41:230–237. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Richardson PG, Baz R, Wang M, Jakubowiak
AJ, Laubach JP, Harvey RD, Talpaz M, Berg D, Liu G, Yu J, et al:
Phase 1 study of twice-weekly ixazomib, an oral proteasome
inhibitor, in relapsed/refractory multiple myeloma patients. Blood.
124:1038–1046. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Thompson JL: Carfilzomib: A
second-generation proteasome inhibitor for the treatment of
relapsed and refractory multiple myeloma. Ann Pharmacother.
47:56–62. 2013. View Article : Google Scholar : PubMed/NCBI
|