Introduction
Hepatocellular carcinoma (HCC) is one of the leading
causes of cancer-associated mortality worldwide (1). HCC is often detected at an advanced
stage and exhibits poor prognosis (2). The pathogenesis of HCC is incompletely
understood, and an effective treatment for HCC remains a
requirement. As such, it is of great importance to elucidate the
molecular mechanisms underlying the biological behavior of HCC
cells.
Autophagy is a catabolic process that is induced
under various conditions of stress (3). Despite its diverse roles proposed by
several studies, evidence suggests that autophagy is critical for
cell survival (4), and aberrant
autophagic activity is implicated in the development, progression
and drug resistance of HCC (5–7). Mammalian
target of rapamycin (mTOR) tethers several upstream signals to
exert an inhibitory effect on autophagy (8). Inhibition of mTOR signaling is one of
the potential strategies for HCC therapy (9,10);
however, the role of autophagy in this context is not fully
understood.
MicroRNAs (miRNAs/miRs) have emerged as a class of
key regulators in multiple types of cancers (11,12). mRNAs
canonically match with the 3′untranslated region (UTR) of targeted
mRNAs, which consequently results in gene silencing (13). A broad spectrum of miRNAs have been
implicated in the diagnostic and therapeutic strategies for HCC
(14). It has been reported that
miR-7 is downregulated in various types of cancer and functions as
a tumor suppressor (15,16). Previous studies have identified a
number of oncogenes to be targeted by miR-7 (17–20). In
HCC, it exhibits anticancer activity by causing cell cycle arrest
via targeting critical genes in cell cycle progression (17). However, whether miR-7 regulates other
important cell machinery, such as autophagy, remains unknown. In
particular, miR-7 contributes to the inhibition of tumor growth and
metastasis by modulating the phosphoinositide 3-kinase/protein
kinase B (Akt)/mTOR signaling pathway (21), a major signaling pathway involved in
the regulation of autophagy induction, indicating its potential
involvement in autophagy regulation.
In the present study it was identified that miR-7 is
significantly downregulated in HCC tissues compared with the normal
adjacent tissue. Supplementation of miR-7 in HepG2 cancer cells
inhibited the proliferation of the cells. In addition to the
proliferation inhibition effect of miR-7, autophagy was identified
to be induced by miR-7 at the same time, and inhibition of mTOR was
responsible for the autophagy induction. Most notably, suppression
of autophagy enhanced the proliferation inhibition effect of miR-7.
Therefore, the results of the present study revealed a novel
mechanism by which miR-7 regulates autophagy, and modulation of
autophagy may be a feasible approach to enhance the antitumor
activity of miR-7.
Materials and methods
Clinical samples
A total of 17 HCC tissues (age, 32–65; 9 males and 8
females) and paired adjacent tissues were obtained under the
supervision of the ethics committee of the local hospital (Yantai
City Hospital for Infectious Diseases, Shandong, China) between
March 2015 and June 2015. All the patients did not receive any
chemotherapy prior to surgery. The diagnoses of HCC were
histologically confirmed by three independent pathologists, and the
negative resection margin was confirmed during the surgery. Tissues
were snap-frozen in liquid nitrogen. All patients provided written
informed consent for the study.
Cell culture and 3-methyladenine
(3-MA) treatment
The human HCC HepG2 and Huh-7 cell lines and human
embryonic kidney HEK293-T cell line used in the present study were
obtained from the American Type Culture Collection (Manassas, VA,
USA). The cells were maintained in Dulbecco's modified Eagle's
medium (DMEM; Hyclone; GE Healthcare, Logan, UT, USA) with 10%
fetal bovine serum (FBS; Hyclone; GE Healthcare). Cells were
cultured at 37°C in a humidified atmosphere containing 5%
CO2, and the medium was changed every 3 days. 3-MA was
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and
dissolved in the culture medium, and a 5 mM final concentration was
applied to cells to inhibit autophagy. For autophagy flux assay,
co-treatment with 20 µM chloroquine (C6628; Sigma-Aldrich; Merck
KGaA) was applied.
Transfection
The miRNA mimic for miR-7 (miR10004553-1-5) and its
exact antisense inhibitor (miR200004553-1-5) were obtained from
Guangzhou RiboBio Co., Ltd. (Guangzhou, China). The nucleotides
were transfected using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) at a final concentration
of 50 nM, according to the manufacturer's protocol. miRNAs were
diluted in DMEM and incubated with a proper amount (10 µl for one
6-well plate) of Lipofectamine 2000 at 37°C for 20 min. The
Lipofectamine 2000-miRNA mixture was then added to each well of a
6-well plate; after 12 h of incubation in serum-free conditions,
the medium was refreshed with complete medium containing 10%
FBS.
Cell proliferation assay
To determine the cell proliferation, a Cell Counting
Kit-8 (CCK-8; Wuhan Boster Biological Technology, Ltd., Wuhan,
China) was used. The experiment was conducted according to the
manufacturer's protocol. The absorbance value at 450 nm was
determined using a plate reader as a measure of proliferative
activity.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Following treatment, cells were collected and lysed
with 1 ml TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Chloroform (0.2 ml) was then added to the tube and mixed
vigorously with TRIzol reagent. RNA was precipitated using
isopropyl alcohol from the upper phase and washed using 75%
ethanol. The RNA samples were resolved in nuclease-free
double-distilled water (Promega Corporation, Madison, WI, USA).
miR-7 was then reverse-transcribed using a specific stem-loop
primer, followed by a SYBR Green-based amplification protocol.
Reverse transcription was performed with the GoScript reverse
transcription kit (Promega Corporation), at 25°C for 5 min, and
then 42°C for 1 h. PCR was performed using the GoTaq aPCR Master
Mix (Promega Corporation), which included the DNA polymerase. The
thermocycling conditions were as follows: 95°C for 20 sec, 58°C for
20 sec and 72°C for 20 sec, for 40 cycles. Small nuclear RNA U6 was
used as an internal control. The primer sets for miR-7 and U6 were
all obtained from Guangzhou RiboBio Co., Ltd. (Guangzhou, China;
cat. no., miRQ0000252-1-2). All experiments were repeated three
times. Quantification was performed using the 2−ΔΔCq
method (22).
Western blot analysis
Total protein from the treated cells was obtained by
homogenizing cells with radioimmunoprecipitation assay buffer
(Thermo Fisher Scientific, Inc.). The protein concentration was
quantified by a bicinchoninic acid kit (Thermo Fisher Scientific,
Inc.). Protein (~40 µg) was loaded on 15% SDS-PAGE gels. Following
electrophoresis, the protein was blotted onto a polyvinylidene
fluoride membrane. The membrane was blocked with non-fat milk at
37°C for 1 h, and incubated with primary antibodies [LC3 (cat. no.,
3868; dilution, 1:1,000), mTOR (cat. no., 2983; dilution, 1:1,000),
p62 (cat. no., 8025; dilution, 1:1,000), p-mTOR (cat. no., 5536;
dilution, 1:1,000) and β-actin (cat. no., TA310155; dilution,
1:2,000)] at 4°C for 16 h. Following 5 washes of 5 min with PBS
containing 0.5% Tween-20, membranes were incubated with horseradish
peroxidase (HRP)-conjugated secondary antibodies (goat anti-rabbit
HRP (Santa Cruz Biotechnology, Inc., Dallas, TX, USA; cat. no.,
sc-2005; dilution, 1:5,000) and goat anti-mouse HRP (Santa Cruz
Biotechnology, Inc.; cat. no., sc-2004; dilution, 1:5,000) at room
temperature for 1 h, followed by visualization using an enhanced
chemiluminescence detection kit (GE Healthcare Life Sciences,
Little Chalfont, UK), according to the manufacturer's protocol.
Antibodies against 1A/1B-light chain 3 (LC3), mTOR, phosphorylated
mTOR (p-mTOR) and p62 were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Antibody against β-actin was
purchased from OriGene Technologies, Inc. (Beijing, China).
Luciferase activity assay
A 500 bp fragment containing the putative binding
site of miR-7 was amplified from the cDNA of mTOR, the fragment of
mTOR 3′UTR was then inserted into the 3′UTR of PGL-3 firefly
reporter (Promega Corporation). HEK293-T cells grown in 24-well
plates were transfected with promoter Renilla
luciferase-thymidine kinase (internal control), PGL3-mTOR 3′UTR and
miR-7 or miR-7 + miR-7 inhibitor, using Lipofectamine 2000 (Thermo
Fisher Scientific, Inc.). The luciferase activity was detected
after 36 h using the substrates provided in the Dual Luciferase
Reporter Assay kit (Promega Corporation), according to the
manufacturer's protocol. The firefly luciferase activity was
normalized to Renilla luciferase activity, and the relative
luciferase activity in each group was obtained by normalizing the
value of (firefly luciferase activity)/(Renilla luciferase
activity) to that of the negative control (NC)-transfected
group.
Immunofluorescent staining
Cells (2×104/ml) were seeded onto coverslips and
cultured in 24-well plates. Immediately after transfection, cells
were incubated at 37°C for 48 h. The cells on coverslips were then
fixed with PBS containing 4% paraformaldehyde, and quickly
penetrated with 0.3% Triton X-100 on ice for 5 min. The coverslips
were blocked with 5% bovine serum albumin (Sigma-Aldrich; Merck
KGaA) for 1 h at room temperature. Primary antibody against LC3
(Cell Signaling Technology, Inc.; cat. no., 3868; dilution, 1:200)
was then added to the coverslips. Cells were incubated with primary
antibody in a humid chamber overnight at 4°C, and the coverslips
were then washed with PBS containing Triton X100 (0.5%). Alexa
Fluor® 594-conjugated anti-rabbit secondary antibody
(cat. no., R37117; Thermo Fisher Scientific, Inc.) was added and
incubated at room temperature for 1 h to label the primary antibody
for LC3. Cells were counterstained with DAPI and observed by
fluorescence microscopy in five fields of view (Nikon Corporation,
Tokyo, Japan; magnification, ×400).
Statistical analysis
Data are expressed as the mean ± standard deviation.
A paired Student's t-test was used to determine the differential
expression of miR-7 in tumor tissues and normal tissues. One-way
analysis of variance followed by Dunnett's test was used for
multiple comparisons. P<0.05 (two-tailed) was considered to
indicate a statistically significant difference. Statistical
analysis was performed using SPSS version 19 software (IBM SPSS,
Armonk, NY, USA).
Results
miR-7 expression is downregulated in
HCC
Previous studies have demonstrated that miR-7
functions as a tumor suppressor and is downregulated in various
types of cancer, including lung cancer and gastric cancer (19,20,23). The
present study compared the expression of miR-7 in tumor samples and
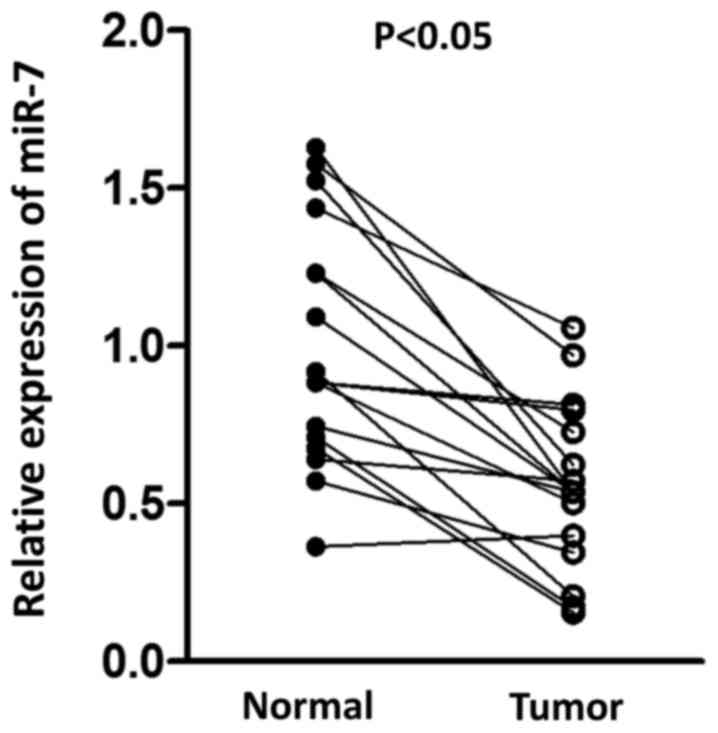
paired normal tissue. As presented in Fig. 1, a significant decrease in miR-7 was
observed in tumor tissue compared with normal tissue, suggesting
the potential antitumor role for miR-7 in HCC.
miR-7 inhibits the proliferation of
HCC cells
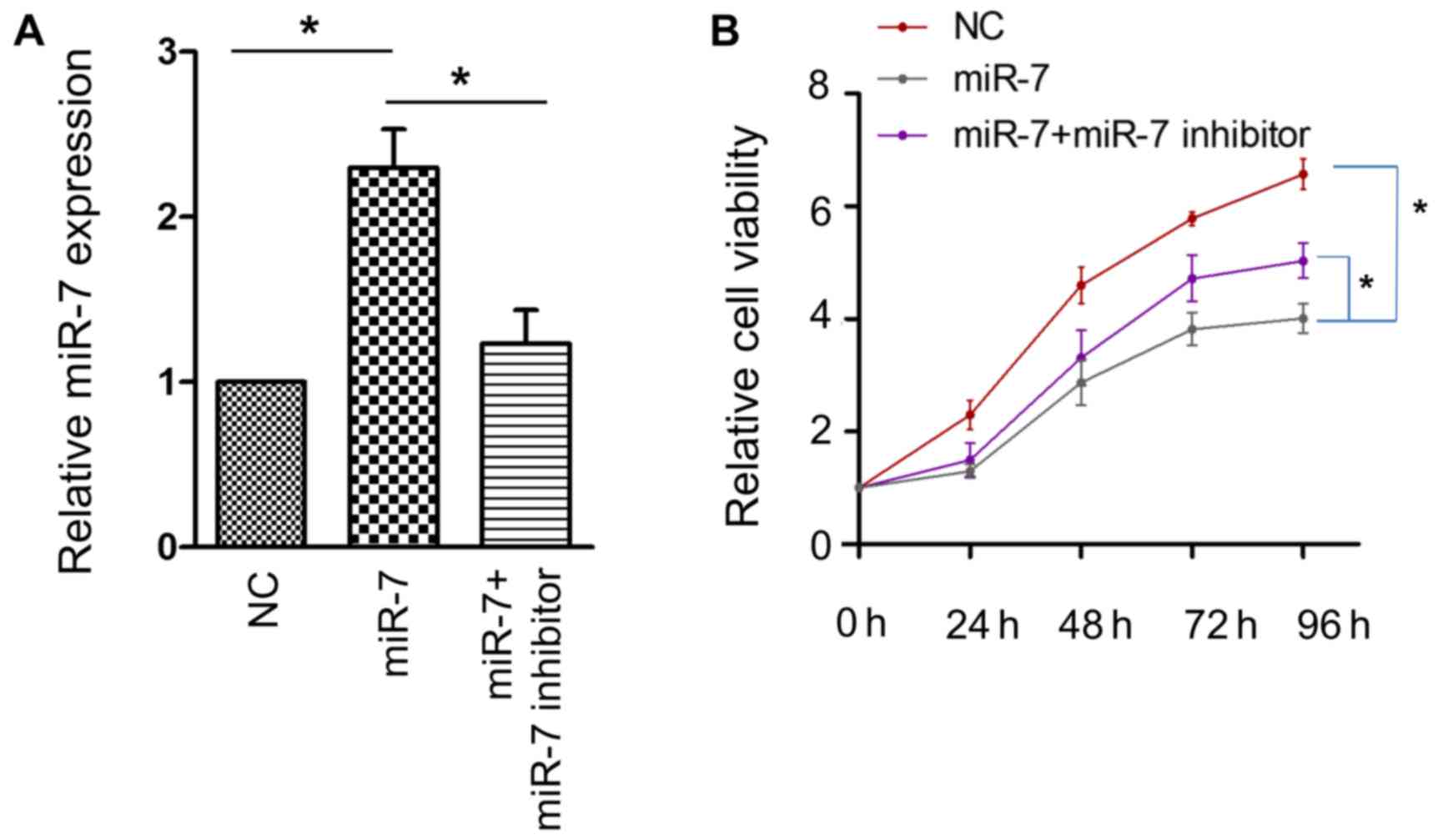
To ascertain the role of miR-7 in HCC, a CCK-8 assay
was employed to examine whether miR-7 affects the proliferative
activity of the HCC HepG2 cell line. The efficacy of transfection
was confirmed using RT-PCR (Fig. 2A).
A significant attenuation of cell proliferative activity was
observed when miR-7 was overexpressed, and this effect was
partially reversed by miR-7 inhibitor (Fig. 2B). These results confirmed that miR-7
serves an antitumor role in HCC.
Overexpression of miR-7 enhances
autophagy
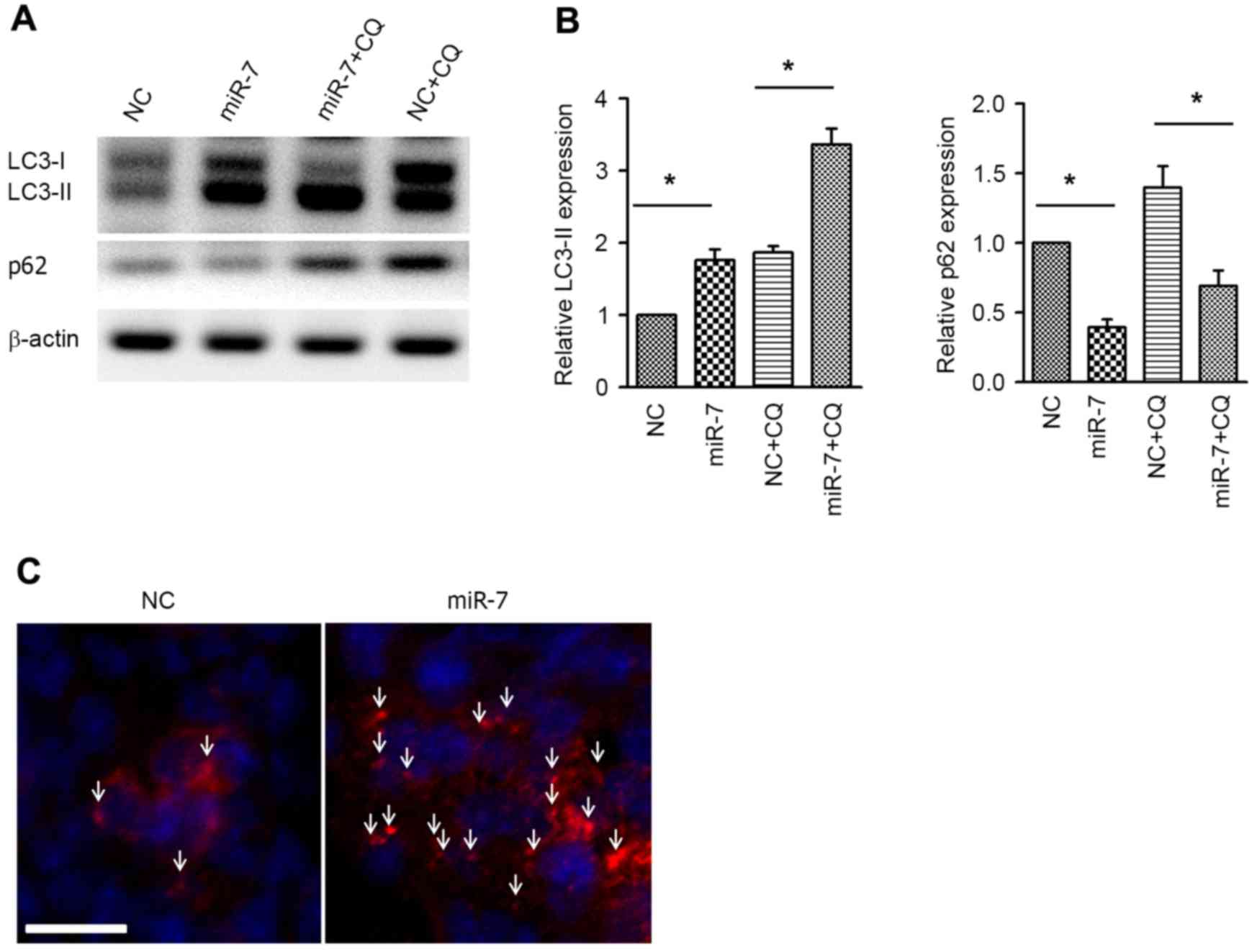
Next, the autophagic activity was examined by
western blot analysis of the autophagy markers LC3 and p62.
Overexpression of miR-7 resulted in increased expression of
LC3-phosphatidylethanolamine conjugate (LC3-II), and, accordingly,
p62, which is a specific substrate of autophagy (24), was downregulated (Fig. 3A and B). Co-administration of
chloroquine (CQ) was used to conduct the autophagy flux assay.
Similar to the aforementioned results, the LC3-II level was also
increased in the miR-7 group in the presence of CQ, indicating the
increased autophagy flux (Fig. 3A and
B). Immunofluorescent staining of LC3 was performed to
visualize the autophagosome, and it was revealed that the
autophagosome was aggregated in miR-7-transfected cells (Fig. 3C). These results suggested that
overexpression enhances autophagic activity in HCC cells.
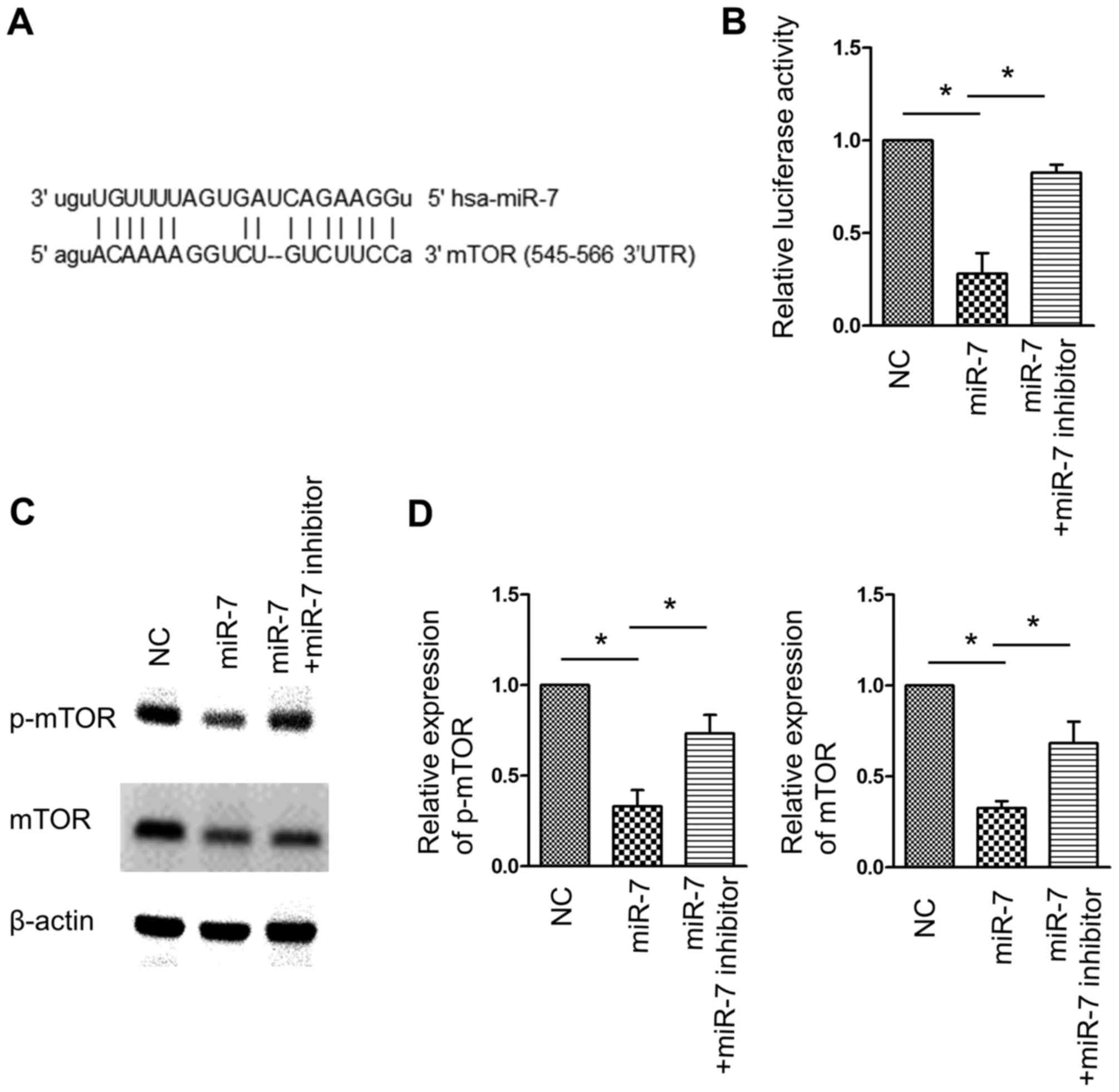
mTOR is a target of miR-7
As mTOR is a canonical negative regulator of
autophagy, it was hypothesized that miR-7-associated autophagy
induction was associated with this pathway. A region in the 3′UTR
of the mRNA of mTOR was identified to comprise the seed sequence
matching miR-7 (Fig. 4A). A
luciferase reporter assay demonstrated that miR-7 overexpression
inhibited the luciferase activity of the reporter carrying mTOR
3′UTR, whereas co-transfection of miR-7 and miR-7 inhibitor
significantly reversed this effect (Fig.
4B). Western blot analysis confirmed the inhibition of mTOR
expression by miR-7, and consequently, p-mTOR, which represents the
kinase activity of mTOR, was also inhibited by miR-7 (Fig. 4C and D). These data indicated that
mTOR is a target of miR-7, and inhibition of mTOR may contribute to
miR-7-activated autophagy.
Inhibition of autophagy enhances the
proliferation inhibition activity of miR-7
To investigate the exact role of autophagy in HCC,
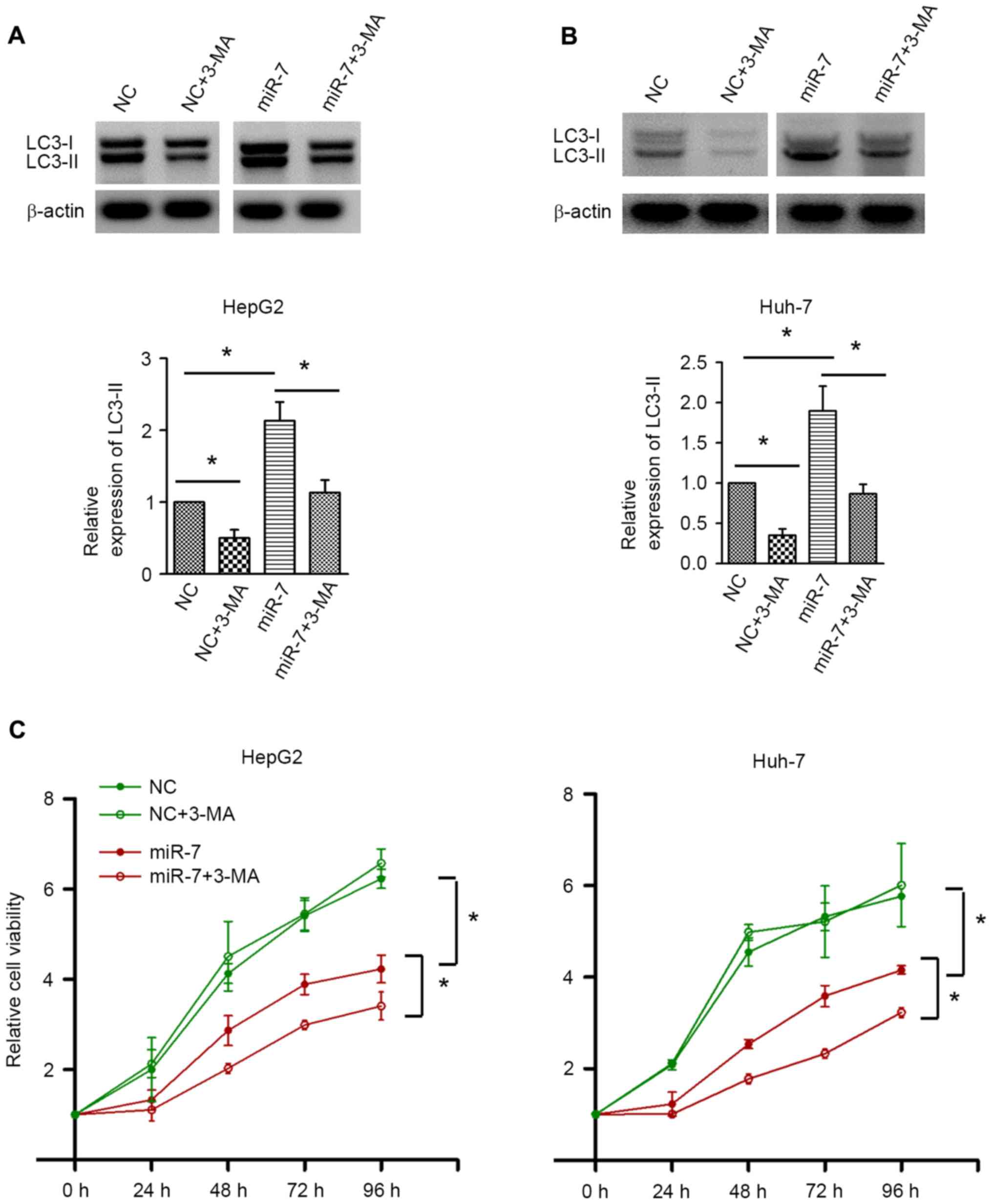
autophagy was inhibited by 3-MA. Western blot analysis confirmed
the autophagic inhibition by 3-MA in HepG2 and Huh-7 cells
(Fig. 5A and B). 3-MA treatment did
not result in significant alterations in cell proliferation in the
NC-transfected group. By contrast, inhibition of autophagy by 3-MA
significantly decreased the cell proliferative activity upon miR-7
overexpression (Fig. 5C). These data
indicated that autophagy induced by miR-7 counteracts the
proliferation inhibition effect of miR-7, and inhibition of
autophagy may enhance the antitumor effect of miR-7.
Discussion
HCC is one of the leading causes of
cancer-associated mortality (11).
Owing to the relatively low sensitivity to chemotherapy drugs, the
prognosis of advanced HCC is poor (25). Understanding the underlying molecular
mechanism will assist in the development of alternative effective
strategies for HCC. Recently, the pharmaceutical value of miRNAs
has drawn wide attention in cancer treatment (26,27). A
spectrum of miRNAs that are involved in cancer cell behavior have
demonstrated therapeutic potential in HCC (16). The present study identified that
miR-7, one of the downregulated miRNAs in HCC (21), functions as a tumor suppressor.
Overexpression of miR-7 was able to significantly suppress the
proliferation of HCC cells, and importantly, it was accompanied by
increased autophagy. Furthermore, it was demonstrated that mTOR, a
negative regulator of autophagy, is a direct target of miR-7.
Pharmacological inhibition of autophagy using 3-MA enhanced the
anti-proliferative activity of miR-7, indicating that the autophagy
inhibitor may serve as the adjuvant drug to enhance the efficacy of
miR-7 in the treatment of HCC.
Previous studies have depicted the crucial role of
miRNAs in hepatocarcinogenesis (14,28).
miRNAs have been incorporated into a number of pathways to regulate
cell proliferation, apoptosis, transdifferentiation and drug
resistance. miR-7 has been demonstrated in a previous study to
directly impede the normal function of several oncogenes in various
types of cancer (16). A number of
critical oncogenes, including epidermal growth factor receptor
(EGFR), B-cell lymphoma-2, Akt and Krüppel-like factor-4 have been
validated as the target of miR-7 (18,20,21),
suggesting that miR-7 functions as the concurrent upstream
regulator of multiple pathways that regulate cell survival. Fang
et al (21) reported that
p70S6 kinase and mTOR are targeted by miR-7, which
potentially provides the molecular basis for miR-7-based therapy
for HCC. In the present study, it was confirmed that miR-7 was
downregulated in HCC, and consistent with the previous study
(21), miR-7 overexpression exerted a
significant proliferation inhibition action. Considering that mTOR
is critical for the nutrient balance and multiple cellular events
of cancer cells (10), the
association between mTOR and miR-7 was then investigated.
Consistent with the results of Fang et al (21), the present study demonstrated that
mTOR was a target of miR-7.
In addition to examining the role of miR-7 in the
proliferative activity of HCC cells, the present study focused on
other critical processes. Autophagy is a lysosomal degradation
process, through which diverse essential substances, including
amino acids, lipids and nucleotides, are recycled (3,5). It has
been reported that autophagy at the physiological level is
beneficial for cell survival, and uncontrolled autophagy may lead
to cell death (4). Currently, studies
on the role of autophagy in HCC are inconsistent. Luo et al
(29) demonstrated that proteasome
26S subunit non-ATPase 10-induced autophagy promoted cell survival
against starvation and chemotherapy. Tian et al (30) reported that autophagy is required for
the prevention of hepatocarcinogenesis and the development of HCC.
These studies suggested that autophagy may be a survival mechanism
of HCC cells. By contrast, other studies also demonstrated that
autophagy inhibits tumorigenesis in HCC (31–33).
Therefore, the exact role of autophagy in the development and
progression of HCC is possibly context- and pathway-dependent.
There are a few studies demonstrating the regulation of autophagy
by miRNAs (34,35). He et al (36) demonstrated that miR-21 inhibits
autophagy via the Akt/mTOR signaling pathway, which is important
for sorafenib resistance of HCC cells. The present study identified
that autophagy was significantly activated upon miR-7
overexpression. Similar to miR-21, a significant suppression of
mTOR by miR-7 was observed, suggesting that the mTOR signaling
pathway may also confer the induction of autophagy by miR-7. More
importantly, inhibition of autophagy by 3-MA sensitized cells to
miR-7-induced proliferation inhibition. These data indicated that
autophagy may function as an adaptive survival mechanism to
overcome the adverse effect of miR-7 on cancer cells. Notably,
Tazawa et al (37)
demonstrated that genetically engineered oncolytic adenovirus kills
lung cancer cells through an autophagic cell death signaling
pathway, which is mediated by the E2F transcription
factor1/miR-7/EGFR axis. In this study, inhibition of autophagy
partially rescued the decrease in cell viability caused by miR-7
overexpression, indicating an opposite role for autophagy in their
system. This discrepancy may be a result of several differences in
the stimulation type and the gene expression profile of the
recipient cells. However, the mechanism for this phenomenon
requires clarification in the future. Nonetheless, the results of
the present study revealed that miR-7-induced autophagy is
cytoprotective in the scenario of HCC.
In conclusion, the results of the present study
revealed a novel mTOR-dependent mechanism by which miR-7 regulates
autophagy. In addition, inhibition of autophagy enhanced the
proliferation inhibition effect of miR-7. The present study
suggested that modulating the levels of autophagy may be a feasible
tool to enhance the therapeutic efficacy of miR-7 in the treatment
of HCC.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Balogh J, Victor D III, Asham EH,
Burroughs SG, Boktour M, Saharia A, Li X, Ghobrial RM and Monsour
HP Jr: Hepatocellular carcinoma: a review. J Hepatocell Carcinoma.
3:41–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Puri P and Chandra A: Autophagy modulation
as a potential therapeutic target for liver diseases. J Clin Exp
Hepatol. 4:51–59. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kumar A, Singh UK and Chaudhary A:
Targeting autophagy to overcome drug resistance in cancer therapy.
Future Med Chem. 7:1535–1542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Du H, Yang W, Chen L, Shi M, Seewoo V,
Wang J, Lin A, Liu Z and Qiu W: Role of autophagy in resistance to
oxaliplatin in hepatocellular carcinoma cells. Oncol Rep.
27:143–150. 2012.PubMed/NCBI
|
|
8
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bupathi M, Kaseb A, Meric-Bernstam F and
Naing A: Hepatocellular carcinoma: Where there is unmet need. Mol
Oncol. 9:1501–1509. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang Z, Jin W, Jin H and Wang X: mTOR in
viral hepatitis and hepatocellular carcinoma: Function and
treatment. Biomed Res Int. 2014:7356722014.PubMed/NCBI
|
|
11
|
Inamura K: Diagnostic and therapeutic
potential of MicroRNAs in lung cancer. Cancers (Basel). 9:pii: E49.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cătană CS, Pichler M, Giannelli G, Mader
RM and Berindan-Neagoe I: Non-coding RNAs, the Trojan horse in
two-way communication between tumor and stroma in colorectal and
hepatocellular carcinoma. Oncotarget. 8:29519–29534.
2017.PubMed/NCBI
|
|
13
|
Faller M and Guo F: MicroRNA biogenesis:
There's more than one way to skin a cat. Biochim Biophys Acta.
1779:663–667. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yao M, Wang L, Yao Y, Gu HB and Yao DF:
Biomarker-based MicroRNA therapeutic strategies for hepatocellular
carcinoma. J Clin Transl Hepatol. 2:253–258. 2014.PubMed/NCBI
|
|
15
|
Horsham JL, Kalinowski FC, Epis MR, Ganda
C, Brown RA and Leedman PJ: Clinical potential of microRNA-7 in
cancer. J Clin Med. 4:1668–1687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kalinowski FC, Brown RA, Ganda C, Giles
KM, Epis MR, Horsham J and Leedman PJ: microRNA-7: A tumor
suppressor miRNA with therapeutic potential. Int J Biochem Cell
Biol. 54:312–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Hu S, Zhang X, Wang L, Zhang X,
Yan B, Zhao J, Yang A and Zhang R: MicroRNA-7 arrests cell cycle in
G1 phase by directly targeting CCNE1 in human hepatocellular
carcinoma cells. Biochem Biophys Res Commun. 443:1078–1084. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiong S, Zheng Y, Jiang P, Liu R, Liu X
and Chu Y: MicroRNA-7 inhibits the growth of human non-small cell
lung cancer A549 cells through targeting BCL-2. Int J Biol Sci.
7:805–814. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luo J, Li H and Zhang C: MicroRNA-7
inhibits the malignant phenotypes of non-small cell lung cancer
in vitro by targeting Pax6. Mol Med Rep. 12:5443–5448.
2015.PubMed/NCBI
|
|
20
|
Chang YL, Zhou PJ, Wei L, Li W, Ji Z, Fang
YX and Gao WQ: MicroRNA-7 inhibits the stemness of prostate cancer
stem-like cells and tumorigenesis by repressing KLF4/PI3K/Akt/p21
pathway. Oncotarget. 6:24017–24031. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fang Y, Xue JL, Shen Q, Chen J and Tian L:
MicroRNA-7 inhibits tumor growth and metastasis by targeting the
phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma.
Hepatology. 55:1852–1862. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xie J, Chen M, Zhou J, Mo MS, Zhu LH, Liu
YP, Gui QJ, Zhang L and Li GQ: miR-7 inhibits the invasion and
metastasis of gastric cancer cells by suppressing epidermal growth
factor receptor expression. Oncol Rep. 31:1715–1722.
2014.PubMed/NCBI
|
|
24
|
Bitto A, Lerner CA, Nacarelli T, Crowe E,
Torres C and Sell C: P62/SQSTM1 at the interface of aging,
autophagy, and disease. Age (Dordr). 36:96262014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu AX: Systemic treatment of
hepatocellular carcinoma: Dawn of a new era? Ann Surg Oncol.
17:1247–1256. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gandellini P, Doldi V and Zaffaroni N:
microRNAs as players and signals in the metastatic cascade:
Implications for the development of novel anti-metastatic
therapies. Semin Cancer Biol. Mar 23–2017.(Epub ahead of Print).
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Denaro N, Merlano MC, Russi EG and Lo
Nigro C: Non coding RNAs in head and neck squamous cell carcinoma
(HNSCC): A clinical perspective. Anticancer Res. 34:6887–6896.
2014.PubMed/NCBI
|
|
28
|
Karagonlar ZF, Korhan P and Atabey N:
Targeting c-Met in cancer by MicroRNAs: Potential therapeutic
applications in hepatocellular carcinoma. Drug Dev Res. 76:357–367.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luo T, Fu J, Xu A, Su B, Ren Y, Li N, Zhu
J, Zhao X, Dai R, Cao J, et al: PSMD10/gankyrin induces autophagy
to promote tumor progression through cytoplasmic interaction with
ATG7 and nuclear transactivation of ATG7 expression. Autophagy.
12:1355–1371. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tian Y, Kuo CF, Sir D, Wang L,
Govindarajan S, Petrovic LM and Ou JH: Autophagy inhibits oxidative
stress and tumor suppressors to exert its dual effect on
hepatocarcinogenesis. Cell Death Differ. 22:1025–1034. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang GM, Jiang QH, Cai C, Qu M and Shen
W: SCD1 negatively regulates autophagy-induced cell death in human
hepatocellular carcinoma through inactivation of the AMPK signaling
pathway. Cancer Lett. 358:180–190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Y, Wu J, Lin B, Li X, Zhang H, Ding
H, Chen X, Lan L and Luo H: Galangin suppresses HepG2 cell
proliferation by activating the TGF-β receptor/Smad pathway.
Toxicology. 326:9–17. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ,
Tsai TF, Lin YJ, Wu CT and Liu HS: Autophagy suppresses
tumorigenesis of hepatitis B virus-associated hepatocellular
carcinoma through degradation of microRNA-224. Hepatology.
59:505–517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gozuacik D, Akkoc Y, Ozturk DG and Kocak
M: Autophagy-Regulating microRNAs and cancer. Front Oncol.
7:652017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu L, Liao JZ, He XX and Li PY: The role
of autophagy in hepatocellular carcinoma: Friend or foe.
Oncotarget. Apr 18–2017.(Epub ahead of print).
|
|
36
|
He C, Dong X, Zhai B, Jiang X, Dong D, Li
B, Jiang H, Xu S and Sun X: MiR-21 mediates sorafenib resistance of
hepatocellular carcinoma cells by inhibiting autophagy via the
PTEN/Akt pathway. Oncotarget. 6:28867–28881. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tazawa H, Yano S, Yoshida R, Yamasaki Y,
Sasaki T, Hashimoto Y, Kuroda S, Ouchi M, Onishi T, Uno F, et al:
Genetically engineered oncolytic adenovirus induces autophagic cell
death through an E2F1-microRNA-7-epidermal growth factor receptor
axis. Int J Cancer. 131:2939–2950. 2012. View Article : Google Scholar : PubMed/NCBI
|