Introduction
Glioma commonly occurs in the brain or spine, and is
a particularly lethal solid tumor that arises from support cells in
the central nervous system. Glioma can be classified into the
following types: Astrocytic tumors, oligodendrogliomas and
oligoastrocytomas (1). In addition,
patients with glioma often suffer from fatigue, motor difficulties,
drowsiness, communication difficulties, headaches and visual
problems (2). A previous study
reported that the median survival time of patients with the most
malignant form of glioma is only 14.6 months, and the 5-year
survival rate is only 9.8% (3). The
high rates of morbidity and mortality associated with glioma are
due to its chemoradiation resistance, and high proliferative and
invasive capabilities (4).
In addition to childhood leukemia, glioma is the
most common type of tumor in children, and accounts for 80% of
malignant brain tumors (5). There is
a peak incidence of glioma between the ages of 0 and 8 years
(6). Patients with glioma that have
survived surgery are more affected by their tumor and its resection
than previously appreciated (7). In
addition, in patients with childhood brain tumors treated with
radiotherapy, memory deficits reflect general cognitive dysfunction
(8). At present, the standard
approach for treating glioma in children is chemotherapy; however,
the safety and effectiveness of chemotherapy in children remains a
problem.
Understanding the regulation of the pathological
activities of glioma is important for its eradication. Research
regarding glioma pathogenesis is expected to provide novel methods
of adjuvant therapy for the treatment of glioma (9). With scientific improvements, the concept
of epigenetics offers a potential method for cancer therapy.
Epigenetics refers to stable alteration in gene expression with no
underlying modifications in genetic sequence (10); DNA methylation is central to the
aberrant epigentics associated with cancer (11). DNA methylation is a biochemical
process where a methyl group is added to the CpG islands of DNA
nucleotides by DNA methyltransferases in mammals. DNA methylation
of CpG dinucleotides is considered a distinct inhibitory pathway
that may mediate stable silencing (12). CpG islands are located on promoter
regions, and the sequences that surround promoter regions. It has
previously been reported that methylation at the CpG islands may
induce tumorigenesis, since the abnormal expression of genes is
involved in tumor development (13).
Costello et al (14) reported
that transcriptional repression of a tumor suppressor gene,
cyclin-dependent kinase inhibitor 2A, was mediated by aberrant
methylation of the CpG island in glioma. A previous study also
demonstrated that promoter methylation of DNA repair genes is
associated with the tumorigenesis and development of glioma
(15). However, despite extensive
research, the exact mechanisms underlying how DNA methylation
induces glioma remain unclear.
The present study compared DNA methylation and
differentially expressed genes (DEGs) between glioma and normal
tissues using Gene Expression Omnibus (GEO) data. Subsequently, the
important genes and pathways in glioma were screened by enrichment
analysis of transcriptional regulatory networks and sub-pathways.
The present study aimed to determine the possible molecular
mechanisms and the aberrant promoter methylation of
tumor-associated genes in glioma, via identification of key genes
and pathways. The results may provide a theoretical basis and
identify potential molecular biological therapies for the treatment
of glioma in children.
Materials and methods
Data sources
GSE50021 DNA microarray data (16) for gene expression profiling and
GSE50022 dual-channel DNA methylation data (16) were downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo/). GSE50021
contains 45 samples, including 35 samples from patients with glioma
(average age 1.008±1.190491 years) and 10 autopsy samples of normal
human brain tissue. Data from the chip (Illumina HumanHT-12 WG-DASL
v4.0 Expression Beadchip; Illumina, San Diego, CA, USA) were
annotated based on the GPL13938 platform. GSE50022 contains 28
samples from patients with glioma (average age, 0.9433333±0.781596
years). Data from the chip (Illumina HumanMethylation450 BeadChip
[UBC enhanced annotation v1.0]; Illumina) were annotated based on
the GPL16304 platform.
Preprocessing of gene expression
profiles
The expression values for genes with various capture
probes were used to determine a mean value for each gene. The
K-nearest neighbour (KNN) impute missing values module was used to
impute missing values by assigning gene expression values based on
the nearest neighbors of the gene (k=10). All expression
values were normalized using quantile normalization, as implemented
in the normalize.quantiles function of the preprocessCore R package
(17). Expression values were
displayed using a box plot.
Preprocessing of DNA methylation
data
For probes with missing values in the original
methylation index matrix the KNN impute missing values module was
used to impute the missing values by assigning methylation gene
expression values based on the nearest neighbors of the gene
(k=10).
Screening of differentially expressed
genes (DEGs)
The probe identifiers were converted into gene
names. The DEG analysis was conducted using limma package
(http://www.bioconductor.org).
|log2FC|>1 and P<0.05 were set as the cut-offs to screen for
DEGs. DEGs were initially identified following an analysis using
Student's t-test. The identified DEGs were considered
glioma-related genes in children.
Screening of key methylated sites
According to the annotation information, probes were
screened to determine which methylated sites were located near
DEGs. Subsequently, the preliminarily reserved methylated sites
were screened, and ≥80% of samples with a methylation index of ≥0.8
were reserved. Due to the lack of clear definition of the promoter
regions of genes, 50 kb on either side of the transcription start
site was considered the promoter region. Finally, the methylated
sites were re-screened, and methylated sites were reserved that
were located 50 kb on either side of the transcription start site.
After screening three times, the remaining methylated sites were
considered key methylated sites, which were located in the promoter
region of genes and have a high methylation index, thus potentially
influencing the expression of downstream genes.
Screening of significant transcription
factors
Using the transcription factor binding sites (TFBS)
prediction tool from University of California, Santa Cruz (UCSC;
http://genome.ucsc.edu) the methylated sites were
screened in TFBS. A transcription factor regulatory network was
generated using Cytoscape (http://cytoscape.org/), which is an open source
desktop tool for integrating, visualizing and analyzing data in the
context of biological networks. Enrichment analysis of potential
downregulated genes influenced by methylation was performed using
the Database for Annotation, Visualization and Integrated Discovery
(http://david.abcc.ncifcrf.gov/).
Analysis of risk sub-pathways
A hypergeometric test for risk sub-pathways was
performed by application of the R package iSubpathwayMiner
(http://cran.r-project.org/web/packages/iSubpathwayMiner/).
All genes that were mapped to sub-pathways with a significant
enrichment were reported. These pathways were used to identify
glioma-related pathways.
Results
Preprocessing of gene expression
profiles and DNA methylation data
To explore glioma-related genes in children, 19,252
genes were identified from 45 samples following preprocessing of
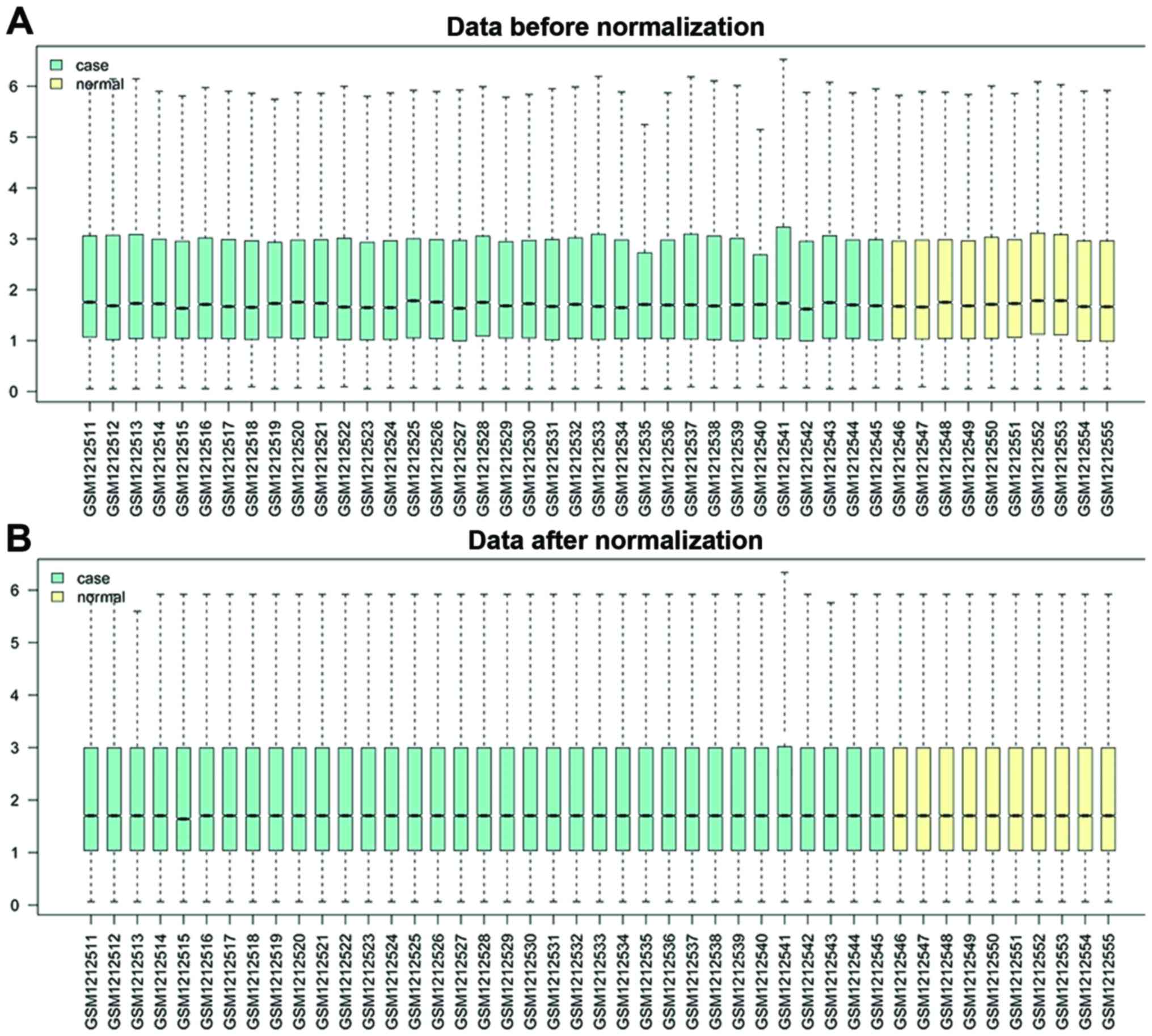
gene expression profiles. As shown in Fig. 1 expression boxplots were generated
before and after normalization. The original expression datasets
from all conditions were processed into expression estimates using
the median polish algorithm. As shown in a box plot (Fig. 1B), the median of different samples was
almost on the same line after normalization, which shows an
excellent degree of standardization.
Data located in the X/Y and mitochondrial
chromosomes were removed. Subsequently, a methylation index array
identified 371,920 sites from the 28 samples, following imputation
of missing values.
Screening of DEGs
DEGs were analyzed using limma package. Using the
selective conditions (|log2FC|>1 and P<0.05), 58 DEGs were
identified, all of which were downregulated.
Identification of DNA methylation in
the promoter regions of downregulated genes
TFBS methylation has a strong impact on the
regulatory role of transcription factors and the normal expression
of downstream genes, thus affecting body functioning. DNA
methylation in the promoter region is an important aspect of DNA
methylation in cancer (18,19).
The 58 downregulated DEGs identified in the present
study were overlapped with DNA methylation data; 1,123 methylation
sites were revealed to be associated with the 58 downregulated
genes. The sites were distributed evenly over the 22 autosomes. The
methylation index of the 1,123 methylation sites was analyzed, and
≥80% of samples with a methylation index of ≥0.8 were reserved.
This resulted in the reservation of 168 methylation sites. To
further analyze the 168 methylation sites, methylated sites located
50 kb on either side of the transcription start sites were
reserved. This resulted in the reservation of 119 methylation sites
located on the transcription start sites of 43 genes. These 43
genes are considered potential glioma susceptibility genes in
children. Finally, 17 methylation sites located on 33 TFBS were
identifed, which may regulate 17 of the downregulated target genes.
These results were determined using the TFBS prediction tool from
UCSC (Table I).
 | Table I.Key methylation sites. |
Table I.
Key methylation sites.
| ID | Chromosome | Mapinfo | tfbs_start | tfbs_end | Transcription
factor |
Distance_closest_TSS |
Closest_TSS_gene_name |
|---|
| cg06191091 | chr17 | 30583855 | 30583848 | 30583862 | USF | −9339 | RHBDL3 |
| cg02629157 | chr9 | 138670609 | 138670546 | 138670568 | TCF11 | 25013 | KCNT1 |
| cg11709150 | chr1 | 2440438 | 2440431 | 2440444 | TCF11 | 10256 | PLCH2 |
| cg04585209 | chr11 | 6292311 | 6292257 | 6292272 | TAXCREB | 306 | CCKBR |
| cg10707626 | chr3 | 51747098 | 51747027 | 51747051 | STAT5A | 6018 | GRM2 |
| cg06191091 | chr17 | 30583855 | 30583849 | 30583860 | SREBP1 | −9339 | RHBDL3 |
| cg12603173 | chr11 | 64508421 | 64508409 | 64508423 | RREB1 | −66 | RASGRP2 |
| cg11025960 | chr3 | 51749188 | 51749177 | 51749195 | RFX1 | 8108 | GRM2 |
| cg10692302 | chr3 | 51747227 | 51747224 | 51747245 | PPARG | 6147 | GRM2 |
| cg02629157 | chr9 | 138670609 | 138670558 | 138670569 | POU6F1 | 25013 | KCNT1 |
| cg11014582 | chr6 | 76333727 | 76333675 | 76333696 | PAX6 | −852 | LMO7 |
| cg04341461 | chr1 | 2410006 | 2409978 | 2410006 | PAX5 | −1616 | PLCH2 |
| cg10692302 | chr3 | 51747227 | 51747222 | 51747252 | PAX4 | 6147 | GRM2 |
| cg04625615 | chr15 | 41788368 | 41788310 | 41788330 | P53 | 2313 | ITPKA |
| cg07200386 | chr8 | 22079169 | 22079113 | 22079135 | OLF1 | 10682 | PHYHIP |
| cg11014582 | chr6 | 76333727 | 76333676 | 76333683 | NKX25 | −852 | LMO7 |
| cg09864712 | chr16 | 726786 | 726720 | 726749 | MYOGNF1 | 712 | RHBDL1 |
| cg06191091 | chr17 | 30583855 | 30583848 | 30583862 | MYCMAX | −9339 | RHBDL3 |
| cg00810908 | chr3 | 13612319 | 13612306 | 13612320 | MEIS1AHOXA9 | 2080 | FBLN2 |
| cg11025960 | chr3 | 51749188 | 51749181 | 51749190 | LMO2COM | 8108 | GRM2 |
| cg03358506 | chr8 | 22058702 | 22058688 | 22058703 | ISRE | 31149 | PHYHIP |
| cg07776629 | chr16 | 57989122 | 57989116 | 57989129 | IRF2 | 15898 | CNGB1 |
| cg07776629 | chr16 | 57989122 | 57989116 | 57989129 | IRF1 | 15898 | CNGB1 |
| cg10692302 | chr3 | 51747227 | 51747225 | 51747244 | HNF4 | 6147 | GRM2 |
| cg06632557 | chr11 | 61313548 | 61313495 | 61313505 | HMX1 | −3678 | SYT7 |
| cg00155846 | chr9 | 138011566 | 138011506 | 138011522 | HAND1E47 | 14081 | OLFM1 |
| cg11025960 | chr3 | 51749188 | 51749181 | 51749190 | GATA3 | 8108 | GRM2 |
| cg11025960 | chr3 | 51749188 | 51749179 | 51749193 | GATA1 | 8108 | GRM2 |
| cg04625615 | chr15 | 41788368 | 41788310 | 41788321 | GATA3 | 2313 | ITPKA |
| cg05392169 | chr9 | 138011814 | 138011802 | 138011816 | FOXO3 | 14329 | OLFM1 |
Analysis of transcriptional regulatory
networks
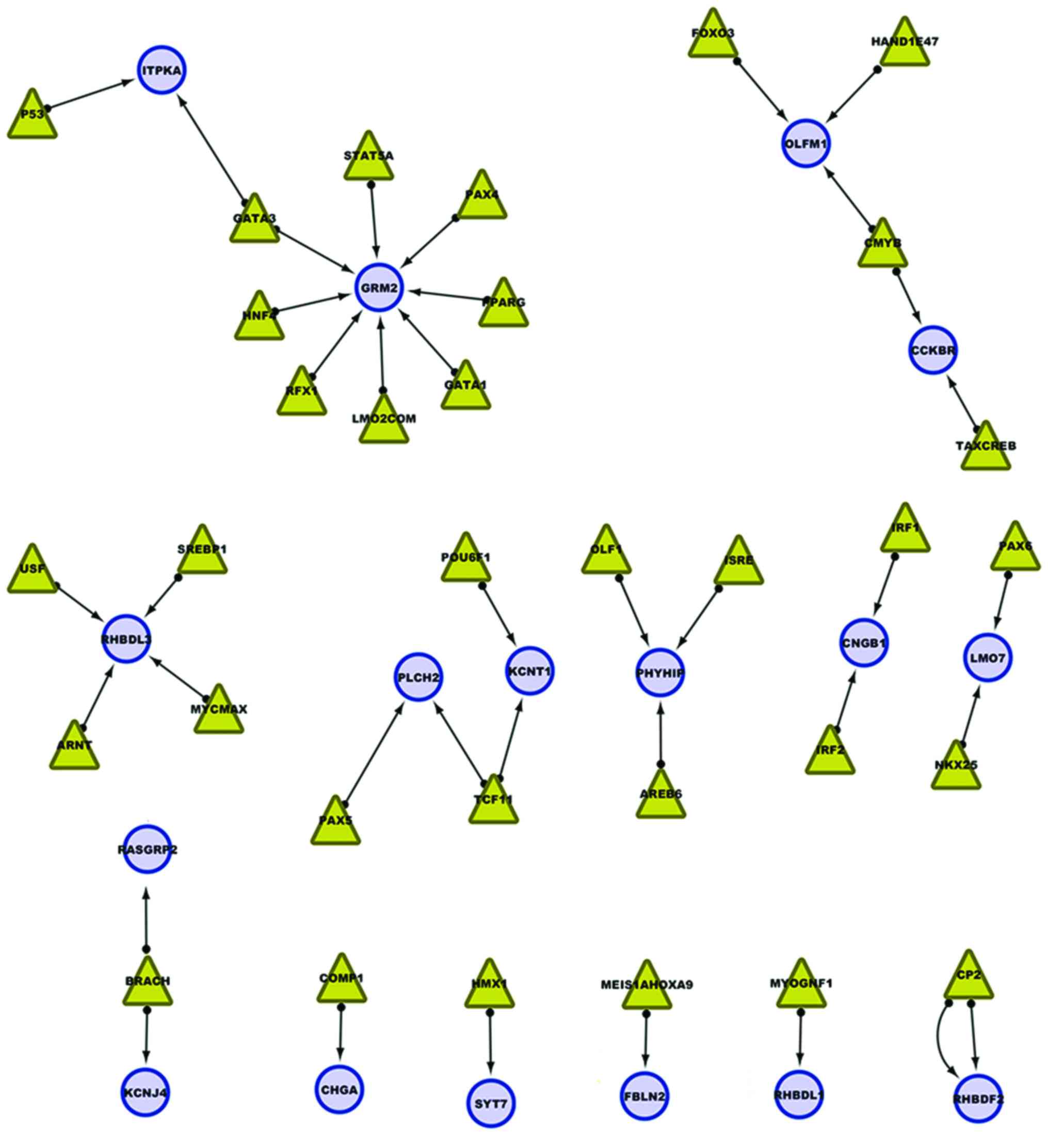
To construct a transcription factor regulatory
network, predictive models of transcriptional regulatory networks
were generated using Cytoscape. A transcriptional regulatory
network including 33 transcription factors and target genes is
presented in Fig. 2. The eight
methylation sites in the TFBS of glutamate metabotropic receptor 2
(GRM2) potentially influenced the binding of eight transcription
factors to TFBS. It may therefore be hypothesized that the
downregulation of GRM2 is caused by methylation of these TFBS. In
addition, methylation sites in the TFBS of rhomboid, veinlet-like 3
(Drosophila) (RHBDL3) resulted in the failure of four
transcription factors binding with TFBS, thus affecting
transcriptional regulation. Furthermore, the transcription factor
CP2 regulates rhomboid 5 homolog 2 (Drosophila) (RHBDF2)
expression; in the present study, two methylation sites were
detected in TFBS of CP2, which may influence transcriptional
regulation of RHBDF2.
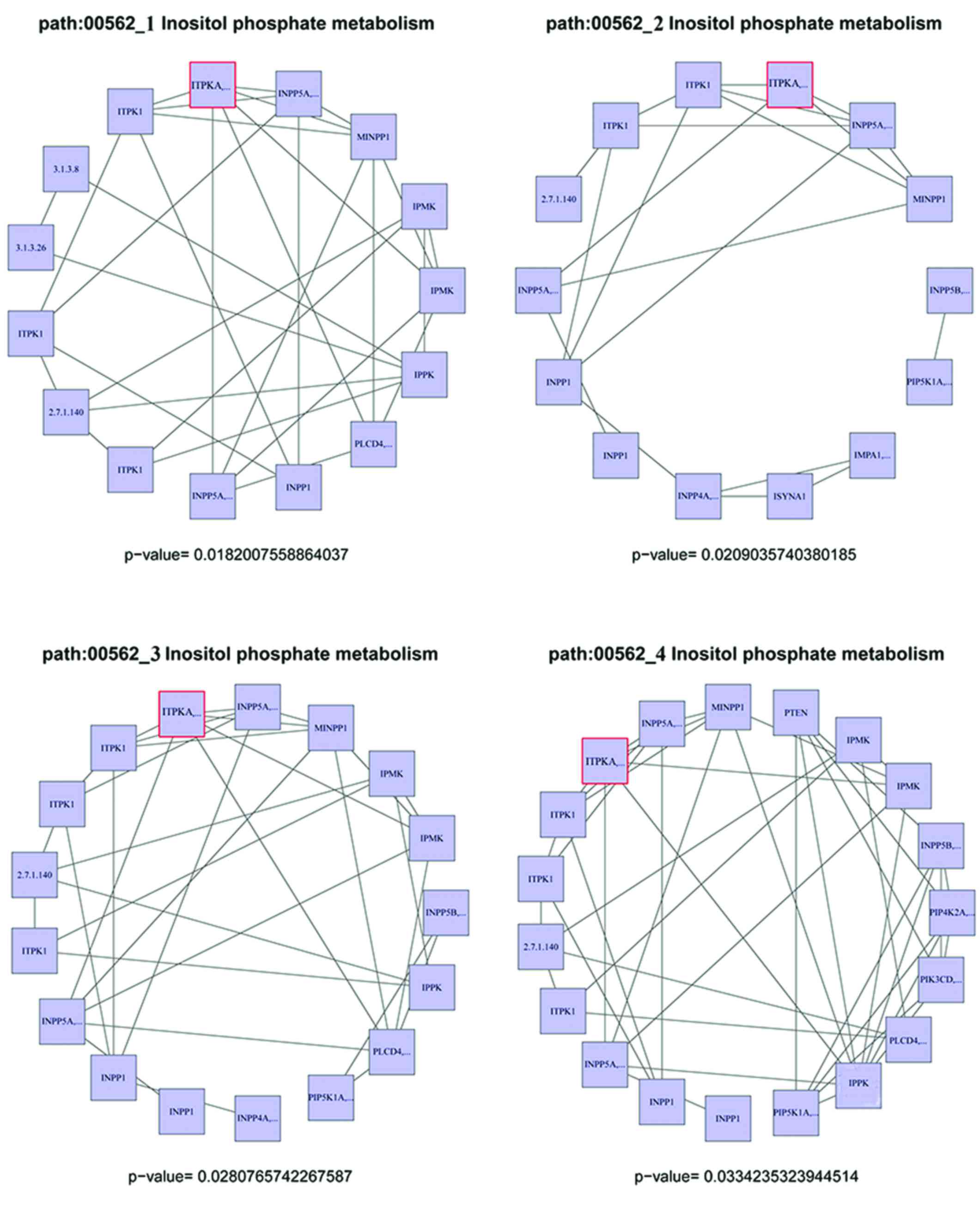
Functional analysis of key genes
To generate the downregulated target gene-related
risk sub-pathways, a hypergeometric test was performed using R
package iSubpathwayMiner. Four sub-pathways were generated, which
were associated with inositol phosphate metabolism (hsa00562). As
shown in Fig. 3 the
inositol-trisphosphate 3-kinase protein (EC 2.7.1.127), which is
coded by the ITPKA gene (marked with a red rim), appeared in all of
the risk sub-pathways. In conclusion, ITPKA was considered to be
the gene most associated with the risk of glioma in children.
Discussion
Bioinformatics is an interdisciplinary field that
develops methods and software tools for understanding biological
data. The identification of candidate genes and single nucleotide
polymorphisms are common uses of bioinformatics to understand the
genetic basis of disease (20). The
present study identified 17 methylation sites located on 33 TFBS,
which may regulate the 17 downregulated target genes. GRM2 was
identified as one of the 17 downregulated target genes.
Furthermore, ITPKA was revealed to be the gene most associated with
glioma risk in children. The protein coded by the ITPKA gene
appeared in all risk sub-pathways, thus suggesting that ITPKA is
the gene most associated with glioma risk, and inositol phosphate
metabolism is the key pathway associated with glioma in
children.
Glioma is the most common type of primary brain
tumor, which is associated with a poor prognosis for patients due
to its aggressive growth behavior and highly invasive nature
(21). Abnormal metabolic networks
are closely associated with the pathogenesis of glioma. A metabolic
pathway is a series of chemical reactions, which are catalyzed by
enzymes, where the product of one enzyme acts as a substrate for
the next. Abnormal gene expression of specific enzymes may affect
other enzymatic genes in the network. Therefore, identification of
disease-related genes in the metabolic pathway is of great
importance.
The correct targeting, localization, regulation and
signaling of metabotropic glutamate receptor 2 (mGluR2), which is
coded by the GRM2 gene, represents a major mechanism underlying the
complex function of neuronal networks (22). Dysfunction of mGluR and associated
proteins is associated with neurodegenerative and neuropsychiatric
disorders. Several types of glioma have been reported to release
high levels of glutamate, which promotes malignancy. A previous
study reported that activation of mGluR2 supports the growth of
human glioma cells in culture, and that antagonists of mGluR2
should be assessed for their ability to reduce tumour growth in
vivo (23). Arcella et al
(24) demonstrated that mGlu2/3
receptor antagonists are of potential use in the experimental
treatment of malignant gliomas. Furthermore, ectopic expression of
ITPKA in lung cancer cells has been shown to increase their
metastatic potential, since the protein exhibits two
actin-regulating activities (25).
The ITPKA gene has also been reported to be hypermethylated in
human patients with normal karyotype acute myeloid leukemia
(26). Furthermore, the remaining 15
downregulated genes identified in the present study may be
beneficial for the generation of a target therapy for the treatment
of patients with glioma.
In conclusion, the present study proposed a novel
method for extracting DEGs from gene expression datasets combined
with DNA methylation data, and applied it to the occurrence of
glioma in children. Results of the functional analysis revealed
that ITPKA was considered the gene most associated with the risk of
glioma, and inositol phosphate metabolism may serve an important
role in glioma in children. The identification of specific genes
may improve understanding regarding the pathogenesis and possible
therapeutic targets of glioma in children.
References
|
1
|
Brat DJ, Scheithauer BW, Fuller GN and
Tihan T: Newly codified glial neoplasms of the 2007 WHO
classification of tumours of the central nervous system:
Angiocentric glioma, pilomyxoid astrocytoma and pituicytoma. Brain
Pathol. 17:319–324. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Osoba D, Brada M, Prados MD and Yung WK:
Effect of disease burden on health-related quality of life in
patients with malignant gliomas. Neuro Oncol. 2:221–228. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al: Effects of radiotherapy with concomitant and
adjuvant temozolomide versus radiotherapy alone on survival in
glioblastoma in a randomised phase III study: 5-year analysis of
the EORTC-NCIC trial. Lancet Oncol. 10:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Van Meir EG, Hadjipanayis CG, Norden AD,
Shu HK, Wen PY and Olson JJ: Exciting new advances in
neuro-oncology: The avenue to a cure for malignant glioma. CA
Cancer J Clin. 60:166–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Paul SP, Perrow R and Webster MA: Brain
tumours in children: Reducing time to diagnosis. Emerg Nurse.
22:32–36; quiz 37. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pollack IF: Brain tumors in children. N
Engl J Med. 331:1500–1507. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Turner CD, Chordas CA, Liptak CC,
Rey-Casserly C, Delaney BL, Ullrich NJ, Goumnerova LC, Scott RM,
Begley HC, Fletcher WJ, et al: Medical, psychological, cognitive
and educational late-effects in pediatric low-grade glioma
survivors treated with surgery only. Pediatr Blood Cancer.
53:417–423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reimers TS, Mortensen EL and Schmiegelow
K: Memory deficits in long-term survivors of childhood brain tumors
may primarily reflect general cognitive dysfunctions. Pediatr Blood
Cancer. 48:205–212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu Ct and Morris JR: Genes, genetics, and
epigenetics: A correspondence. Science. 293:1103–1105. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Portela A and Esteller M: Epigenetic
modifications and human disease. Nat Biotechnol. 28:1057–1068.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Goll MG and Bestor TH: Eukaryotic cytosine
methyltransferases. Annu Rev Biochem. 74:481–514. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Herman JG, Latif F, Weng Y, Lerman MI,
Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM, et al:
Silencing of the VHL tumor-suppressor gene by DNA methylation in
renal carcinoma. Proc Natl Acad Sci USA. 91:9700–9704. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Costello JF, Berger MS, Huang HS and
Cavenee WK: Silencing of p16/CDKN2 expression in human gliomas by
methylation and chromatin condensation. Cancer Res. 56:2405–2410.
1996.PubMed/NCBI
|
|
15
|
Skiriute D, Vaitkiene P, Saferis V,
Asmoniene V, Skauminas K, Deltuva VP and Tamasauskas A: MGMT,
GATA6, CD81, DR4, and CASP8 gene promoter methylation in
glioblastoma. BMC Cancer. 12:2182012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Buczkowicz P, Hoeman C, Rakopoulos P,
Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E,
Bartels U, et al: Genomic analysis of diffuse intrinsic pontine
gliomas identifies three molecular subgroups and recurrent
activating ACVR1 mutations. Nat Genet. 46:451–456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bolstad BM: preprocessCore: A collection
of pre-processing functions. R Package. Version 1.22.0. 2014.
|
|
18
|
Tekpli X, Landvik NE, Anmarkud KH, Skaug
V, Haugen A and Zienolddiny S: DNA methylation at promoter regions
of interleukin 1B, interleukin 6, and interleukin 8 in non-small
cell lung cancer. Cancer Immunol Immunother. 62:337–345. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Spiegl-Kreinecker S, Pirker C, Filipits M,
Lötsch D, Buchroithner J, Pichler J, Silye R, Weis S, Micksche M,
Fischer J and Berger W: O6-Methylguanine DNA methyltransferase
protein expression in tumor cells predicts outcome of temozolomide
therapy in glioblastoma patients. Neuro Oncol. 12:28–36. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Véron A, Blein S and Cox DG: Genome-wide
association studies and the clinic: A focus on breast cancer.
Biomark Med. 8:287–296. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kleihues P and Sobin LH: World Health
Organization classification of tumors. Cancer. 88:28872000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Enz R: Metabotropic glutamate receptors
and interacting proteins: Evolving drug targets. Curr Drug Targets.
13:145–156. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
D'Onofrio M, Arcella A, Bruno V, Ngomba
RT, Battaglia G, Lombari V, Ragona G, Calogero A and Nicoletti F:
Pharmacological blockade of mGlu2/3 metabotropic glutamate
receptors reduces cell proliferation in cultured human glioma
cells. J Neurochem. 84:1288–1295. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arcella A, Carpinelli G, Battaglia G,
D'Onofrio M, Santoro F, Ngomba RT, Bruno V, Casolini P, Giangaspero
F and Nicoletti F: Pharmacological blockade of group II
metabotropic glutamate receptors reduces the growth of glioma cells
in vivo. Neuro Oncol. 7:236–245. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schröder D, Rehbach C, Seyffarth C,
Neuenschwander M, Kries JV and Windhorst S: Identification of a new
membrane-permeable inhibitor against
inositol-1,4,5-trisphosphate-3-kinase A. Biochem Biophys Res
Commun. 439:228–234. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sonnet M, Claus R, Becker N, Zucknick M,
Petersen J, Lipka DB, Oakes CC, Andrulis M, Lier A, Milsom MD, et
al: Early aberrant DNA methylation events in a mouse model of acute
myeloid leukemia. Genome Med. 6:342014. View Article : Google Scholar : PubMed/NCBI
|