Introduction
Most non-small cell lung cancers (NSCLC) are already
at an advanced stage when diagnosed and are therefore past the
optimal timing for surgical resection (1). Seeking effective treatments for these
types of cases is particularly important. Docetaxel is a
semi-synthetic analogue of paclitaxel, which is widely used as a
therapeutic agent in advanced NSCLC (2,3). Docetaxel
shows survival benefit when it is used as single agent or in
combination with other drugs including chemotherapeutics or
vascular endothelial growth factor inhibitors (4–7). However,
patients with NSCLC generally develop resistance to docetaxel, and
the underlying mechanisms of acquired resistance to docetaxel are
not fully understood (8).
The expression of the multidrug resistance (MDR)
phenotype is a main mechanism involved in resistance to taxanes
(9–12). ABCB1 (P-glycoprotein) is an
ATP-binding cassette (ABC) drug pump (13), and is currently the most extensively
studied MDR-related transporter protein (14,15),
mediating the ATP-dependent efflux of a wide range of hydrophobic
drugs such as taxanes (16–18). Several therapeutic agents targeting
ABCB1 are available (18). Inhibitors
of ABCB1, which reverse the ABCB1 efflux pump, have been studied
for more than twenty years, and third-generation drugs, such as
elacridar (GF120918), have been developed (9,19). They
specifically and potently inhibit ABCB1 and generally do not alter
the plasma pharmacokinetics of simultaneously administered
antitumor agents, and therefore show potential for combined
application with anticancer drugs to combat chemotherapeutic
resistance (19–21).
Cellular phenotypes in addition to ABC molecules
have been shown to be associated with multidrug resistance
(22). Epithelial-mesenchymal
transition (EMT) phenotype, loss of epithelial characteristics
(E-cadherin), and acquisition of mesenchymal properties (vimentin,
fibronectin, or N-cadherin), have been shown to play a crucial role
in drug resistance of cancer cells against conventional
therapeutics including taxane, vincristine, oxaliplatin, as well as
epidermal growth factor receptor (EGFR)-targeted agents (23–25).
Cancer stem cell (CSC) features also contribute to this drug
resistance (26,27). CSCs drive both continued expansion of
malignant cells and resistance to chemotherapy (28–30).
However, CSCs in lung cancer remain a subject of ongoing research,
and specific markers have not yet been identified. Jiang et
al (31) reported that ALDH1 is a
lung tumor stem cell-associated marker and that ALDH1-positive
cells are highly resistant to chemotherapeutic drugs.
In the present study, we investigated the mechanisms
of docetaxel resistance in NSCLC, and examined the effects of the
ABCB1 inhibitor elacridar in combination with docetaxel in
docetaxel-resistant lung cancer cells.
Materials and methods
Cell lines and reagents
Three NSCLC cell lines (H1299, HCC827, and HCC4006)
were used in this study, including one wild-type EGFR cell line
(H1299) and two EGFR-mutant cell lines HCC827 (exon19del E746-A750)
and HCC4006 (exon19del L747-E749). These cell lines were obtained
from Adi F. Gazdar, MD (Hamon Center for Therapeutic Oncology
Research and Department of Pathology, University of Texas
Southwestern Medical Center at Dallas, Dallas, TX). These cell
lines were proven to have individual genetic origins by using the
Powerplex 1.2 system (Promega, Madison, WI, USA) at the University
of Texas Southwestern Medical Center at Dallas. All cell lines were
cultured in RPMI-1640 media supplemented with 10% fetal bovine
serum. They were grown in a humidified incubator with 5%
CO2 at 37°C. Docetaxel-resistant sublines (H1299-DR,
HCC4006-DR, and HCC827-DR) were established by their parental cells
which were cultured with stepwise escalation of concentrations of
docetaxel from 0.1 to 100 nmol/l for about 9 months.
Docetaxel, gefitinib, afatinib, and AZD9291 were
purchased from Selleck Chemicals (Selleck Chemicals, Houston, TX,
USA). Elacridar and Tween 80 (polysorbate 80) were purchased from
Sigma, Inc. (Sigma-Aldrich, St Louis, MO, USA).
Determination of cell
proliferation
Cells were seeded into 96-well plates at a density
of 2×103 cells/well with or without drugs for 72 h and
the sensitivities to the drugs were determined by using a modified
MTS assay with CellTiter 96 Aqueous One Solution Reagent (Promega),
as previously described (32). The
anti-proliferative effects are shown as IC50, which is the
concentration of the drug required to inhibit cell proliferation by
50%.
Western blot analysis
Resistant cells were cultured in 6 cm dishes for 24
h, and then treated with dimethyl sulfoxide (DMSO) as control, 100
nM docetaxel, and 100 nM docetaxel combined with 0.25 µg/ml
elacridar for 48 h. The total cell lysates were extracted with
lysis buffer, a mixture of RIPA buffer, phosphatase inhibitor
cocktails 2 and 3 (Sigma-Aldrich), and Complete Mini (Roche, Basel,
Switzerland). The primary antibodies were as follows: Monoclonal
anti-PARP (46D11) (Catalog #9532), anti-E-cadherin (24E10) (#3195),
anti-vimentin (D21H3) (#5741), and anti-ABCB1/MDR1 (E1Y7B) (#13342)
(Cell Signaling Technology, Beverly, MA, USA). Monoclonal
anti-actin antibody (#MAB1501R), used as an equal loading control,
was purchased from Merck Millipore (Billerica, MA, USA). The
following secondary antibodies were used: Goat anti-rabbit
(#sc-2030) or anti-mouse (#sc-2031) immunoglobulin G
(IgG)-conjugated horseradish peroxidase (Santa Cruz Biotechnology,
Dallas, TX). To detect specific signals, the membranes were
examined using the ECL Prime Western Blotting Detection System (GE
Healthcare, Amersham, UK) and LAS-3000 (Fujifilm, Tokyo,
Japan).
mRNA and siRNA expression analysis by
qRT-PCR
Total RNA was extracted by using RNeasy Mini Kit
(Qiagen, Valencia, CA, USA) and transcribed into cDNA using
High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems,
Foster City, CA, USA) according to the manufacturer's instructions.
mRNA expression analysis by quantitative reverse transcription PCR
(qRT-PCR) was conducted on cDNA by using TaqMan probes and the
TaqMan Universal PCR Master Mix (Applied Biosystems). PCR
amplification was conducted on an ABI StepOne Real-Time PCR
Instrument (Applied Biosystems) and gene expression was calculated
using the comparative CT method. Three replicates per sample were
assayed for each gene. To quantify the relative changes in gene
expression, the (−ΔΔCQ) method was used and
reactions were normalized to endogenous control gene
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression levels
in mRNA expression analysis.
Transient transfection
Cells were reverse-transfected with 10 nM small
interfering RNAs (siRNAs) mixed with Lipofectamine RNAiMAX
(Invitrogen, Carlsbad, CA, USA). The validated siRNAs specific for
ABCB1 (si-ABCB1#1, 5-CGA UAC AUG GUU UUC CGA U-3; si-ABCB1#2, 5-GGC
UUG CUG UAA UUA CCC A-3) and nonspecific scrambled siRNAs (si-Scr)
were purchased from Applied Biosystems. Twenty-four hours after
reverse-transfection, the transfected-cells could be used for
further experiment according to the manufacturer's protocol. The
indicated drugs were added, and cell viability was measured by
using the MTS assay after an additional 72 h or made the RNA and
protein extraction for real-time PCR and western blot
detection.
Results
All established cell lines showed
resistance not only to docetaxel but also to EGFR-tyrosine kinase
inhibitors
In order to understand the mechanisms of resistance
to docetaxel in NSCLC, we first established docetaxel-resistant
cell lines. Three cell lines (H1299, HCC827, and HCC4006) were
exposed to docetaxel using the stepwise escalation method. Then,
the resistance of the established cell lines to docetaxel was
confirmed by an MTS assay (Table I).
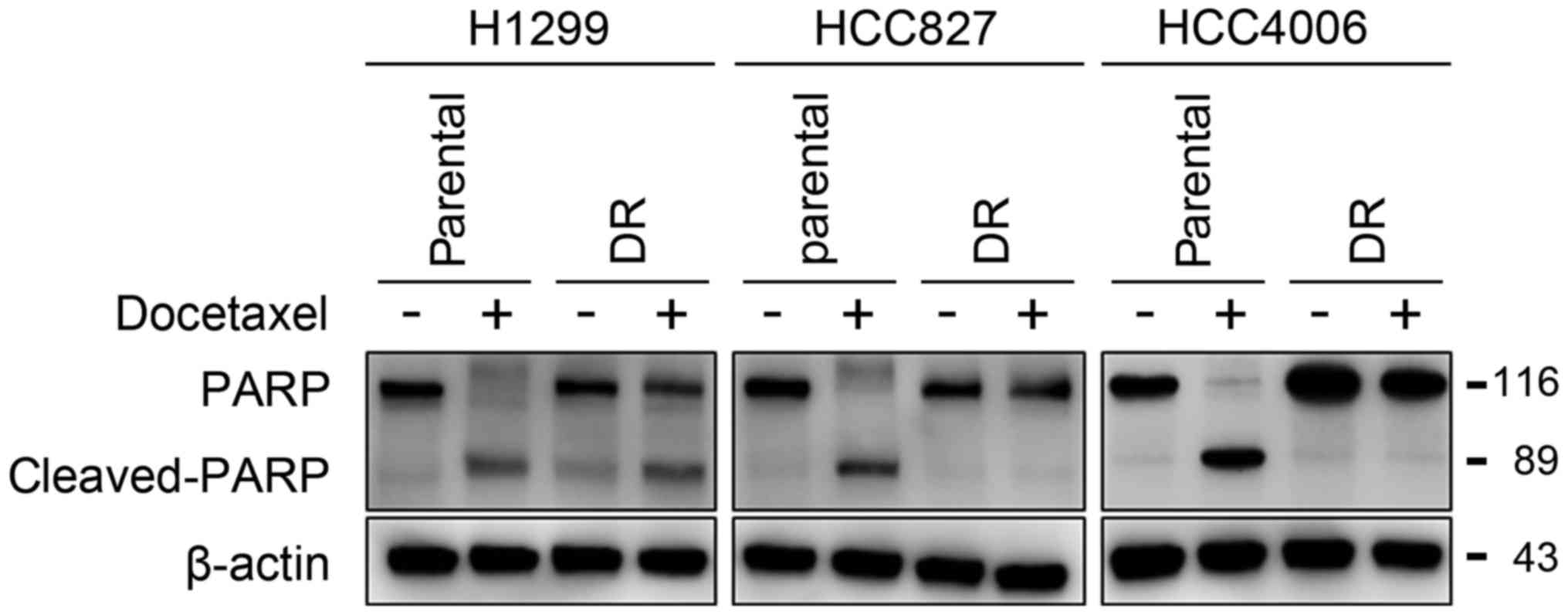
The IC50 values against docetaxel of all the established
docetaxel-resistant cells were significantly higher compared with
their corresponding parental cells. Moreover, the results were
confirmed by western blot analysis for expression of the apoptosis
marker, cleaved PARP, and showed that docetaxel treatment
effectively induced apoptosis in parental cells, but not in
resistant cells (Fig. 1).
 | Table I.IC50 values against each
drug of parental and docetaxel resistant cell lines. |
Table I.
IC50 values against each
drug of parental and docetaxel resistant cell lines.
| Cell Line |
EGFR-mutation | Docetaxel (nM) | Gefitinib (µM) | Afatinib (µM) | AZD9291 (µM) |
|---|
| H1299 | Wild-type | 3.2 | >10 | 4.3 | 5.8 |
| H1299-DR | Wild-type | 272.1 | >10 | 5.7 | 5.5 |
| HCC827 | Exon 19 del
(E746-A750) | 0.3 | 0.0005 | <0.001 | <0.001 |
| HCC827-DR | Exon 19 del
(E746-A750) | >1000 | 5.0 | 1.6 | 2.5 |
| HCC4006 | Exon 19 del
(L747–A750, P ins) | 0.02 | 0.031 | 0.002 | 0.025 |
| HCC4006-DR | Exon 19 del
(L747–A750, P ins) | >1000 | 8.3 | 2.7 | 3.1 |
Next, we explored sensitivity to EGFR-tyrosine
kinase inhibitors (TKIs) (gefitinib, afatinib, and AZD9291) both in
parental and resistant cell lines. As shown in Table I, the docetaxel-resistant cells with
the EGFR mutation showed greater resistance to EGFR-TKIs
than their parental cells. The wild-type EGFR cell lines,
H1299 parental and H1299-DR, were both insensitive to EGFR-TKI
treatment. Although we also treated cells with chemotherapeutic
agents such as cisplatin and pemetrexed, no obvious differences
between the parental and resistant cells were observed (data not
shown).
All resistant cells overexpressed
ABCB1, and HCC827-DR and HCC4006-DR exhibited a CSC-like marker and
EMT features
Overexpression of ABCB1 is known to be the most
common mechanism of cellular resistance to cytotoxic agents. Thus,
to explore the mechanism of resistance to docetaxel, expression of
ABCB1, ALDH1, and EMT-related markers was examined by real-time PCR
and Western blotting in both parental and resistant cell lines. As
shown in Fig. 2A, docetaxel-resistant
cells highly expressed ABCB1. Moreover, HCC827-DR overexpressed
ALDH1, showing a CSC-like marker. To further investigate whether
the acquisition of docetaxel resistance induced specific molecular
changes consistent with EMT, western blot analysis was performed.
As shown in Fig. 2B, lower expression
of E-cadherin (epithelial marker) at the protein level was observed
in HCC4006-DR cells compared to parental HCC4006 cells.
Elacridar, a third-generation ABCB1
inhibitor, overcomes docetaxel resistance, but not to EGFR-TKIs
resistance
Having identified that ABCB1 is overexpressed in
docetaxel-resistant cells, we examined whether suppression of ABCB1
leads to improved docetaxel resistance. We chose elacridar to
inhibit the drug pump function of ABCB1 by competitively combining
with ABCB1. Elacridar markedly recovered sensitivity to docetaxel,
but not to gefitinib (Table II). The
IC50 value of H1299-DR was 9.4 nM at 0.25 µg/ml of
elacridar, which was higher than that of parental cells (3.2 nM);
thus elacridar only partially recovered the sensitivity to
docetaxel. These cells may contain a resistance mechanism other
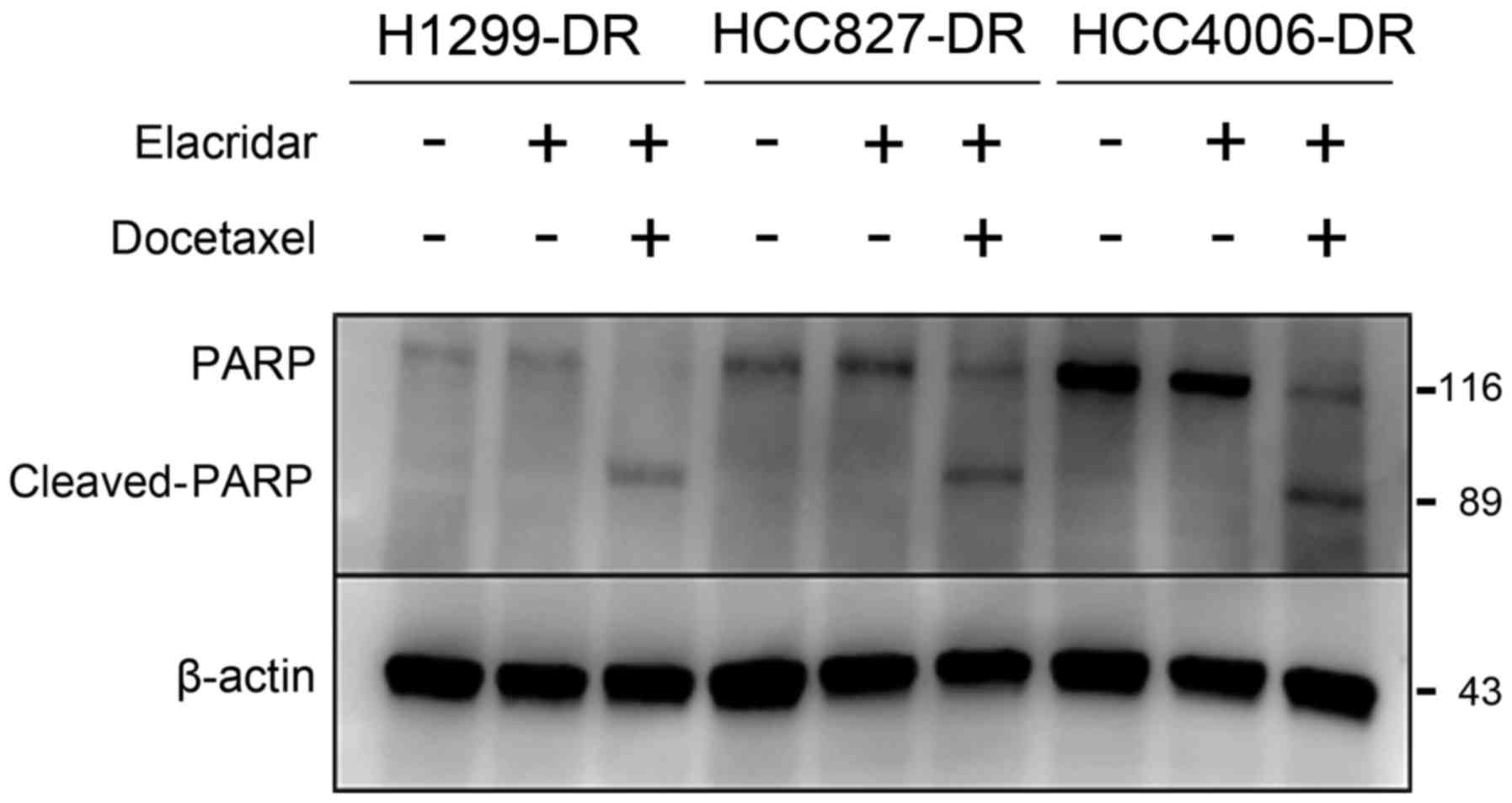
than ABCB1. The expression of the apoptosis marker cleaved-PARP
increased in the resistant cells only when both docetaxel and
elacridar were administered in combination (Fig. 3). These results suggest that elacridar
restored the sensitivity to docetaxel.
| Table II.IC50 values against
docetaxel or gefitinib of docetaxel-resistant cell lines, with or
without elacridar treatment. |
Table II.
IC50 values against
docetaxel or gefitinib of docetaxel-resistant cell lines, with or
without elacridar treatment.
|
| Docetaxel (nM) | Gefitinib (µM) |
|---|
|
|
|
|
|---|
| Elacridar (0.25
µg/ml) | − | + | − | + |
|---|
| H1299DR | 272.1 | 9.4 | >10 | >10 |
| HCC827DR | >1000 | 21.9 | 5.0 | 1.6 |
| HCC4006DR | >1000 | 0.3 | 8.3 | 4.7 |
siRNA-mediated knockdown of ABCB1
sensitized resistant cells to docetaxel
To further investigate the function of ABCB1 in the
docetaxel resistance mechanism, siRNA-mediated suppression of ABCB1
expression was examined in the docetaxel resistant cell lines. The
efficacy of ABCB1 knockdown was confirmed by real-time PCR and
Western blot analysis (Fig. 4A and
B); siRNA-mediated knockdown of ABCB1 partially sensitized
resistant cells to docetaxel (Fig. 4C
and Table III). In contrast, ABCB1
depletion did not restore gefitinib sensitivity in
docetaxel-resistant cells (Table
III).
| Table III.IC50 values against
docetaxel or gefitinib of docetaxel resistant cell lines, with or
without si-RNA mediated knockdown of ABCB1. |
Table III.
IC50 values against
docetaxel or gefitinib of docetaxel resistant cell lines, with or
without si-RNA mediated knockdown of ABCB1.
|
| Docetaxel (nM) | Gefitinib (µM) |
|---|
|
|
|
|
|---|
|
| si-Scr | si-ABCB1#1 | si-ABCB1#2 | si-Scr | si-ABCB1#1 | si-ABCB1#2 |
|---|
| H1299-DR | 590.0 |
25.8 |
12.6 | >10 | >10 | >10 |
| HCC827-DR | 679.4 | 301.4 | 382.2 | 1.2 | 0.67 | 0.43 |
| HCC4006-DR | 995.8 | 290.2 | 328.6 | 5.7 | 6.0 | 4.2 |
Discussion
In the present study, we established
docetaxel-resistant NSCLC cell lines and analyzed their resistance
mechanisms in order to understand the biological mechanisms of
chemo-resistance in NSCLC cells and to identify reversion
opportunities. Several mechanisms for resistance to taxanes have
already been reported, such as MDR, CSC-like, and EMT-related
marker overexpression (9,25,31,33). The
main mechanisms involved in resistance to taxanes are expression of
the MDR phenotype and the alteration of their cellular target,
namely the tubulin/microtubule system (11,34).
Several putative mechanisms of resistance, including alteration of
the signaling pathways, altered regulation of the cell cycle, and
altered control of apoptosis and cell death signals, have also been
described (10,12). Our study confirmed that ABCB1, an MDR
molecule, was overexpressed in experimentally established cells.
Overexpression of ABCB1 was one of the causes of docetaxel
resistance, confirmed by using elacridar and siRNA against ABCB1.
However, restoration of sensitivity to docetaxel by ABCB1 siRNA was
not perfect. This may be due to incomplete suppression of ABCB1
gene expression in our system, and we therefore inferred that
overexpression of ABCB1 is not the only mechanism of docetaxel and
multidrug resistance.
We tested EMT- and CSC-related markers in the
resistant cell lines because we have recently reported that a
gefitinib-resistant cell line was also resistant to docetaxel and
exhibited both EMT features and CSC properties (32). E-cadherin and vimentin are EMT-related
markers (25), and the expression
levels of E-cadherin were slightly lower in HCC4006-DR than in
parental cells. The functional implications of EMT include enhanced
mobility, invasion, and resistance to apoptotic stimuli (35,36).
Moreover, tumor cells can acquire CSC features, secondary tumor
initiating, and chemo-resistance properties through EMT (37–39). CSCs,
which generate tumors with self-renewal and differentiation
abilities, are believed to be highly resistant to conventional
chemotherapies owing to various crucial features, including high
expression of ABC transporter proteins (40) and ALDH activity (41). HCC827-DR cells overexpressed ALDH1, a
CSC-like feature. ABC transporters, including ABCB1, have also been
reported to be implicated in promoting CSC-like properties
(42).
Notably, EGFR-mutant docetaxel-resistant
cells showed cross-resistance to EGFR-TKIs. To our knowledge, we
found first that docetaxel treatment led to EGFR-TKIs resistance in
EGFR-mutant NSCLC cells. However, ABCB1 expression was not
related to sensitivity to EGFR-TKI because elacridar or siRNA
against ABCB1 had no effect on sensitivity to EGFR-TKI. We were
unable to clarify the mechanism of resistance to EGFR-TKI because
well-known mechanisms, such as T790M mutation or MET amplification,
were not observed in these cell lines. Although HCC4006-DR cells
did not show increased expression of other CSC-like markers, we
recently reported a similar observation, that EGFR-TKI resistant
cells exhibiting both EMT features and CSC properties with
overexpression of ABCB1 were resistant to anti-microtuble agents
(32). Mizuuchi et al
(43) recently reported similar
findings that acquired EGFR-TKI resistance cells became more
resistant to anti-microtuble agents. They suggested that this
‘collateral’ cross-resistance to EGFR-TKIs and anti-microtubule
agents resulted from two distinct mechanisms, both of which were
thought to be a cause of or to result from EMT. Further
investigation is required to clarify detailed mechanisms causing
this cross-resistance.
Based on our results, elacridar showed a strong
effect on docetaxel-resistant NSCLC cells. Elacridar, a
third-generation inhibitor of ABCB1, is a non-competitive inhibitor
that functions by changing the active allosteric site of ABCB1
(19,44). It shows a relatively minor influence
on other members of the ABC family and on the pharmacokinetics of
the anti-tumor drugs in clinical use (45).
In conclusion, we have demonstrated that
docetaxel-resistant NSCLC cells showed multi-resistance to
docetaxel as well as EGFR-TKIs. Molecular analyses showed that all
of these resistant cell lines highly expressed ABCB1, and ABCB1
played an important role in acquired resistance to docetaxel in
lung cancer. Furthermore, HCC827-DR and HCC4006-DR cells exhibited
a CSC-like marker and EMT features, respectively. Thus, elacridar
could overcome resistance to docetaxel, suggesting that development
of ABCB1 inhibitors show great promise for clinical use to overcome
multi-drug resistance.
Acknowledgements
We thank Ms. Fumiko Isobe (Department of Thoracic,
Breast and Endocrinological Surgery, Okayama University Graduate
School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama,
Japan) for her technical assistance.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
MDR
|
multidrug resistance
|
|
ABC
|
ATP-binding cassette
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
EGFR
|
epidermal growth factor receptor
|
|
CSC
|
Cancer stem cell
|
|
IC50
|
50% inhibitory concentrations
|
|
DR
|
docetaxel-resistant
|
|
TKI
|
tyrosine kinase inhibitor
|
|
GAPDH
|
gene glyceraldehyde-3-phosphate
dehydrogenase
|
|
Scr
|
scrambled
|
References
|
1
|
Wu L, Chang W, Zhao J, Yu Y, Tan X, Su T,
Zhao L, Huang S, Liu S and Cao G: Development of autoantibody
signatures as novel diagnostic biomarkers of non-small cell lung
cancer. Clin Cancer Res. 16:3760–3768. 2010.PubMed/NCBI
|
|
2
|
Rigas JR and Kelly K: Current treatment
paradigms for locally advanced non-small cell lung cancer. J Thorac
Oncol. 2:(Suppl 2). S77–S85. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Choy H, Pyo H, Kim JS and MacRae R: Role
of taxanes in the combined modality treatment of patients with
locally advanced non-small cell lung cancer. Expert Opin
Pharmacother. 2:963–974. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wakelee H, Ramalingam S and Belani CP:
Docetaxel in advanced non-small cell lung cancer. Expert Rev
Anticancer Ther. 5:13–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stinchcombe TE: Recent advances in the
treatment of non-small cell and small cell lung cancer. F1000Prime
Rep. 6:1172014. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Garon EB, Ciuleanu TE, Arrieta O, Prabhash
K, Syrigos KN, Goksel T, Park K, Gorbunova V, Kowalyszyn RD, Pikiel
J, et al: Ramucirumab plus docetaxel versus placebo plus docetaxel
for second-line treatment of stage IV non-small-cell lung cancer
after disease progression on platinum-based therapy (REVEL): A
multicentre, double-blind, randomised phase 3 trial. Lancet.
384:665–673. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoh K, Hosomi Y, Kasahara K, Yamada K,
Takahashi T, Yamamoto N, Nishio M, Ohe Y, Koue T, Nakamura T, et
al: A randomized, double-blind, phase II study of ramucirumab plus
docetaxel vs. placebo plus docetaxel in Japanese patients with
stage IV non-small cell lung cancer after disease progression on
platinum-based therapy. Lung Cancer. 99:186–193. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lage H: An overview of cancer multidrug
resistance: A still unsolved problem. Cell Mol Life Sci.
65:3145–3167. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Myer MS, Joone G, Chasen MR and van
Rensburg CE: The chemosensitizing potential of GF120918 is
independent of the magnitude of P-glycoprotein-mediated resistance
to conventional chemotherapeutic agents in a small cell lung cancer
line. Oncol Rep. 6:217–218. 1999.PubMed/NCBI
|
|
10
|
Galletti E, Magnani M, Renzulli ML and
Botta M: Paclitaxel and docetaxel resistance: Molecular mechanisms
and development of new generation taxanes. ChemMedChem. 2:920–942.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Orr GA, Verdier-Pinard P, McDaid H and
Horwitz SB: Mechanisms of Taxol resistance related to microtubules.
Oncogene. 22:7280–7295. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Luqmani YA: Mechanisms of drug resistance
in cancer chemotherapy. Med Princ Pract. 14:(Suppl 1). S35–S48.
2005. View Article : Google Scholar
|
|
13
|
Gottesman MM and Ambudkar SV: Overview:
ABC transporters and human disease. J Bioenerg Biomembr.
33:453–458. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sarkadi B, Homolya L, Szakács G and Váradi
A: Human multidrug resistance ABCB and ABCG transporters:
Participation in a chemoimmunity defense system. Physiol Rev.
86:1179–1236. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Higgins CF: Multiple molecular mechanisms
for multidrug resistance transporters. Nature. 446:749–757. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sharom FJ: ABC multidrug transporters:
Structure, function and role in chemoresistance. Pharmacogenomics.
9:105–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Y, Yuan H, Yang K, Xu W, Tang W and Li
X: The structure and functions of P-glycoprotein. Curr Med Chem.
17:786–800. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Szakács G, Váradi A, Ozvegy-Laczka C and
Sarkadi B: The role of ABC transporters in drug absorption,
distribution, metabolism, excretion and toxicity (ADME-Tox). Drug
Discov Today. 13:379–393. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Palmeira A, Sousa E, Vasconcelos MH and
Pinto MM: Three decades of P-gp inhibitors: Skimming through
several generations and scaffolds. Curr Med Chem. 19:1946–2025.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu CP, Calcagno AM and Ambudkar SV:
Reversal of ABC drug transporter-mediated multidrug resistance in
cancer cells: Evaluation of current strategies. Curr Mol Pharmacol.
1:93–105. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hubensack M, Müller C, Höcherl P, Fellner
S, Spruss T, Bernhardt G and Buschauer A: Effect of the ABCB1
modulators elacridar and tariquidar on the distribution of
paclitaxel in nude mice. J Cancer Res Clin Oncol. 134:597–607.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kajiyama H, Shibata K, Terauchi M,
Yamashita M, Ino K, Nawa A and Kikkawa F: Chemoresistance to
paclitaxel induces epithelial-mesenchymal transition and enhances
metastatic potential for epithelial ovarian carcinoma cells. Int J
Oncol. 31:277–283. 2007.PubMed/NCBI
|
|
23
|
Min C, Eddy SF, Sherr DH and Sonenshein
GE: NF-kappaB and epithelial to mesenchymal transition of cancer. J
Cell Biochem. 104:733–744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sabbah M, Emami S, Redeuilh G, Julien S,
Prévost G, Zimber A, Ouelaa R, Bracke M, De Wever O and Gespach C:
Molecular signature and therapeutic perspective of the
epithelial-to-mesenchymal transitions in epithelial cancers. Drug
Resist Updat. 11:123–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ren J, Chen Y, Song H, Chen L and Wang R:
Inhibition of ZEB1 reverses EMT and chemoresistance in
docetaxel-resistant human lung adenocarcinoma cell line. J Cell
Biochem. 114:1395–1403. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu C and Alman BA: Side population cells
in human cancers. Cancer Lett. 268:1–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sugano T, Seike M, Noro R, Soeno C, Chiba
M, Zou F, Nakamichi S, Nishijima N, Matsumoto M, Miyanaga A, et al:
Inhibition of ABCB1 overcomes cancer stem cell-like properties and
acquired resistance to MET inhibitors in non-small cell lung
cancer. Mol Cancer Ther. 14:2433–2440. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
O'Flaherty JD, Barr M, Fennell D, Richard
D, Reynolds J, O'Leary J and O'Byrne K: The cancer stem-cell
hypothesis: Its emerging role in lung cancer biology and its
relevance for future therapy. J Thorac Oncol. 7:1880–1890. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Berns A: Stem cells for lung cancer? Cell.
121:811–813. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jiang F, Qiu Q, Khanna A, Todd NW, Deepak
J, Xing L, Wang H, Liu Z, Su Y, Stass SA and Katz RL: Aldehyde
dehydrogenase 1 is a tumor stem cell-associated marker in lung
cancer. Mol Cancer Res. 7:330–338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shien K, Toyooka S, Yamamoto H, Soh J,
Jida M, Thu KL, Hashida S, Maki Y, Ichihara E, Asano H, et al:
Acquired resistance to EGFR inhibitors is associated with a
manifestation of stem cell-like properties in cancer cells. Cancer
Res. 73:3051–3061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang YJ, Zhang YK, Kathawala RJ and Chen
ZS: Repositioning of tyrosine kinase inhibitors as antagonists of
ATP-binding cassette transporters in anticancer drug resistance.
Cancers (Basel). 6:1925–1952. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Krishna R and Mayer LD: Multidrug
resistance (MDR) in cancer. Mechanisms, reversal using modulators
of MDR and the role of MDR modulators in influencing the
pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 11:265–283.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Scheel C and Weinberg RA: Phenotypic
plasticity and epithelial-mesenchymal transitions in cancer and
normal stem cells? Int J Cancer. 129:2310–2314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hennessy BT, Gonzalez-Angulo AM,
Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J,
Sahin A, Agarwal R, Joy C, et al: Characterization of a naturally
occurring breast cancer subset enriched in
epithelial-to-mesenchymal transition and stem cell characteristics.
Cancer Res. 69:4116–4124. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gal A, Sjöblom T, Fedorova L, Imreh S,
Beug H and Moustakas A: Sustained TGF beta exposure suppresses Smad
and non-Smad signalling in mammary epithelial cells, leading to EMT
and inhibition of growth arrest and apoptosis. Oncogene.
27:1218–1230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dean M: ABC transporters, drug resistance,
and cancer stem cells. J Mammary Gland Biol Neoplasia. 14:3–9.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Resetkova E, Reis-Filho JS, Jain RK, Mehta
R, Thorat MA, Nakshatri H and Badve S: Prognostic impact of ALDH1
in breast cancer: A story of stem cells and tumor microenvironment.
Breast Cancer Res Treat. 123:97–108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mizuuchi H, Suda K, Sato K, Tomida S,
Fujita Y, Kobayashi Y, Maehara Y, Sekido Y, Nishio K and Mitsudomi
T: Collateral chemoresistance to anti-microtubule agents in a lung
cancer cell line with acquired resistance to erlotinib. PLoS One.
10:e01239012015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Goldman B: Multidrug resistance: Can new
drugs help chemotherapy score against cancer? J Natl Cancer Inst.
95:255–257. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mayur YC, Peters GJ, Prasad VV, Lemo C and
Sathish NK: Design of new drug molecules to be used in reversing
multidrug resistance in cancer cells. Curr Cancer Drug Targets.
9:298–306. 2009. View Article : Google Scholar : PubMed/NCBI
|