Introduction
Tumors are complex tissues composed not only of
tumor cells, but also a repertoire of immune cells that give them
special features to allow tumor growth and metastasis. These
special features, proposed by Hanahan and Weinberg in 2011, provide
the tumor with proliferative signal support, the avoidance of
growth suppressors, cell death circumvention, cell immortality,
angiogenesis, and invasive and metastatic activation. Two
additional features are involved in cancer pathogenesis: The
ability to reprogram the cellular metabolism to support neoplastic
proliferation, and the ability to evade immune recognition and
destruction by T and B lymphocytes, macrophages, and natural killer
(NK) cells (1).
Cancer progression typically requires tumor cells to
acquire the ability to avoid immune detection and destruction.
Thus, understanding the interplay between the tumor, the immune
system and the tumor microenvironment is of pivotal importance to
providing the rationale for designing therapeutic approaches that
trigger specific antitumor immune responses.
Inducing an effective immune response against cancer
by immunotherapeutic intervention is a challenge that depends on
several factors of the tumor and the immune system functioning
together to either eradicate tumors or promote immune evasion. One
of the most important factors in the development of antitumor
immunotherapies is the constitution of the tumor microenvironment
(including immune cell types, cytokine profiles, acidity and
oxygenation levels, molecular signatures) as this may positively or
negatively impact the arrival and cytotoxic activity of effector
cells, thus determining an improved or worse clinical outcome
(2). Therefore, to improve
immunotherapy outcomes, it is important to alter the tumor
microenvironment so that it is permissive for cytotoxic NK and
T-cell activity. Thus, depending on the specific disease phenotype
of the patient, certain therapeutic approaches must be escalated,
while others should be avoided, in order to obtain a desirable
clinical response (3).
Altered HLA (human leukocyte antigen)-I expression
on the tumor cell surface is an early and frequent event that
promotes carcinogenesis, as HLA-I is critical for the immune
recognition of tumor cells and signaling between tumor and immune
cells (4,5). Several studies reported total or partial
loss of classical HLA-I molecule expression in different human
tumors (6,7), with at least 50% of multiple HLA allele
loss caused by loss of heterozygosity (LOH) events (8). Another HLA-mediated strategy used by
tumor cells to avoid recognition by various immune effectors is the
aberrant expression of non-classical HLA-I molecules (HLA-E and
HLA-G), which function as inhibitor ligands for immune-competent
cells, allowing tumor immune escape (9).
As mentioned previously, the complexity of the
alterations to HLA expression in carcinogenesis makes selecting a
therapeutic target to potentiate antitumor immune responses very
difficult. However, correcting these alterations may provide a
first step towards improving the currently available cancer
immunotherapies.
Cancer immune response: Host-protective
while tumor promoting
It is possible to separate tumor associated antigens
(TAAs) into two main classes: Self and tumor-restricted antigens.
Self-antigens include differentiation (including Melan A in
melanoma) and overexpressed antigens (including ErbB2 receptor
tyrosine kinase 2 in colon, breast and lung cancer), whereas
tumor-restricted antigens may be of viral origin (including human
papilloma virus in cervical and throat cancer), from the germ line
(including NY-ESO-1 in melanoma) or neoantigens (for example
mutated antigens, including β-catenin in melanoma) (10). It is possible to induce immunization
against tumor self-antigens, potentially generating an effective
antitumor T-cell and antibody response (11).
During carcinogenesis, innate and adaptive immunity
stimulation occurs. Innate immunity mediates surveillance and tumor
lysis in a rapid and non-specific fashion, whereas adaptive immune
response is more specific; directed by TAAs that induce T-cell
responses and antibody production (12). The main immune effectors of antitumor
innate immunity are NK cells, which serve important functions in
cancer immune surveillance: These cells express a variety of
activating and inhibitory receptors that recognize cellular stress
ligands, as well as major histocompatibility complex class I and
similar molecules. These interactions mediate their tolerance to
healthy self-cells and their cytotoxicity against stressed cells
(13–15).
NK cell cytotoxic activity is either direct or
indirect. Direct killing occurs via antibody-dependent
cell-mediated cytotoxicity, an adaptive immune cell-killing
mechanism mediated by activated NK cells (16), whereas indirect killing occurs through
the secretion of cytokines, which exert antitumor effects via the
stimulation of immune system regulatory components (17).
Although the main adaptive immune effectors capable
of eliminating transformed cells are cytotoxic CD8+
T-cells (18), CD4+
T-cells, through the secretion of a Th1 cytokine profile (19), and B-cells, through the production of
antitumor antibodies, also serve important functions in generating
a powerful antitumor immune response. However, tumor features,
including the nature of tumor antigens (20), immune modulatory factors produced by
tumor and host immune cells, and the existence of regulatory cells
[including regulatory T-cells (T-regs), myeloid derived suppressor
cells (MDSCs) and tumor-associated macrophages (TAMs)] favor tumor
development and progression (21).
The shift from antitumor innate immunity to a
long-lasting adaptive immune response is mediated by lymphoid cells
and their products (22). Genetic and
epigenetic alterations generate tumor antigens that are recognized
by a T-cell's T-cell receptor (TCR) when presented in an
HLA-restricted manner. This recognition leads to T-cell priming,
activation, proliferation, differentiation and cytokine production,
and thus is pivotal for immune response amplitude and quality. Once
tumor antigen recognition occurs, CD28 amplifies TCR signaling to
completely activate T-cells. These activated effector T-cells leave
the lymph nodes in search of tumor cells bearing the cognate
HLA-peptide, leading to tumor cell death by T-cell mediated
cytotoxicity (23). Under normal
physiological conditions, T-cell activation is regulated by a
balance between co-stimulatory and inhibitory signals, which
ultimately leads to an effective immune response.
Immune-suppressing pathway proteins, collectively termed immune
checkpoints, are crucial for the maintenance of self-tolerance and
for protection from tissue damage caused by the immune response
itself (24). Tumors may alter immune
homeostasis, suppressing T-cell activation and effector function,
driving T-cell tolerance by chronic antigenic stimulation, and
simultaneously activating suppressor pathways to prevent T-cell
mediated killing. The immune checkpoint molecules associated with
these phenomena have been demonstrated to be excellent targets for
cancer immune therapy (25).
The best understood co-stimulatory/regulatory
pathways are those mediated by CD80, CD86, cytotoxic
T-lymphocyte-associated protein (CTLA)-4 and programmed cell death
protein (PD)-1. CD80 and CD86 are expressed on the surface of
antigen-presenting cells (APCs), bind to CD28 receptors on the
T-cell surface and induce interleukin (IL)-2 production to support
specific T-cell expansion (26). Once
TCR activation occurs, regulatory signals are generated to limit
the expansion and activation of TCR-triggered T-cells. CTLA-4 and
programmed cell death protein 1 (PD1) are immune checkpoints
capable of limiting T-cell activation in secondary lymphoid organs
and activating T, B and myeloid cells (27,28). Upon
receptor ligation, T-cells stop clonal expansion and cytokine
production (29–31). These regulatory signals compete for
ligands and key substrates with co-stimulatory receptors. For
example, on activated T-cells, CTLA-4 competes with CD28 molecules
for the CD80 and CD86 ligands on APCs to regulate cell cycle
proteins and cytokine expression. Another immune check point with
pivotal relevance in cancer is HLA-G, a tolerogenic non-classical
HLA-I molecule, which binds to CD8 (32), CD160 (33), the inhibitory receptors immunoglobulin
(Ig)-like transcript (ILT)-2 and −4, and killer cell Ig-like
receptor, 2 Ig domains and long cytoplasmic tail 4 (KIR2DL4)
(34,35). Besides ILT-4, all these inhibitory
receptors are widely expressed on lymphoid immune cells, whereas
myeloid immune cells express CD8, ILT-2 and ILT-4 (36). Thus, immune responses are regulated in
order to guarantee an effective immune response while preventing
excessive immune activation.
In cancer, tumor cell plasticity may generate tumors
with low immunogenicity in response to selective immune pressure
exerted by the host immune system, enabling tumor evasion of immune
surveillance. This host-protective, tumor-promoting immunity action
is known as cancer immune editing, a process that occurs in three
sequential phases: i) Elimination; ii) equilibrium and iii) escape
(37). Elimination corresponds to the
initial phase of cancer immune surveillance, in which the immune
system is able to detect and destroy transformed cells, preventing
tumor progression. In this phase, immune effector cells, including
cytotoxic T-lymphocytes (CTLs) and NK cells, are able to recognize
and eliminate tumor cells. Dendritic cells (DC) and CD4+
T-cells are also components of this elimination phase, as they
recognize and kill transformed cells long before they become
clinically apparent, working as extrinsic tumor suppressors
(38). Killing at this phase depends
on i) stress ligand expression, including NK group (NKG) 2D; ii)
TAA recognition in an HLA-restricted manner and iii) and
co-stimulatory signals which completely activate T-cells (39).
Tumor cell variants that survive the elimination
phase enter into an equilibrium stage in which the immune system
controls tumor outgrowth, but the tumor remains clinically
undetectable. Tumor cells with edited immunogenicity eventually
continue growing; tumor dormancy is broken, and the edited tumors,
which exhibit reduced immunogenicity, grow without immune control
and progressively establish an immunosuppressive microenvironment,
becoming clinically apparent (40,41).
Immuno-editing provides tumor cells with a plethora of molecular
tools with which they may control the immune response. The tumor
may recruit all immune cells and once inside, they participate in
dynamic crosstalk with cancer cells to govern tumor development
(Fig. 1). Early eradication or
spontaneous tumor regression, as well as tumor promotion and
development, depend on the nature of immune cells infiltrating the
tumor and on tumor-induced immune factor production (37).
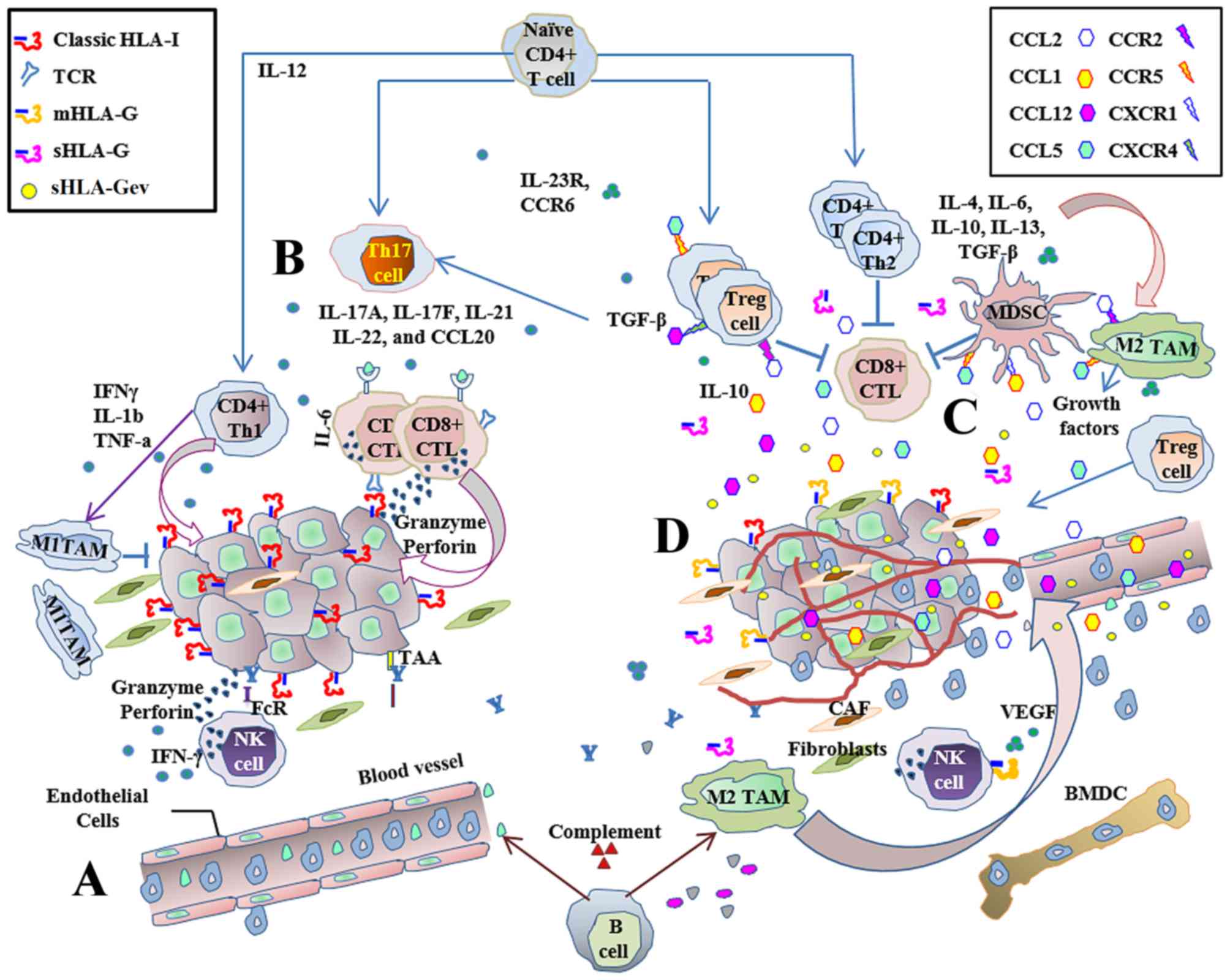 | Figure 1.Immune response to cancer:
Host-protective while tumor-promoting. The innate and adaptive
immune responses are stimulated during carcinogenesis and are
capable of surveillance and tumor lysis. (A) The main antitumor
immune effectors are NK and CD8+ T-cells, which are
capable of responding directly against cancer with cytotoxicity, or
by secreting cytokines. Inflammatory cells infiltrate the tumor and
exert antitumor immune responses. (B) Cytotoxic CD8+
T-cells are the main adaptive immune effectors. CD4+
T-cells help to improve antitumor immune responses through the
secretion of Th1 cytokines. Antitumor immune responses mediated by
CTLs are effective and prevent tumor development in HLA-I positive
tumor cells, but these immune responses are ultimately insufficient
to prevent disease progression. (C) When inflammatory responses
become chronic, regulatory cell populations generate a tolerant
pro-tumor immune response via cytokine secretion and the production
of growth factors. Tumor-promoting activity favors angiogenesis,
invasion and metastasis, and is capable of suppressing adaptive
immunity. (D) Aberrant expression of classical and non-classical
HLA-I contributes to the establishment of an immunosuppressive
microenvironment, promoting tumor growth by controlling immune
stimulation and suppression signals. NK, natural killer; CD,
cluster of differentiation; CTL, cytotoxic T-cell; HLA, human
leukocyte antigen; BMDC, bone marrow-derived cells; CAF,
cancer-associated fibroblast; CCL, C-C motif chemokine ligand; CCR,
C-C motif chemokine receptor; CXCR, C-X-C motif chemokine receptor;
FcR, fragment crystallizable receptor; IFN, interferon; IL,
interleukin; MDSC, myeloid derived suppressor cells; mHLA-G
membrane-bound human leukocyte antigen-G; sHLA-G, soluble human
leukocyte antigen-G; sHLA-Gev extracellular vesicle-associated
soluble human leukocyte antigen-G; TAAs, tumor associated antigens;
TAM, tumor-associated macrophages; TCR, T-cell receptor; TGF-β,
transforming growth factor β; Th, T helper cells; T-reg, regulatory
T-cells. |
Tumor microenvironment: Antitumor and tumor
promoting
The adaptive antitumor immune response is not always
capable of tumor eradication, potentially due to immune evasion
mechanisms including the induction of immunological ignorance and
immunological tolerance, or interactions between tumor cells and
the host immune response. These phenomena may inhibit T-cell
activation and induce tumor resistance against immune attack
(37), and are activated by a number
of mechanisms that will be described.
At the initial tumor growth stage, tissue damage
induces acute Th1 inflammatory responses that favor APC maturation
and innate immune cell polarization, promoting the elimination of
developing tumors. APC maturation initiates adaptive immune
responses mediated by CD4+ and CD8+ T-cells.
Simultaneously, the acute activation of B-cells results in the
induction of soluble mediators, including antigen-specific Igs
capable of complement activation, to coordinate phagocytic or
cytotoxic destruction of damaged cells by innate immune cells
(42). During inflammation,
chemokines control immune cell movement, immune response
polarization and T-cell and dendritic cell interactions, while
cytokines mediate intercellular communication in the immune system
and function as immune regulators (43,44).
Chemokine and cytokine expression profiles modulate the functional
status of the immune system to negatively impact tumor development
and progression.
In cancer development, tumor cells and tumor
infiltrating immune cells produce antitumor and pro-tumor immune
factors, which modulate the tumor immune response. A pro-tumor
effect may dominate through various means: Inflamed tumors express
high levels of pro-inflammatory innate and adaptive immune signals,
as well as several immune-inhibitory factors, including programmed
cell death ligand (PDL) 1 and indoleamine-2, 3-dioxygenase. They
also recruit forkhead box p3 and T-regs to promote immune escape.
Alternatively, non-inflamed tumors express a reduced level of
chemokines, resulting in the poor attraction of CD8+
effector T-cells into the tumor mass and poor effector cell
trafficking (45). In addition, high
levels of vascular markers and high macrophage and fibroblast
infiltration also favor tumor growth (Fig. 2) (46,47). TAMs,
tolerogenic DCs, regulatory T-cells and MDSCs, which are the main
regulatory immune cells recruited by the tumor to create an
environment with anti-inflammatory properties, favor tumor growth
and survival (45). In addition, the
chronic activation of B-cells is deleterious in certain types of
cancer, potentially through the production of IL-10 (48). Thus, the nature of immune cells
infiltrating the tumor serves a fundamental function in the failure
of antitumor immune responses. In breast cancer, immune cell
infiltration was previously demonstrated to correlate with an
improved prognosis, a reduced tumor diameter and longer
recurrence-free survival time (49).
In other types of cancer, high CD4+ T-cell infiltration
was identified to correlate with tumor progression, potentially
because the main tumor infiltrating cells are CD4+ T-reg
cells (50).
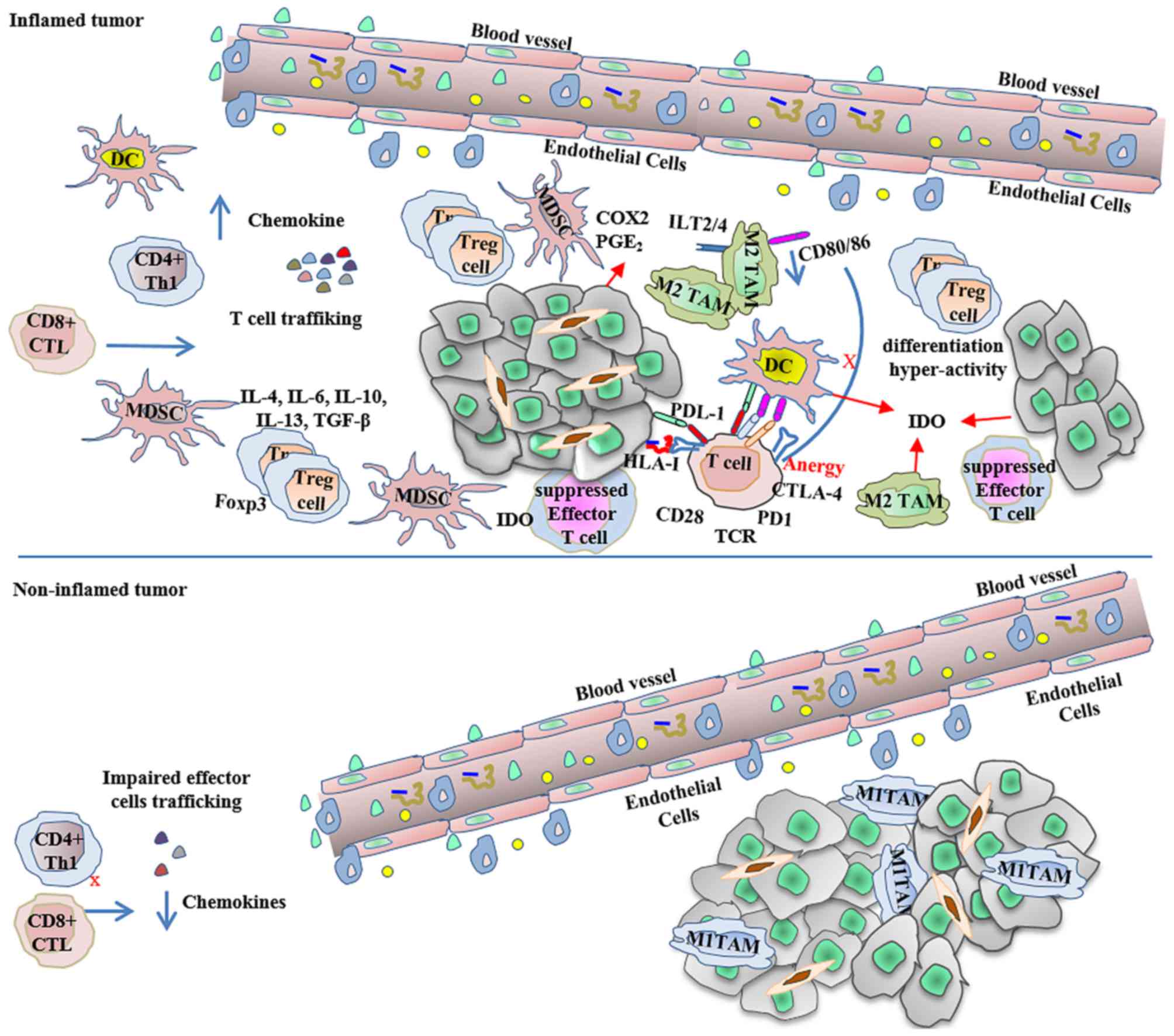 | Figure 2.Inflamed and non-inflamed tumors
escape immune-mediated destruction. As described by Gajewski et
al (44), inflamed tumors express
high levels of pro-inflammatory innate and adaptive signals, as
well as immunoregulatory factors that contribute to the creation of
an immunosuppressive environment, in which a dominant effect of
negative regulation mediates the tumor escape. In contrast,
non-inflamed tumors with poor chemokine production have few
effector cells, abundant macrophages and cancer-associated
fibroblasts, and express high levels of vascular markers, also
allowing tumor escape. CD, cluster of differentiation; COX2,
cytochrome c oxidase 2; CTL, cytotoxic T-cell; CTLA-4, cytotoxic
T-lymphocyte-associated protein 4; DC, dendritic cells; Foxp3,
forkhead box p3; HLA, human leukocyte antigen; IDO, indoleamine-2,
3-dioxygenase; IL, interleukin; ILT, immunoglobulin-like
transcript; MDSC, myeloid derived suppressor cells; PD1, programmed
cell death protein 1; PDL1, programmed cell death ligand 1; PGE2,
prostaglandin E2; TAM, tumor-associated macrophages; TCR, T-cell
receptor; TGF-β, transforming growth factor β; Th, T helper cells;
T-reg, regulatory T-cells. |
Alternatively, IL-10 in the tumor microenvironment
may generate a neoplastic cell phenotype resistant to CTL-mediated
lysis by decreasing transporter associated with antigen processing
(TAP)1/2 expression and function, resulting in low peptide
translocation into the endoplasmic reticulum, thus affecting
HLA-I-mediated antigen presentation (51,52). HLA-I
downregulation and non-classical HLA-I molecule neo-expression
promote immunosuppression and, therefore, tumor immunoescape. A
number of studies have demonstrated that HLA-G, HLA-E and IL-10
expression levels in cancer are associated with tumor progression,
metastasis and a poor prognosis (53–55), and
that the IL-10-positive T-reg cell frequency may be associated with
malignant transformation by contributing to immunosuppression in
the tumor microenvironment (56). Due
to the plethora of possible immunosuppressive features present in a
particular tumor entity, it is necessary to personalize the
selection of the therapeutic targets for cancer treatment to induce
an effective antitumor immune response, thus avoiding the
development of tumor chemo-resistance and a subsequent poor
outcome.
HLA-mediated cancer cell escape
mechanisms
The malignant transformation of cells is often
associated with alterations to gene expression and the antigenic
profile. Alterations in HLA expression (including classical and
non-classical HLA-I and HLA-II) are frequent and early events
during carcinogenesis (4,57). As tumor cells are immunogenic, they
must acquire a plethora of molecular mechanisms to avoid
destruction by CTLs and NK cells. By downregulating classical
HLA-I, they prevent tumor recognition and rejection by CTLs, and by
overexpressing non-classical HLA-I molecules they disable all types
of immune cell involved in tumor recognition and rejection
(including T and B lymphocytes, APCs and NK cells) (58). Conventional changes of HLA expression
in malignant cells include total or allele-specific loss of
classical HLA-I expression and the induction of non-classical HLA-I
and HLA-II expression, potentially due to an immune selection
process that enables the initiation of malignant lesions with an
HLA-altered phenotype, which will be necessary to consider when
designing novel immunotherapies for cancer treatment (59).
HLA expression is crucial for the generation of
adaptive immunity, as tumor antigens are presented in an
HLA-restricted manner to T-cells, activating them and controlling
immune crosstalk (60). Altered HLA
expression on the tumor cell surface has been described in a
variety of human tumors, with percentages ranging from 60–90%
expression in different human tumor types (4,61). These
alterations result in different HLA-altered phenotypes, including
the neo-expression of non-classical HLA-I molecules like HLA-G,
which primarily function as inhibitor ligands for immune-competent
cells (6,7), and HLA-E, which together with HLA-G and
IL-10, is associated with the evasion and progression capacities in
tumor entities including lip squamous cell carcinoma (62). HLA-G and HLA-E exhibit limited
polymorphism, low cell surface expression and restricted tissue
distribution (63). They exert
several immune regulatory functions: HLA-G has immuno-tolerogenic
properties and inhibits CTL and NK cell lytic functions (64), whereas HLA-E may act as an
immuno-tolerogenic or immuno-activating molecule depending on the
NK cell receptor it is attached to. HLA-G inhibits immune cells
from binding to ILT2, ILT4 and KIR2DL4 receptors (65,66),
whereas HLA-E is the major ligand required for the inhibitory NK
cell receptors CD94/NKG2A and CD94/NKG2B expressed in NKs and CTLs
to produce immune tolerance, but also for the CD94/NKG2C activating
receptor expressed on NK cells and cytotoxic T-cells to support
their cytotoxic activity (67,68). Thus,
due to the pivotal immune function of HLA molecules, alterations in
their expression may be the most common evasion mechanism used by
tumor cells to avoid immune responses (39).
It is possible to classify HLA-altered tumor cell
phenotypes into two main groups: Reversible regulatory or
irreversible structural defects. Reversible HLA class I regulatory
defects may occur at any step of synthesis, assembly, transport
and/or molecular surface expression, and are caused by genetic,
epigenetic, transcriptional or post-transcriptional events,
resulting in regulatory abnormalities that it is possible to
recover with cytokine treatment. In contrast, structural defects
caused by mutation events that disrupt HLA-I heavy chain and
β2 microglobulin (β2m) genes are irreversible
(69).
In cancer, HLA class I loss occurs frequently and is
predominantly caused by genetic aberrations in chromosomes 6p21.3
and 15q21 (70). It has been reported
that at least 50% of multiple HLA allele loss is caused by LOH,
which is a frequent mechanism for HLA haplotype loss in various
types of human tumor (71).
Irreversible total HLA-I loss frequently occurs due to the
coincidence of two molecular events: Mutation of one β2m
gene, and the loss of the second copy by LOH. This alteration has
been described in various types of malignancy (72).
In cervical cancer, HLA-I downregulation occurs
early in tumor development and is associated with HLA-G
upregulation. The majority of HLA-G+ tumors also
expresses IL-10, thus suggesting the involvement of IL-10 in the
generation of an immunosuppressive environment, by downregulating
classical HLA-I and upregulating HLA-G expression (73). The HLA-G primary transcript generates
seven different protein isoforms by alternative splicing, including
four membrane-bound isoforms, HLA-G1, G2, G3 and G4, and three
soluble (s)HLA-G5, G6 and G7 isoforms (64). The soluble forms are secreted as free
soluble HLA-G molecules (sHLA-Gfree) or in extracellular vesicles
(sHLA-Gev), enabling tumors to inhibit virtually all immune cells
(Fig. 3) (74,75).
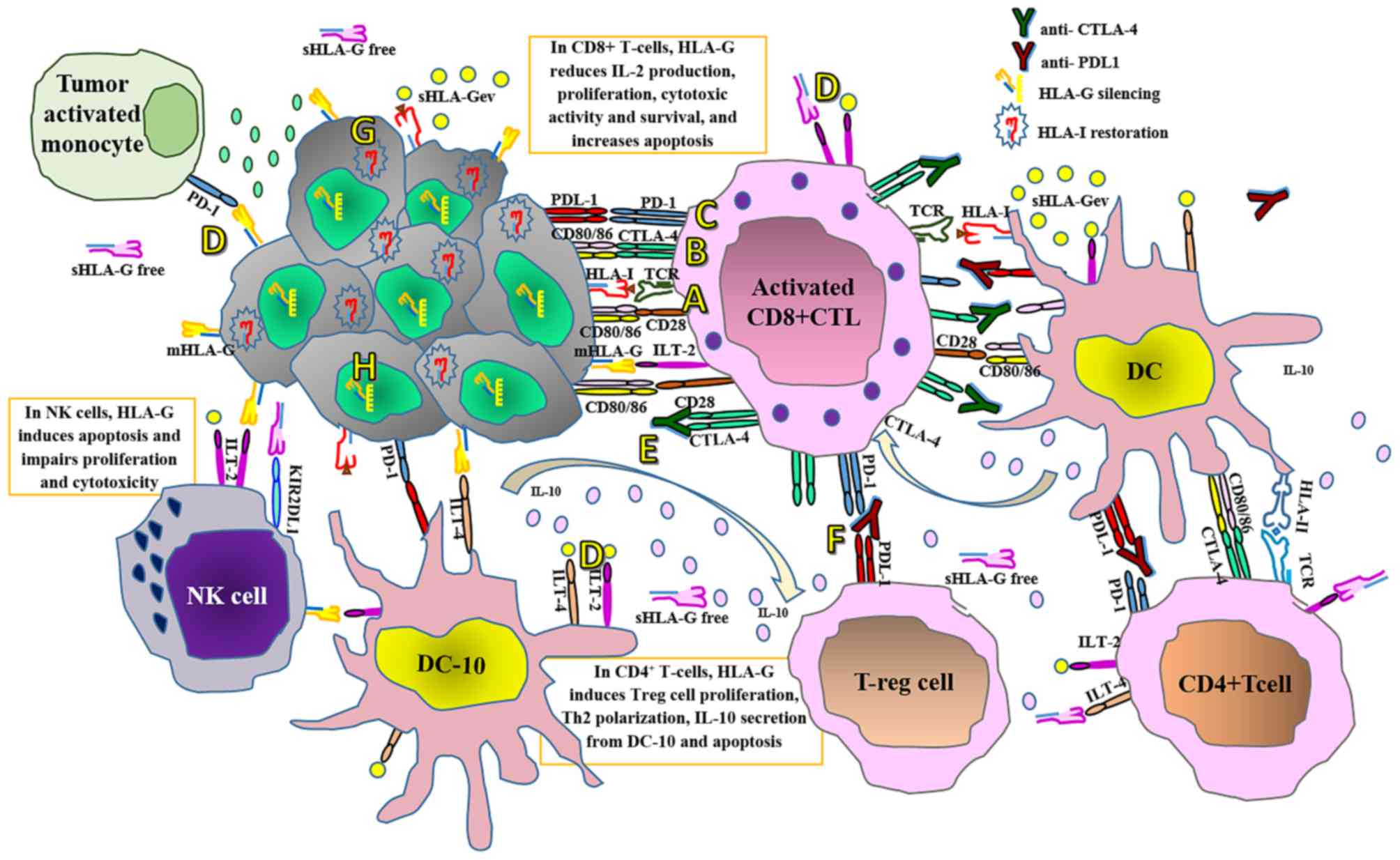 | Figure 3.Current immune checkpoint blockade
therapies and proposed adjuvant therapies for personalized cancer
treatment. (A) T-cells are activated when TCRs bind antigens in a
major histocompatibility complex-restricted manner on antigen
presenting cells, in concert with CD28-CD80/CD86 mediated
co-stimulation. (B) At the tumor site following T-cell activation,
CTLA-4 is translocated on the T-cell surface and competes with CD28
for binding the CD80/CD86 ligands. This interaction delivers an
inhibitory signal, which abrogates T-cell activation and
proliferation. (C) Tumor cells express PDL1 and when this interacts
with PD1 expressed by T-cells and other immune cells, it interferes
with several T-cell signaling pathways that promote the induction
of T-cell anergy, impairing the lytic capacity of T-cells on tumor
cells at the HLA-I antigen-presenting stage. However, PD1 and
CTLA-4 expression depend on T-cell activation that, in turn,
depends on antigen recognition in an HLA-I-restricted manner. (D)
On the other hand, interactions of membrane-bound and soluble HLA-G
isoforms with their specific inhibitory receptors expressed by
immune cells, including ILT-2, ILT-4 and KIR2DL4, impairs virtually
all antitumor immune responses. In contrast to PD1 and CTLA-4,
HLA-G expression does not require T-cell activation. (E) Thus,
although therapy with anti-CTLA-4 monoclonal antibodies impairs the
immunosuppressive CTLA-4 signal, promoting interactions between
CD80/CD86 and CD28 and keeping T-cells activated, and (F) anti-PDL1
therapy may restore the activity of antitumor T-cells that have
become quiescent, (G) tumor cells bearing defective HLA-I
expression may be refractory to these therapeutic approaches.
Targeting the aberrant HLA-I expression at the tumor cell surface
may improve the clinical efficacy of these approaches, and (H)
silencing HLA-G expression or blocking the inhibitory HLA-G
receptors on immune cells may prevent inhibitory signaling and
restore the effector antitumor capacity of immune cells. TCR,
T-cell receptor; CD, cluster of differentiation; CTLA-4, cytotoxic
T-lymphocyte-associated protein 4; PDL1, programmed cell death
ligand 1; PD1, programmed cell death protein 1; HLA, human
leukocyte antigen; ILT, immunoglobulin-like transcript; KIRD2L4,
killer cell immunoglobulin-like receptor, 2 immunoglobulin domains
and long cytoplasmic tail 4; DC-10, interleukin-10-secreting
dendritic cells; DC, dendritic cells; IL, interleukin; mHLA-G,
membrane-bound human leukocyte antigen-G; NK, natural killer;
sHLA-Gev, extracellular vesicle-associated soluble human leukocyte
antigen-G; sHLA-Gfree, free soluble human leukocyte antigen G; Th,
T helper cells; T-reg, regulatory T-cells. |
HLA-G expression by the tumor prevents immune
responses by a variety of strategies, including the prevention of
cell lysis by CTLs and NK cells, the induction of tolerant myeloid
DCs, and the induction of anergic or immunosuppressive
CD4+ and CD8+ T-cells (76). It has also been demonstrated that NK
cells may acquire an immune-suppressive phenotype through
HLA-G+ tumor cell trogocytosis (77) and that sHLA-G exerts immunosuppressive
functions by inducing apoptosis mediated by Fas cell surface death
receptor/Fas ligand in circulating antigen specific T-cells
(32). Furthermore, different sHLA-G
subcomponents exhibit different prognostic impacts on the clinical
outcome of patients with breast cancer treated with neo-adjuvant
chemotherapy (NACT): High levels of sHLA-Gev prior to NACT were
associated with disease progression and stem cell-like circulating
tumor cells, whereas high sHLAGfree levels were associated with
improved clinical outcome. However, total sHLA-G levels, without
considering sHLA-Gfree and sHLA-Gev subcomponents, were not
associated with clinical parameters (65,78).
HLA-G1 and HLA-G5 are the full-length membrane-bound and soluble
isoforms, respectively, and require peptide association for their
correct expression, whereas the other membrane-bound and soluble
isoforms have low stability and have different in vivo
functional activities (9,79).
HLA-II alterations also function in immune escape by
impairing the antigen-presenting capability of peripheral blood
monocytes in patients with acute leukemia (80). An association between HLA-II variants
and breast cancer susceptibility has been suggested in Chinese
breast cancer patients. In this population, HLA-II variants may be
associated with prognosis: The expression of HLA-DQB1 may indicate
a poor prognosis, whereas HLA-DRB5 may be associated with a good
prognosis (81). In addition,
aberrant expression of HLA-DRB1 and HLA-DQB1, which may occur due
to aberrant gene methylation, serves key functions in the
pathogenesis of esophageal squamous cell carcinoma (ESCC), by
influencing immune response to specific tumor epitopes and by
promoting ESCC occurrence and progression (82). Furthermore, in the population of
Guangdong, China, the occurrence of certain HLA-II alleles,
including DPB1*1301, DPB1*0202, DQB1*030302, and DQB1*050301,
occurred with higher frequency in patients with cervical cancer
than in controls, suggesting that they may confer susceptibility to
cervical cancer. On the other hand, the occurrence of the
DRB1*13-DQB1*06 haplotype was significantly lower in patients with
cervical cancer compared with controls, suggesting that this
haplotype may confer a decreased risk of cervical cancer within
this population (83).
Novel immunotherapeutic approaches against
cancer
Current therapies against cancer include
chemotherapy (84), radiation therapy
(85), immunotherapies (86), biological therapies and targeted
therapies. Therapeutic schemes currently in clinical trials include
cryosurgery, hyperthermia and cancer vaccines designed to prevent
(prophylactic) or treat (therapeutic) cancer (84). A large volume of research is being
produced concerning strategies to induce antitumor immunity,
including via innate and adaptive effector mechanisms. The blockade
of immune checkpoints may trigger the antitumor immune response,
while co-stimulatory receptor agonists and inhibitory signals
antagonists may induce antigen-specific T-cell response
amplification, potentially transforming human cancer therapeutics
(24). Currently, a range of
therapeutic agents that exploit this mechanism are in clinical
trials (Table I).
 | Table I.Molecular therapies targeting immune
regulation in cancer. |
Table I.
Molecular therapies targeting immune
regulation in cancer.
| Therapy | Mode of action | Limitations | Target/reagent
type | Indications | PMID |
|---|
| Antibodies |
|
|
|
|
|
|
Trastuzumab | Highly selective
agonism or blockade of extracellular protein-protein immune
pathways. | Expensive and
time-consuming manufacturing and development costs; challenges in
achieving high tumor exposure | HER2 | HER2-positive
breast cancer, HER2-positive advanced gastric cancer | 27526299 |
|
|
|
|
|
| 10211534 |
|
|
|
|
|
| 16328600 |
|
Bevacizumab | Long half-life,
non-immunogenic, includes human or humanized vaccine agonists
(targets include gp100, mucin 1 and MAGE family member A) |
| Vascular
endothelial growth factor | Non-small-cell lung
cancer, colorectal cancer, breast cancer | 18565863 |
|
|
|
|
|
| 26257518 |
|
Cetuximab |
|
| EGFR | Colorectal cancer
and head and neck cancer | 27446583 |
|
|
|
|
|
| 27511844 |
|
|
|
|
|
| 27465221 |
|
Panitumumab |
|
| EGFR | Colorectal
cancer | 27438067 |
|
|
|
|
|
| 27354619 |
|
Rituximab |
|
| CD20 B-cell surface
antigen | Primary mediastinal
B-cell lymphoma, non-Hodgkin's B-cell lymphoma | 27477167 |
|
|
|
|
|
| 27479818 |
|
|
|
|
|
| 27497027 |
|
Ibritumomab tiuxetan |
|
|
|
|
|
|
Alemtuzumab |
|
| CD52 lymphocyte
surface antigen | Refractory chronic
lymphocytic leukemia, T-cell lymphoma | 26489498 |
|
|
|
|
|
| 26201283 |
|
Gemtuzumab ozogamicin |
|
| CD33 leukemic-cell
surface antigen linked to calicheamicin | Acute myeloid
leukemia | 11970767 |
| CT-011
(humanized immunoglobulin G1) |
|
| PD1 | Advanced
hematologic malignancies | 18483370 |
|
Tositumomab |
|
| CD20 B-cell surface
antigen | Non-Hodgkin's
B-cell lymphoma, diffuse large B-cell lymphoma | 26832194 |
|
|
|
|
|
| 26257518 |
|
Ipilimumab Tremelimumab
(CP-675,206) |
|
| CTLA-4 | Metastatic melanoma
Metastatic melanoma, mesothelioma renal cell carcinoma, breast
cancer. | 18838703 |
|
|
|
|
|
| 19052265 |
|
|
|
|
|
| 27042127 |
|
Nivolumab |
|
| PD1 | Advanced
melanoma | 27093328 |
|
|
|
|
|
| 27013881 |
|
|
|
|
|
| 27099755 |
|
Pembrolizumab |
|
|
| Advanced melanoma,
metastatic renal carcinoma |
|
| Recombinant
cytokines |
|
|
|
|
|
|
Denileukin diftitox | Agonism or blockade
of protein-protein immune pathways. (granulocyte-macrophage colony
stimulating factor, IL-7, −12, −15, −18 and −21) | Antigenicity, poor
pharmacokinetics, high toxicity | Recombinant IL-2
and fragments of diphtheria toxin (binds CD25R on T-cells) | Cutaneous T-cell
lymphoma | 26240767 |
|
Aldesleukin |
|
| IL-2 | Melanoma,
renal-cell carcinoma | 27471714 |
|
|
|
|
|
| 25424850 |
|
Interferon α-2a and b |
|
| Recombinant
interferon | Hairy-cell
leukemia, chronic lymphocytic leukemia, Kaposi's sarcoma, melanoma,
non-Hodgkin's lymphoma, multiple myeloma, renal cancer | 14965794 |
|
|
|
|
|
| 26601863 |
|
|
|
|
|
| 7680399 |
| Small
molecules |
|
|
|
|
|
|
Imiquimod | Uniquely suited for
intracellular targets, but equally applicable to cell surface or
extracellular targets | Off-target
activities, dose-limiting toxicities, ineffective at blocking
protein-protein interactions, require daily doses | Toll-like receptor
7 agonist | Basal-cell
carcinoma | 26450707 |
|
Imatinib, nilotinib or
dasatinib |
|
| Abl proto-oncogene,
PDGFR, KIT proto-oncogene | Chronic myeloid
leukemia, gastrointestinal stromal tumors, metastatic chordoma,
chemoresistant Kaposi's sarcoma | 26180502 |
|
|
|
|
|
| 27231512 |
|
|
|
|
|
| 17032555 |
|
|
|
|
|
| 26628884 |
|
|
|
|
|
| 26796903 |
| Gefitinib |
|
| EGFR | Non-small cell lung
cancer | 27212579 |
|
Erlotinib |
|
| EGFR | Non-small cell lung
cancer, advanced pancreatic cancer | 12882624 |
|
|
|
|
|
| 27401642 |
|
Sunitinib |
|
| VEGFR, PDGFR,
FLT3 | Gastrointestinal
stromal tumors, renal cell carcinoma, pancreatic cancer | 15639298 |
|
|
|
|
|
| 27374084 |
|
Sorafenib |
|
| VEGFR, PDGFR,
FLT3 | Clear renal cell
carcinoma, hepatocellular carcinoma | 16425993 |
|
|
|
|
|
| 27487101 |
Although novel immunotherapeutic approaches with a
number of different molecular targets and modes of action are
currently in development, obstacles including difficulties in
immunological monitoring, poor clinical trial design and the
absence of cancer vaccine regulation make it difficult to achieve
an adequate evaluation of effective immune responses following
antitumor therapy. It is possible to overcome these obstacles by
improving patient selection, using combined therapies to impact
several immune signaling pathways simultaneously, identifying novel
biomarkers to evaluate clinical responses and coordinating
immunological monitoring for clinical trials. These improvements
must be achieved prior to successful clinical translation (87). Combined therapies use checkpoint
inhibitors as immunological adjuvants to boost cancer immunotherapy
and vaccines. The inhibition of signaling pathways, including
vascular endothelial growth factor to inhibit angiogenesis,
epidermal growth factor receptor to inhibit proliferating signals
or telomerase to interfere with replicative immortality enablement,
are examples of targeting the hallmarks of cancer (1). Cytokines have potential therapeutic and
preventive applications, but the associated systemic toxicity
limits their use in treating cancer. To overcome this problem,
novel recombinant antibody-cytokine fusion proteins have been
designed to maximize cytokine therapy efficacy by exploiting the
specific tumor-targeting ability of monoclonal antibodies (mAb) and
the immune stimulatory ability of cytokines, to induce antitumor
immune responses while preventing systemic toxicities of cytokine
therapy alone (88).
The blockade of immune checkpoints using human
immune-modulatory mAbs are in preclinical and clinical development.
These mAbs target immune system components rather than the tumor
itself, thus resulting in different responses to anti-CTLA-4
therapy (89), compared with
conventional antitumor mAbs, chemotherapies and immunotherapies
(including vaccines and cytokines) in terms of the pattern of
response; such as the response time to the therapy, duration of
response and adverse event profile (90). Another benefit of anti-immune
checkpoint mAb therapy is that it is possible to use it to treat a
variety of malignancies (91),
including hepatocellular carcinoma, which constitutes a significant
challenge for conventional cancer immunotherapy, as the unique
immune response in the liver favors immune tolerance, impairing the
therapeutic action of immunotherapy (92). The clinical use of these drugs induces
immune-related adverse events, including rashes, colitis,
thyroiditis and hepatitis, and clinical management for these
symptoms typically consists of treatment discontinuation or
symptomatic management with steroids or other immunosuppressive
agents (93).
Activation of the immune system through CTLA-4, PD1
and programmed cell death ligand 1 (PDL1) immune checkpoint
blocking is a promising cancer therapy strategy (91,94). As
the clinical success of targeting PD1/PDL1 or CTLA-4 depends on
blocking the regulatory activities of the receptor-ligand
interactions, it is important to consider that CTLA-4, PD1 and PDL1
expression depend on TCR activation, and that HLA-I expression on
cancer cell surfaces is a prerequisite for a successful T-cell
activation. Thus, lack of HLA-I expression by tumor cells has a
major effect on tumor recognition and the further activation of
T-cells, which remain unstimulated and incapable of recognizing
cancer cells. In this scenario, anti-CTLA-4, -PD1 and -PDL1
therapies would not work (Fig. 3)
(95). Thus, HLA status on the tumor
cell surface must first be assured to determine the suitability of
an immunotherapy treatment based on T-cell activation following TAA
recognition.
Cancer immunotherapy strategies to activate
T-cell-mediated antitumor responses include the use of antibodies
to target inhibitory molecules that impair T-cell cytotoxicity
(25), adoptive cell transfer with
tumor infiltrating lymphocytes expanded in vitro (96), or genetically modified cytotoxic
T-cells (97,98). However, CD8+ T-cells have
been established to recognize and destroy HLA-I positive tumor
cells. As human cancers are frequently characterized by alterations
in HLA-I expression, attempts to treat cancer by increasing the
CD8+ T-cell response will be unsuccessful in patients
harboring tumors with negative or deficient HLA-I expression. Thus,
a requirement for achieving successful clinical responses following
administration of T-cell activation based immunotherapy is, again,
to verify whether these important molecules for T-cell cytotoxicity
are correctly expressed by cancer cells. Such expression would be
an appropriate predictive biomarker to determine which patients
should enter into these treatment schemes (84). Other patients may first require a
neo-adjuvant scheme to restore normal HLA expression on cancer
cells prior to the utilization of T-cell activation or
immune-checkpoint blockade-based immunotherapies.
A range of immunotherapeutic schemes have been
designed to modify the tumor microenvironment to improve the
response to therapy in patients with cancer; however, restoration
of normal HLA-I expression in cancer cells is of pivotal importance
to ensure the immunogenicity of these schemes. Current strategies
to restore normal HLA-I expression work well only when the
molecular mechanism mediating HLA-I downregulation is reversible,
as when HLA-I downregulation is due to heavy chain structural
defects its expression is difficult to correct. Adenovirus-mediated
gene transfer may be a powerful strategy to correct HLA expression,
as human β2m gene transfer to tumor cells negative for
HLA-I following β2m structural alteration has been
demonstrated to restore HLA-I expression on tumor cell surface
(99). The restoration of HLA
expression reestablishes tumor cell immunogenicity, thereby
inducing T-cell activation in a peptide-specific, HLA-restricted
manner, suggesting that gene transfer of the β2m gene
may be a suitable neo-adjuvant therapy prior to T-cell
activation-based immunotherapy in the patients harboring tumors
negative for HLA-I due to β2m structural alteration
(100). Table I further summarizes the main molecular
therapies targeting immune regulation in cancer.
Conclusions
The benefit of conventional therapies is often
limited by collateral damage to normal tissues. Radiotherapy
induces massive cell death and chemotherapy toxicity is directed
against all actively proliferating cells. During massive cell
death, CD8+ T-cells specific for tumor antigens undergo
repeated TCR stimulation due to the persistence of TAAs.
Chronically stimulated T-cells gradually lose their ability to
secrete IL-2, tumor necrosis factor-α and interferon-γ, and are
finally eliminated by apoptosis in a process known as T-cell
exhaustion, which is characterized by the overexpression of
inhibitory receptors (101). PD1,
CTLA-4, lymphocyte-activation gene 3, T-cell Ig and mucin domain-3
and T-cell immunoreceptor with Ig and ITIM domains, among others
(102), dampen the stimulation of an
effective antitumor immune response by immunotherapeutic drugs.
However, in patients with an exhausted immune system, blocking of
these receptors leads to T-cell activation, suggesting that the
restoration of a non-exhausted immune context may improve immune
activation. Previous results have indicated that T-cell exhaustion
is reversible, which may have profound implications for cancer
treatment (103).
Novel immune-based therapies for cancer include
adoptive cell therapy, tumor vaccines, cytokines or the inhibition
of immune suppressive mechanisms including with immune checkpoint
inhibitors, and the depletion of T-regs or MDSCs. The search for
targets for the design and improvement of novel therapies should
include the search for biomarkers to measure therapeutic activity
and evaluate potential synergy among different immune-therapeutic
modalities (104). However, as a
large number of signaling pathways are typically associated with
carcinogenesis, it is probable that a single therapeutic agent
inhibiting one molecular target in a given tumor will not be
sufficient to eradicate the entire tumor mass. Advances in
knowledge of antitumor immune responses have been facilitated by
the development of targeted therapies for cancer control, including
anti-CTLA-4 antibodies, the therapeutic success of which suggests
that immunotherapy may achieve long-lasting and durable antitumor
immune responses in patients with cancer (105).
Blocking immune checkpoints may restore immune
function in certain scenarios, depending on the HLA phenotype. In
tumors with normal HLA-I expression, inhibitors of PD1 or
anti-CTLA-4 mAbs function as PD1 and CTLA-4 expression depend on
T-cell activation that, in turn, is dependent on HLA-restricted
antigen recognition. Therefore, tumors bearing defective HLA-I
expression may be refractory to these therapies due to their
inability to present TAAs to CTLs. Reestablishment of normal HLA
expression on the tumor cell surface by gene therapy may improve
the clinical impact of anti-CTLA-4 and PD1 immunotherapies and
restoring HLA-I expression may be an adjuvant therapy not only for
TCR-stimulation-based immunotherapies, but also therapy based on
checkpoint blocking. The combination of immunotherapy with
conventional therapy, for example chemotherapy, has been
demonstrated to produce a significant increase in the clinical
response of patients with cancer, despite the toxicity caused by
chemotherapy to immune system cells (106–108).
Another important factor associated with HLA is the
aberrant expression of HLA-G, as most tumors neo-express HLA-G at
various stages of their evolution and HLA-G neo-expression
deactivates all antitumor immune responses. As plasmatic (free or
vesicular) and membrane-bound (m)HLA-G expression is significantly
increased in most cancer types and associated with poor prognosis,
it is possible to use the levels of membrane-bound human leukocyte
antigen-G on tumor and immune cells and/or sHLA-G (free or as part
of extracellular vesicles) in plasma as diagnostic and prognostic
tools in cancer patients. Furthermore, HLA-G may also serve as a
therapeutic target for blocking mAbs or interfering RNAs (109).
HLA-G expression, in contrast with CTLA-4 and PD1,
does not depend on T-cell activation and is capable of blocking
antitumor immune responses by inhibiting all immune effectors, from
APC activation to effector priming, as well as blocking activated
CTL and NK cell function. Taking into account all the scientific
evidence concerning the function of HLA loss of expression, it is
possible to speculate that a cancer therapy targeting aberrant
HLA-I expression would restore T-cell recognition of tumor cells
and thus improve the clinical response to immunotherapies based on
CTLA-4, PD1 and PDL1 expression. In addition, silencing of HLA-G
expression or blocking the inhibitory ILT-2/4 receptors on immune
cells may prevent inhibitory signaling and restore the antitumor
effector capacity of immune cells. It may be possible to extend
HLA-based clinical applications to the design of promising tools
not only for diagnostic application to improve immunotherapeutic
management of the disease, but also as prognostic markers for the
clinical outcome of therapies, including NACT. A variety of studies
focused on targeted therapies to modify the immunoregulatory nature
of the tumor microenvironment are underway, and perhaps in the
future, early diagnosis will allow early immunotherapeutic
treatment, thus leading to improved survival rates.
Acknowledgements
The author sincerely thanks Dr Laura Sanchez, Dr
Silvia Serrano, Dr Alba Lucia Combita and Dr Nataly Cruz from the
Cancer Biology Research Group at the National Cancer Institute of
Colombia in Bogotá, Colombia, for their careful review of the
manuscript. The present study received financial support through an
INC/DNP grant (grant no. 41030610-109).
Glossary
Abbreviations
Abbreviations:
|
APC
|
antigen-presenting cells
|
|
β2m
|
β2 microglobulin
|
|
BMDC
|
bone marrow derived cells
|
|
CD
|
cluster of differentiation
|
|
CTL
|
cytotoxic T-cell
|
|
CTLA-4
|
cytotoxic T-lymphocyte-associated
protein 4
|
|
DC
|
dendritic cells
|
|
ESCC
|
esophageal squamous cell carcinoma
|
|
HLA
|
human leukocyte antigen
|
|
Ig
|
immunoglobulin
|
|
IL
|
interleukin
|
|
ILT
|
immunoglobulin-like transcript
|
|
KIR2DL4
|
killer cell immunoglobulin-like
receptor, 2 immunoglobulin domains and long cytoplasmic tail 4
|
|
LOH
|
loss of heterozygosity
|
|
mAb
|
monoclonal antibody
|
|
MDSC
|
myeloid derived suppressor cells
|
|
mHLA-G
|
membrane-bound human leukocyte
antigen-G
|
|
NACT
|
neo-adjuvant chemotherapy
|
|
NK
|
natural killer
|
|
NKG
|
natural killer group
|
|
PD1
|
programmed cell death protein 1
|
|
PDL1
|
programmed cell death ligand 1
|
|
sHLA-G
|
soluble human leukocyte antigen-G
|
|
sHLA-Gev
|
extracellular vesicle-associated
soluble human leukocyte antigen-G
|
|
sHLA-Gfree
|
free soluble human leukocyte antigen
G
|
|
TAAs
|
tumor associated antigens
|
|
TAM
|
tumor-associated macrophages
|
|
TCR
|
T-cell receptor
|
|
Th
|
T helper cells
|
|
T-reg
|
regulatory T-cells
|
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grivennikov SI, Greten FR and Karin M:
Immunity, Inflammation and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Van den Boorn JG and Hartmann G: Turning
tumors into vaccines: Co-opting the innate immune system. Immunity.
39:27–37. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Campoli M and Ferrone S: HLA antigen
changes in malignant cells: Epigenetic mechanisms and biologic
significance. Oncogene. 27:5869–5885. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chang CC, Campoli M and Ferrone S:
Classical and nonclassical HLA class I antigen and NK
Cell-activating ligand changes in malignant cells: Current
challenges and future directions. Adv Cancer Res. 93:189–234. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Garrido F, Cabrera T, Concha A, Glew S,
Ruiz-Cabello F and Stern PL: Natural history of HLA expression
during tumour development. Immunol Today. 14:491–499. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garrido F, Ruiz-Cabello F, Cabrera T,
Pérez-Villar JJ, López-Botet M, Duggan-Keen M and Stern PL:
Implications for immunosurveillance of altered HLA class I
phenotypes in human tumours. Immunol Today. 18:89–95. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koopman LA, Corver WE, van der Slik AR,
Giphart MJ and Fleuren GJ: Multiple genetic alterations cause
frequent and heterogeneous human histocompatibility leukocyte
antigen class I loss in cervical cancer. J Exp Med. 191:961–976.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moreau P, Rousseau P, Rouas-Freiss N, Le
Discorde M, Dausset J and Carosella ED: HLA-G protein processing
and transport to the cell surface. Cell Mol Life Sci. 59:1460–1466.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Adams JL, Smothers J, Srinivasan R and
Hoos A: Big opportunities for small molecules in immuno-oncology.
Nat Rev Drug Discov. 14:603–622. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Srinivasan R and Wolchok JD: Tumor
antigens for cancer immunotherapy: Therapeutic potential of
xenogeneic DNA vaccines. J Transl Med. 2:122004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Waldhauer I and Steinle A: NK cells and
cancer immunosurveillance. Oncogene. 27:5932–5943. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jaeger BN and Vivier E: Natural killer
cell tolerance: Control by self or self-control? Cold Spring Harb
Perspect Biol. 4:pii: a007229. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pegram HJ, Andrews DM, Smyth MJ, Darcy PK
and Kershaw MH: Activating and inhibitory receptors of natural
killer cells. Immunol Cell Biol. 89:216–224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yokoyama WM and Kim S: Licensing of
natural killer cells by self-major histocompatibility complex class
I. Immunol Rev. 214:143–154. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clynes RA, Towers TL, Presta LG and
Ravetch JV: Inhibitory Fc receptors modulate in vivo cytotoxicity
against tumor targets. Nat Med. 6:443–496. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hayakawa Y and Smyth MJ: Innate immune
recognition and suppression of tumors. Adv Cancer Res. 95:293–322.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lennerz V, Fatho M, Gentilini C, Frye RA,
Lifke A, Ferel D, Wölfel C, Huber C and Wölfel T: The response of
autologous T cells to a human melanoma is dominated by mutated
neoantigens. Proc Natl Acad Sci USA. 102:16013–16018. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Perez-Diez A, Joncker NT, Choi K, Chan WF,
Anderson CC, Lantz O and Matzinger P: CD4 cells can be more
efficient at tumor rejection than CD8 cells. Blood. 109:5346–5354.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gotter J, Brors B, Hergenhahn M and
Kyewski B: Medullary epithelial cells of the human thymus express a
highly diverse selection of tissue-specific genes colocalized in
chromosomal clusters. J Exp Med. 199:155–166. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gerloni M and Zanetti M: CD4 T cells in
tumor immunity. Springer Semin Immunopathol. 27:37–48. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Palm NW and Medzhitov R: Pattern
recognition receptors and control of adaptive immunity. Immunol
Rev. 227:221–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Smith-Garvin JE, Koretzky GA and Jordan
MS: T cell activation. Annu Rev Immunol. 27:591–619. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pardoll DM: The blockade of immune
checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
de Coaña Pico Y, Choudhury A and Kiessling
R: Checkpoint blockade for cancer therapy: Revitalizing a
suppressed immune system. Trends Mol Med. 21:482–491. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carreno BM and Collins M: The B7 family of
ligands and its receptors: New pathways for costimulation and
inhibition of immune responses. Annu Rev Immunol. 20:29–53. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nishimura H, Okazaki T, Tanaka Y, Nakatani
K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N
and Honjo T: Autoimmune dilated cardiomyopathy in PD-1
receptor-deficient mice. Science. 291:319–322. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Waterhouse P, Penninger JM, Timms E,
Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H and Mak TW:
Lymphoproliferative disorders with early lethality in mice
deficient in Ctla-4. Science. 270:985–988. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chemnitz JM, Parry RV, Nichols KE, June CH
and Riley JL: SHP-1 and SHP-2 associate with immunoreceptor
tyrosine-based switch motif of programmed death 1 upon primary
human T cell stimulation, but only receptor ligation prevents T
cell activation. J Immunol. 173:945–954. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kirchhof MG, Chau LA, Lemke CD, Vardhana
S, Darlington PJ, Márquez ME, Taylor R, Rizkalla K, Blanca I,
Dustin ML and Madrenas J: Modulation of T cell activation by
stomatin-like protein 2. J Immunol. 181:1927–1936. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Teft WA, Kirchhof MG and Madrenas J: A
molecular perspective of CTLA-4 function. Annu Rev Immunol.
24:65–97. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Contini P, Ghio M, Poggi A, Filaci G,
Indiveri F, Ferrone S and Puppo F: Soluble HLA-A,-B,-C and -G
molecules induce apoptosis in T and NK CD8+ cells and inhibit
cytotoxic T cell activity through CD8 ligation. Eur J Immunol.
33:125–134. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Le Bouteiller P, Fons P, Herault JP, Bono
F, Chabot S, Cartwright JE and Bensussan A: Soluble HLA-G and
control of angiogenesis. J Reprod Immunol. 76:17–22. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Colonna M, Samaridis J, Cella M, Angman L,
Allen RL, O'Callaghan CA, Dunbar R, Ogg GS, Cerundolo V and Rolink
A: Human myelomonocytic cells express an inhibitory receptor for
classical and nonclassical MHC class I molecules. J Immunol.
160:3096–3100. 1998.PubMed/NCBI
|
|
35
|
Rajagopalan S and Long EO: A human
histocompatibility leukocyte antigen (HLA)-G-specific receptor
expressed on all natural killer cells. J Exp Med. 189:1093–1100.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pankratz S, Ruck T, Meuth SG and Wiendl H:
CD4(+)HLA-G(+) regulatory T cells: Molecular signature and
pathophysiological relevance. Hum Immunol. 77:727–733. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim R, Emi M and Tanabe K: Cancer
immunoediting from immune surveillance to immune escape.
Immunology. 121:1–14. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vesely MD, Kershaw MH, Schreiber RD and
Smyth MJ: Natural innate and adaptive immunity to cancer. Annu Rev
Immunol. 29:235–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Monjazeb AM, Zamora AE, Grossenbacher SK,
Mirsoian A, Sckisel GD and Murphy WJ: Immunoediting and antigen
loss: Overcoming the achilles heel of immunotherapy with antigen
non-specific therapies. Front Oncol. 3:1972013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Khong HT and Restifo NP: Natural selection
of tumor variants in the generation of ‘tumor escape’ phenotypes.
Nat Immunol. 3:999–1005. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Koebel CM, Vermi W, Swann JB, Zerafa N,
Rodig SJ, Old LJ, Smyth MJ and Schreiber RD: Adaptive immunity
maintains occult cancer in an equilibrium state. Nature.
450:903–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gunderson AJ and Coussens LM: B cells and
their mediators as targets for therapy in solid tumors. Exp Cell
Res. 319:1644–1649. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Esquivel-Velázquez M, Ostoa-Saloma P,
Palacios-Arreola MI, Nava-Castro KE, Castro JI and Morales-Montor
J: The role of cytokines in breast cancer development and
progression. J Interferon Cytokine Res. 35:1–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lippitz BE: Cytokine patterns in patients
with cancer: A systematic review. Lancet Oncol. 14:e218–e228. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gajewski TF, Meng Y, Blank C, Brown I,
Kacha A, Kline J and Harlin H: Immune resistance orchestrated by
the tumor microenvironment. Immunol Rev. 213:131–145. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gajewski TF, Fuertes M, Spaapen R, Zheng Y
and Kline J: Molecular profiling to identify relevant immune
resistance mechanisms in the tumor microenvironment. Curr Opin
Immunol. 23:286–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mahoney KM and Atkins MB: Prognostic and
predictive markers for the new immunotherapies. Oncology (Williston
Park). 28 Suppl 3:S39–S48. 2014.
|
|
48
|
Wong SC, Puaux AL, Chittezhath M, Shalova
I, Kajiji TS, Wang X, Abastado JP, Lam KP and Biswas SK: Macrophage
polarization to a unique phenotype driven by B cells. Eur J
Immunol. 40:2296–2307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Loi S, Sirtaine N, Piette F, Salgado R,
Viale G, Van Eenoo F, Rouas G, Francis P, Crown JP, Hitre E, et al:
Prognostic and predictive value of tumor-infiltrating lymphocytes
in a phase III randomized adjuvant breast cancer trial in
node-positive breast cancer comparing the addition of docetaxel to
doxorubicin with doxorubicin-based chemotherapy: BIG 02–98. J Clin
Oncol. 31:860–867. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tanchot C, Terme M, Pere H, Tran T,
Benhamouda N, Strioga M, Banissi C, Galluzzi L, Kroemer G and
Tartour E: Tumor-infiltrating regulatory T cells: Phenotype, role,
mechanism of expansion in situ and clinical significance. Cancer
Microenviron. 6:147–157. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Petersson M, Charo J, Salazar-Onfray F,
Noffz G, Mohaupt M, Qin Z, Klein G, Blankenstein T and Kiessling R:
Constitutive IL-10 production accounts for the high NK sensitivity,
low MHC class I expression, and poor transporter associated with
antigen processing (TAP)-1/2 function in the prototype NK target
YAC-1. J Immunol. 161:2099–2105. 1998.PubMed/NCBI
|
|
52
|
Salazar-Onfray F, Charo J, Petersson M,
Freland S, Noffz G, Qin Z, Blankenstein T, Ljunggren HG and
Kiessling R: Down-regulation of the expression and function of the
transporter associated with antigen processing in murine tumor cell
lines expressing IL-10. J Immunol. 159:3195–3202. 1997.PubMed/NCBI
|
|
53
|
Chen CJ, Sung WW, Su TC, Chen MK, Wu PR,
Yeh KT, Ko JL and Lee H: High expression of interleukin 10 might
predict poor prognosis in early stage oral squamous cell carcinoma
patients. Clin Chim Acta. 415:25–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Goncalves AS, Wastowski IJ, Capeletti LR,
Sacono NT, Cortez AP, Valadares MC, Silva TA and Batista AC: The
clinicopathologic significance of the expression of HLA-G in oral
squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral
Radiol. 117:361–368. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Levy EM, Bianchini M, Von Euw EM, Barrio
MM, Bravo AI, Furman D, Domenichini E, Macagno C, Pinsky V,
Zucchini C, et al: Human leukocyte antigen-E protein is
overexpressed in primary human colorectal cancer. Int J Oncol.
32:633–641. 2008.PubMed/NCBI
|
|
56
|
Gasparoto TH, de Souza Malaspina TS,
Damante JH, de Mello EF Jr, Ikoma MR, Garlet GP, Costa MR,
Cavassani KA, da Silva JS and Campanelli AP: Regulatory T cells in
the actinic cheilitis. J Oral Pathol Med. 43:754–760. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mendez R, Aptsiauri N, Del Campo A, Maleno
I, Cabrera T, Ruiz-Cabello F, Garrido F and Garcia-Lora A: HLA and
melanoma: Multiple alterations in HLA class I and II expression in
human melanoma cell lines from ESTDAB cell bank. Cancer Immunol
Immunother. 58:1507–1515. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ferns DM, Heeren AM, Samuels S, Bleeker
MC, de Gruijl TD, Kenter GG and Jordanova ES: Classical and
non-classical HLA class I aberrations in primary cervical squamous-
and adenocarcinomas and paired lymph node metastases. J Immunother
Cancer. 4:782016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Campoli M and Ferrone S: HLA antigen and
NK cell activating ligand expression in malignant cells: A story of
loss or acquisition. Semin Immunopathol. 33:321–334. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nilsson Lynge L, Djurisic S and Hviid TV:
Controlling the immunological crosstalk during conception and
pregnancy: HLA-G in reproduction. Front Immunol.
5:1982014.PubMed/NCBI
|
|
61
|
Cabrera T, López-Nevot MA, Gaforio JJ,
Ruiz-Cabello F and Garrido F: Analysis of HLA expression in human
tumor tissues. Cancer Immunol Immunother. 52:1–9. 2003.PubMed/NCBI
|
|
62
|
Goncalves AS, Oliveira JP, Oliveira CF,
Silva TA, Mendonca EF, Wastowski IJ and Batista AC: Relevance of
HLA-G, HLA-E and IL-10 expression in lip carcinogenesis. Hum
Immunol. 77:785–790. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Reimers MS, Engels CC, Putter H, Morreau
H, Liefers GJ, van de Velde CJ and Kuppen PJ: Prognostic value of
HLA class I, HLA-E, HLA-G and Tregs in rectal cancer: A
retrospective cohort study. BMC Cancer. 14:4862014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Rouas-Freiss N, Moreau P, Ferrone S and
Carosella ED: HLA-G proteins in cancer: Do they provide tumor cells
with an escape mechanism? Cancer Res. 65:10139–10144. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Carosella ED, Moreau P, Le Maoult J, Le
Discorde M, Dausset J and Rouas-Freiss N: HLA-G molecules: From
maternal-fetal tolerance to tissue acceptance. Adv Immunol.
81:199–252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
LeMaoult J, Zafaranloo K, Le Danff C and
Carosella ED: HLA-G up-regulates ILT2, ILT3, ILT4, and KIR2DL4 in
antigen presenting cells, NK cells, and T cells. FASEB J.
19:662–664. 2005.PubMed/NCBI
|
|
67
|
Braud VM, Allan DS, O'Callaghan CA,
Söderström K, D'Andrea A, Ogg GS, Lazetic S, Young NT, Bell JI,
Phillips JH, et al: HLA-E binds to natural killer cell receptors
CD94/NKG2A, B and C. Nature. 391:795–799. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Braud VM, Aldemir H, Breart B and Ferlin
WG: Expression of CD94-NKG2A inhibitory receptor is restricted to a
subset of CD8+ T cells. Trends Immunol. 24:162–164. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Garrido F, Cabrera T and Aptsiauri N:
“Hard” and “soft” lesions underlying the HLA class I alterations in
cancer cells: Implications for immunotherapy. Int J Cancer.
127:249–256. 2010.PubMed/NCBI
|
|
70
|
Vermeulen CF, Jordanova ES,
Zomerdijk-Nooijen YA, ter Haar NT, Peters AA and Fleuren GJ:
Frequent HLA class I loss is an early event in cervical
carcinogenesis. Hum Immunol. 66:1167–1173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Brady CS, Bartholomew JS, Burt DJ,
Duggan-Keen MF, Glenville S, Telford N, Little AM, Davidson JA,
Jimenez P, Ruiz-Cabello F, et al: Multiple mechanisms underlie HLA
dysregulation in cervical cancer. Tissue Antigens. 55:401–411.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Maleno I, Aptsiauri N, Cabrera T, Gallego
A, Paschen A, López-Nevot MA and Garrido F: Frequent loss of
heterozygosity in the β2-microglobulin region of chromosome 15 in
primary human tumors. Immunogenetics. 63:65–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rodriguez JA, Galeano L, Palacios DM,
Gómez C, Serrano ML, Bravo MM and Combita AL: Altered HLA class I
and HLA-G expression is associated with IL-10 expression in
patients with cervical cancer. Pathobiology. 79:72–83. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Carosella ED, Rouas-Freiss N, Tronik-Le
Roux D, Moreau P and LeMaoult J: HLA-G: An immune checkpoint
molecule. Adv Immunol. 127:33–144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Seliger B, Ritz U and Ferrone S: Molecular
mechanisms of HLA class I antigen abnormalities following viral
infection and transformation. Int J Cancer. 118:129–138. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ristich V, Liang S, Zhang W, Wu J and
Horuzsko A: Tolerization of dendritic cells by HLA-G. Eur J
Immunol. 35:1133–1142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Caumartin J, Favier B, Daouya M, Guillard
C, Moreau P, Carosella ED and LeMaoult J: Trogocytosis-based
generation of suppressive NK cells. EMBO J. 26:1423–1433. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
König L, Kasimir-Bauer S, Hoffmann O,
Bittner AK, Wagner B, Manvailer LF, Schramm S, Bankfalvi A, Giebel
B, Kimmig R, et al: The prognostic impact of soluble and vesicular
HLA-G and its relationship to circulating tumor cells in
neoadjuvant treated breast cancer patients. Hum Immunol.
77:791–799. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Bainbridge DR, Ellis SA and Sargent IL:
The short forms of HLA-G are unlikely to play a role in pregnancy
because they are not expressed at the cell surface. J Reprod
Immunol. 47:1–16. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Gong FL, Feng XW and Grosse-Wilde H:
Impaired antigen-presenting capability of monocytes correlated with
their decreased expression of HLA-II antigens in patients with
myeloid leukemia. J Tongji Med Univ. 13:65–70. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yang XX, Pan HZ, Li PY, Li FX, Xu WW, Wu
YS, Yao GY and Li M: HLA class II variants in Chinese breast cancer
patients. Asian Pac J Cancer Prev. 12:3075–3079. 2011.PubMed/NCBI
|
|
82
|
Hu JM, Li L, Chen YZ, Liu C, Cui X, Yin L,
Yang L, Zou H, Pang L, Zhao J, et al: HLA-DRB1 and HLA-DQB1
methylation changes promote the occurrence and progression of
Kazakh ESCC. Epigenetics. 9:1366–1373. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Liang J, Xu A, Xie Y, Awonuga AO and Lin
Z: Some but not all of HLA-II alleles are associated with cervical
cancer in Chinese women. Cancer Genet Cytogenet. 187:95–100. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
National Cancer Institute. Chemotherapy
and you U.S. Department of health and human services national
institutes of health. 2011.
|
|
85
|
Taylor A and Powell ME:
Intensity-modulated radiotherapy-what is it? Cancer Imaging.
4:68–73. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Galluzzi L, Vacchelli E, Bravo-San Pedro
JM, Buqué A, Senovilla L, Baracco EE, Bloy N, Castoldi F, Abastado
JP, Agostinis P, et al: Classification of current anticancer
immunotherapies. Oncotarget. 5:12472–12508. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Copier J, Dalgleish AG, Britten CM, Finke
LH, Gaudernack G, Gnjatic S, Kallen K, Kiessling R, Schuessler-Lenz
M, Singh H, et al: Improving the efficacy of cancer immunotherapy.
Eur J Cancer. 45:1424–1431. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Young PA, Morrison SL and Timmerman JM:
Antibody-cytokine fusion proteins for treatment of cancer:
Engineering cytokines for improved efficacy and safety. Semin
Oncol. 41:623–636. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Robert C, Thomas L, Bondarenko I, O'Day S,
Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, et al:
Ipilimumab plus dacarbazine for previously untreated metastatic
melanoma. N Engl J Med. 364:2517–2526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Attia P, Phan GQ, Maker AV, Robinson MR,
Quezado MM, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE,
et al: Autoimmunity correlates with tumor regression in patients
with metastatic melanoma treated with anti-cytotoxic T-lymphocyte
antigen-4. J Clin Oncol. 23:6043–6053. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Fong L and Small EJ: Anti-cytotoxic
T-lymphocyte antigen-4 antibody: The first in an emerging class of
immunomodulatory antibodies for cancer treatment. J Clin Oncol.
26:5275–5283. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Aerts M, Benteyn D, Van Vlierberghe H,
Thielemans K and Reynaert H: Current status and perspectives of
immune-based therapies for hepatocellular carcinoma. World J
Gastroenterol. 22:253–261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Boasberg P, Hamid O and O'Day S:
Ipilimumab: Unleashing the power of the immune system through
CTLA-4 blockade. Semin Oncol. 37:440–449. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ugurel S, Röhmel J, Ascierto PA, Flaherty
KT, Grob JJ, Hauschild A, Larkin J, Long GV, Lorigan P, McArthur
GA, et al: Survival of patients with advanced metastatic melanoma:
The impact of novel therapies. Eur J Cancer. 53:125–134. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Thor Straten P and Garrido F: Targetless T
cells in cancer immunotherapy. J Immunother Cancer. 4:232016.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Vavrova K, Vrabcova P, Filipp D,
Bartunkova J and Horvath R: Generation of T cell effectors using
tumor cell-loaded dendritic cells for adoptive T cell therapy. Med
Oncol. 33:1362016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Fesnak AD, June CH and Levine BL:
Engineered T cells: The promise and challenges of cancer
immunotherapy. Nat Rev Cancer. 16:566–581. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Redeker A and Arens R: Improving adoptive
T cell therapy: The particular role of t cell costimulation,
cytokines, and post-transfer vaccination. Front Immunol. 7:3452016.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Del Campo AB, Aptsiauri N, Méndez R,
Zinchenko S, Vales A, Paschen A, Ward S, Ruiz-Cabello F,
González-Aseguinolaza G and Garrido F: Efficient recovery of HLA
class I expression in human tumor cells after beta2-microglobulin
gene transfer using adenoviral vector: Implications for cancer
immunotherapy. Scand J Immunol. 70:125–135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Del Campo AB, Carretero J, Muñoz JA,
Zinchenko S, Ruiz-Cabello F, González-Aseguinolaza G, Garrido F and
Aptsiauri N: Adenovirus expressing β2-microglobulin recovers HLA
class I expression and antitumor immunity by increasing T-cell
recognition. Cancer Gene Ther. 21:317–332. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Penaloza-MacMaster P, Kamphorst AO,
Wieland A, Araki K, Iyer SS, West EE, O'Mara L, Yang S, Konieczny
BT, Sharpe AH, et al: Interplay between regulatory T cells and PD-1
in modulating T cell exhaustion and viral control during chronic
LCMV infection. J Exp Med. 211:1905–1918. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Wherry EJ: T cell exhaustion. Nat Immunol.
12:492–499. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Pauken KE and Wherry EJ: Overcoming T cell
exhaustion in infection and cancer. Trends Immunol. 36:265–276.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Page DB, Postow MA, Callahan MK, Allison
JP and Wolchok JD: Immune modulation in cancer with antibodies.
Annu Rev Med. 65:185–202. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Mellman I, Coukos G and Dranoff G: Cancer
immunotherapy comes of age. Nature. 480:480–489. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Okamoto M, Kobayashi M, Yonemitsu Y, Koido
S and Homma S: Dendritic cell-based vaccine for pancreatic cancer
in Japan. World J Gastrointest Pharmacol Ther. 7:133–138. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wang Z, Liu Y, Zhang Y, Shang Y and Gao Q:
MDSC-decreasing chemotherapy increases the efficacy of
cytokine-induced killer cell immunotherapy in metastatic renal cell
carcinoma and pancreatic cancer. Oncotarget. 7:4760–4769. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zhao X, Ji CY, Liu GQ, Ma DX, Ding HF, Xu
M and Xing J: Immunomodulatory effect of DC/CIK combined with
chemotherapy in multiple myeloma and the clinical efficacy. Int J
Clin Exp Pathol. 8:13146–13155. 2015.PubMed/NCBI
|
|
109
|
Amiot L, Ferrone S, Grosse-Wilde H and
Seliger B: Biology of HLA-G in cancer: A candidate molecule for
therapeutic intervention? Cell Mol Life Sci. 68:417–431. 2011.
View Article : Google Scholar : PubMed/NCBI
|














