Introduction
Human cytochrome P450 2J2 (CYP2J2) epoxygenase is an
enzyme that metabolizes arachidonic acid and linoleic acid to four
regioisomeric epoxyeicosatrienoic acids (EETs), namely 14,15-EET,
11,12-EET, 5,6-EET and 8,9-EET (1).
This enzyme is widely expressed in multiple human tissues,
including the liver, heart, lung, pancreas and bladder, and in
endothelial cells (2–4). CYP2J2 and its metabolites exert numerous
pathophysiological roles, including regulation of ion channel
activity in cardiomyocytes (5),
inhibition of inflammation (6),
inhibition of apoptosis (7) and
recovery of endothelial cells from hypoxic injury (8). CYP2J2 is also highly expressed in
various cancer cell lines and tissues, including hepatocellular
carcinoma (9). Elevated CYP2J2
messenger RNA and protein levels have been observed in diverse
human cancer cell lines and human cancer tissues (9). Several studies have implicated CYP2J2
and its EET metabolites in the pathological development of human
cancers, including solid tumors and hematological malignancies
(10–12). Additionally, overexpression of CYP2J2
and elevated EET levels promote tumor malignancy, while the
selective inhibition of CYP2J2 attenuates these effects (9,13).
However, the precise role of CYP2J2 in hepatocellular carcinoma
cells is still poorly understood.
Each year, hepatocellular carcinoma is diagnosed in
>500,000 people worldwide (14).
Liver cancer is the fifth most common cancer in men and the seventh
in women (14). Although its
incidence is highly variable in different geographic areas,
hepatocellular carcinoma is one of the leading causes of mortality
in the world, being among the most common cancers in both Eastern
and Western countries (15). A more
detailed understanding of the pathophysiological role of CYP2J2 may
lead to the development of new therapeutic strategies to alter the
pathogenesis of this disease (8,16).
Cancer cells often deregulate the cell cycle and
undergo uncontrolled cell proliferation (17). Almost all anticancer chemotherapy
strategies inhibit the proliferation of tumor cells by targeting
cell cycle mechanisms to arrest cells and induce apoptosis
(17,18). Additionally, the effectiveness of
chemotherapy is limited by drug resistance (19). The present study investigated the
potential role of CYP2J2 in cancer cell proliferation and drug
resistance in hepatocellular carcinoma HepG2 cells. The results
will provide a better understanding of the role of CYP2J2 in cancer
and will help to develop more effective anticancer treatment
strategies.
Materials and methods
Reagents
The HepG2 hepatocellular carcinoma cell line was
purchased from the Korean Cell Line Bank (Seoul, Korea). HEK 293T
cells were obtained from the American Type Culture Collection
(Manassas, VA, USA). Cell Counting kit (CCK)-8 was purchased from
Dojindo Molecular Technologies, Inc. (Kumamoto, Japan). X-tremeGENE
HP DNA Transfection Reagent was acquired from Roche Applied Science
(Penzberg, Germany). Human CYP2J2 complementary DNA
(cDNA)-containing plasmid, Trypan blue solution (0.4%) and
Pierce® ECL Western Blotting Substrate were acquired
from Thermo Fisher Scientific, Inc. (Waltham, MA, USA), while
Hyclone™ fetal bovine serum (FBS) was acquired from GE
Healthcare Life Sciences (Logan, UT, USA). Anti-cyclin D1 (cat. no.
sc-8396), anti-cyclin E (cat. no. sc-25303), anti-cyclin-dependent
kinase (Cdk)2 (cat. no. sc-6248), anti-Cdk4 (cat. no. sc-749),
anti-B-cell lymphoma (Bcl)-2 associated X protein (Bax) (cat. no.
sc-493), anti-Bcl-2 (cat. no. sc-7382), anti-caspase-3 (cat. no.
sc-373730), goat anti-rabbit immunoglobulin (Ig)G (cat. no.
sc-2004) and goat anti-mouse IgG (cat. no. sc-2005) antibodies were
supplied by Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Anti-phosphorylated-Akt (Ser473) (cat. no. 4060) and
anti-total Akt (cat. no. 4691) antibodies were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA).
Penicillin-Streptomycin Solution 100X, LY294002 and doxorubicin
hydrochloride were obtained from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany).
Cell culture
HepG2 cells were maintained in Dulbecco's modified
Eagle's medium (DMEM) high glucose (4.5 g/l; Thermo Fisher
Scientific Inc.), supplemented with 10% FBS and 1%
Penicillin-Streptomycin Solution 100X, at 37°C in a humidified
atmosphere (5% CO2). One day prior to the experiments,
the cells were incubated with fresh DMEM without FBS.
Establishment of a
CYP2J2-overexpressing stable HepG2 cell line
Lentiviral expression constructs containing cDNA
encoding human CYP2J2 were generated. This transgene cassette
employs the cytomegalovirus (CMV) immediate-early promoter
(20). The lentiviral vector
containing the puromycin resistance gene (CMV-eGFP-IRES-Puro
plasmid) and the packaging vectors (VSV-G expressing envelop
plasmid and another plasmid containing gag, pol and
rev genes) were kindly provided by Dr. Yibing Qyang (Yale
Cardiovascular Research Center, Yale School of Medicine, USA). In
brief, the CMV-CYP2J2-IRES-Puro lentiviral vector was generated by
substitution of eGFP sequence in CMV-eGFP-IRES-Puro plasmid with
CYP2J2 sequence. CMV-CYP2J2 and control CMV-GFP viruses were
generated by transfection of the lentiviral vectors
(CMV-CYP2J2-IRES-Puro or CMV-eGFP-IRES-Puro plasmids) and packaging
vectors into HEK 293T cells using X-tremeGene HP DNA transfection
reagent (Roche Applied Science, Penzburg, Germany) at 37°C and 5%
CO2 for 24 h. Virus-containing medium was collected
every day for 3 days following transfection, and concentrated by
ultracentrifugation at 55,200 × g and 4°C for 2 h (Hitachi, Ltd.,
Tokyo, Japan). HepG2 cells were exposed to concentrated
virus-containing medium for 24 h at 37°C, followed by 2 days of
culture in basal medium. Virus-infected cells were selected by
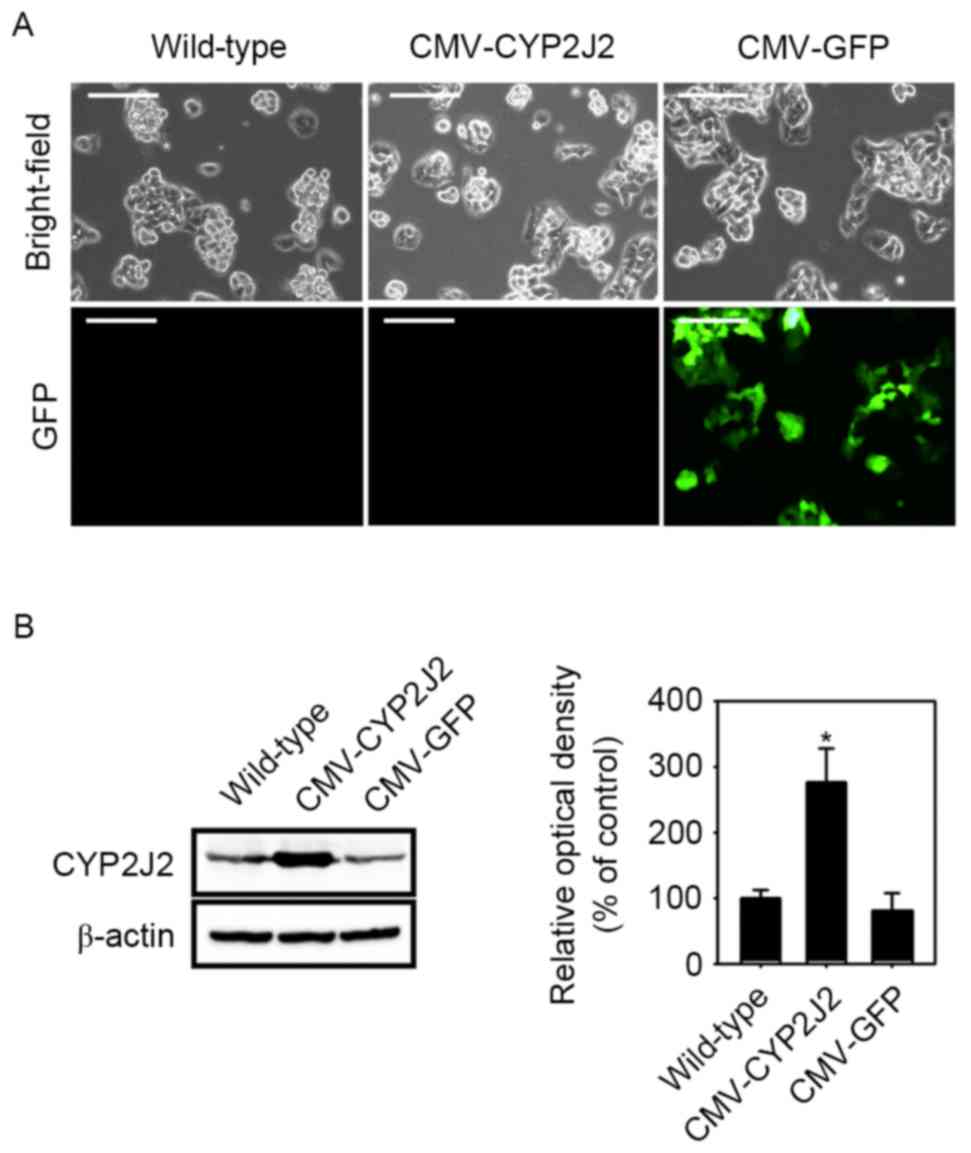
treatment with puromycin (2 µg/ml) for 1 week. Images of each cell
lines were obtained by fluorescence microscopy at magnification,
×200. (Leica DM IL LED Fluo; Leica Microsystems, Inc., Buffalo
Grove, IL, USA).
Cell counting assay
Wild-type HepG2 cells, CMV-CYP2J2-transfected HepG2
cells and CMV-GFP-transfected HepG2 cells were plated at a density
of 1×105 cells/35-mm dish and cultured at 37°C and 5%
CO2. The number of viable cells was counted using the
Trypan Blue exclusion method according to the protocol of the
manufacturer, in triplicate for each group, at 24, 48 and 72 h
after cell plating.
CCK-8 assay
A total of 5×103 wild-type HepG2 cells
and CMV-CYP2J2-transfected HepG2 cells were cultured in 96-well
plates (BD Biosciences, Franklin Lakes, NJ, USA). Following
culture, the cells with or without doxorubicin for 24 h, the CCK-8
solution was then added to each well at 1:10 dilution, followed by
further incubation at 37°C for 3 h. Absorbance was measured at 450
nm using a microplate reader (BioTek Instruments, Inc., Winooski,
VT, USA).
Western blot analysis
Wild-type HepG2 cells, CMV-CYP2J2-transfected HepG2
cells or CMV-GFP-transfected HepG2 cells were cultured in the
presence or absence of LY294002 for 8 h or doxorubicin for 24 h and
the cells were directly lysed in culture dishes with
radioimmunoprecipitation assay buffer (Boston BioProducts, Ashland,
MA, USA) supplemented with a protease and phosphatase inhibitor
cocktail mixture (cat. no. 88668; Thermo Fisher Scientific, Inc.).
Cell lysates (20 µg) were separated using 10 or 12% SDS-PAGE and
then transferred to polyvinylidene fluoride membranes (Merck KGaA).
The blots were washed with TBS containing Tween-20 (TBST) [10 mM
Tris-HCl (pH 7.6), 150 mM NaCl and 0.1% Tween-20], blocked with 5%
skimmed milk in TBST for 1 h at room temperature and incubated for
12 h at 4°C with the primary antibodies at 1:1,000 dilution. Next,
the membranes were washed with TBST and incubated with horseradish
peroxidase-conjugated goat anti-rabbit or goat anti-mouse IgG
antibodies (1:5,000 dilution) for 12 h at 4°C. The bands were
visualized using Pierce® ECL Western Blotting Substrate
(cat. no. 32209; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. β-actin was used as an internal
control.
Statistical analysis
All results are expressed as the mean ± standard
error of the mean using SigmaPlot v11.0 software (Systat Software
Inc., San Jose, CA, USA). Differences between two mean values were
analyzed by the Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results and Discussion
Effect of CYP2J2 overexpression on
cell proliferation and cell cycle regulatory protein
expression
To investigate the role of CYP2J2 on cell
proliferation in HepG2 cells, stable HepG2 cell lines
overexpressing CYP2J2 (CMV-CYP2J2) and GFP (CMV-GFP) were
established using CMV-CYP2J2 or CMV-GFP lentiviruses, respectively.
The expression levels of CYP2J2 protein in the stable cell lines
were examined by western blot analysis. CMV-GFP virus was used as a
positive control. CYP2J2 expression was significantly increased by
infection with CMV-CYP2J2 virus in comparison with that observed in
wild-type HepG2 cells (Fig. 1). To
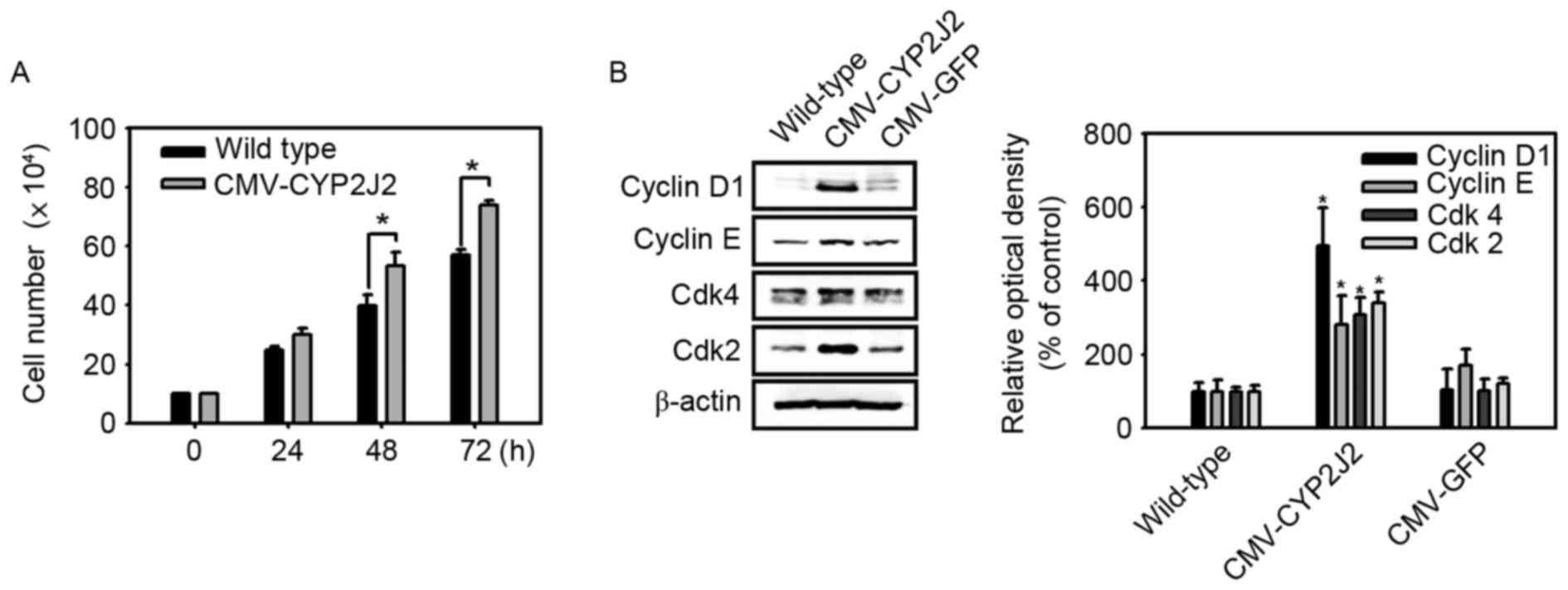
examine the effect of CYP2J2 on HepG2 cell proliferation, the
numbers of wild-type and CMV-CYP2J2-transfected HepG2 cells in
growth medium were counted at 24-h intervals. The rate of
proliferation of CMV-CYP2J2-transfected HepG2 cells was
significantly higher than that of wild-type HepG2 cells (Fig. 2A). The core of the molecular machinery
that drives the cell cycle is the family of Cdks and their
regulatory subunits, which are known as cyclins (21). Cdk-cyclin complexes are activated at
precise points of the cell cycle through multiple levels of
control, including complex assembly or expression levels (22). Therefore, the present study examined
the expression levels of cyclin D1, cyclin E, Cdk2 and Cdk4
proteins. Cyclin D1, cyclin E, Cdk2 and Cdk4 expression was
significantly increased by overexpression of CYP2J2 (Fig. 2B). These results suggest that
overexpression of CYP2J2 promotes cell proliferation in HepG2
hepatocellular carcinoma cells through increased expression of
cyclin D1, cyclin E, Cdk2 and Cdk4 proteins.
Involvement of the Akt signaling
pathway in CYP2J2-induced cell proliferation
Akt is known to serve a central role in signaling
pathways regulating tumor growth (23,24). The
Akt signaling cascade is frequently dysregulated in multiple types
of cancer and is implicated in tumor aggressiveness (23,24).
Several reports indicated that Akt is activated by interaction with
phosphatidylinositol (3–5)-trisphosphate [PtdIns(3,4,5) P3] via the pleckstrin homology
domain (25,26). PtdIns (3,4,5)P3 is normally generated from
phosphatidylinositol 4,5-bisphosphate by the enzyme
phosphoinositide 3-kinase (PI3K) (27). Therefore, the present study
investigated whether CYP2J2 regulates the activity of Akt, and the
results revealed that Akt phosphorylation was significantly
increased in CMV-CYP2J2-transfected HepG2 cells in comparison with
that in wild-type and CMV-GFP-transfected HepG2 cells (Fig. 3A). To further confirm the role of
CYP2J2 on the activity of Akt, the effect of a pharmacological
inhibitor of PI3K, LY294002, was investigated in wild-type and
CMV-CYP2J2-transfected HepG2 cell lines. As shown in Fig. 3B, wild-type and CMV-CYP2J2-transfected
HepG2 cells were treated with different concentrations of LY294002
(0, 10 and 20 µM), and the results revealed that the
phosphorylation of Akt was suppressed by LY294002 in both cell
lines in a dose-dependent manner. However, the levels of Akt
phosphorylation subsequent to LY294002 treatment were higher in
CMV-CYP2J2-transfected HepG2 cells than in wild-type HepG2 cells
(Fig. 3B). The present study also
determined how the above PI3K inhibitor, LY294002, affects the
proliferation of CMV-CYP2J2-transfected HepG2 cells, and it was
observed that the enhanced cell proliferation in
CMV-CYP2J2-transfected HepG2 cells in the absence of LY294002 (10
µM) was significantly inhibited in the presence of LY294002
(Fig. 3C). These results suggest that
overexpression of CYP2J2 enhances the activity of Akt, and that
CYP2J2-mediated cell proliferation is PI3K/Akt
signaling-dependent.
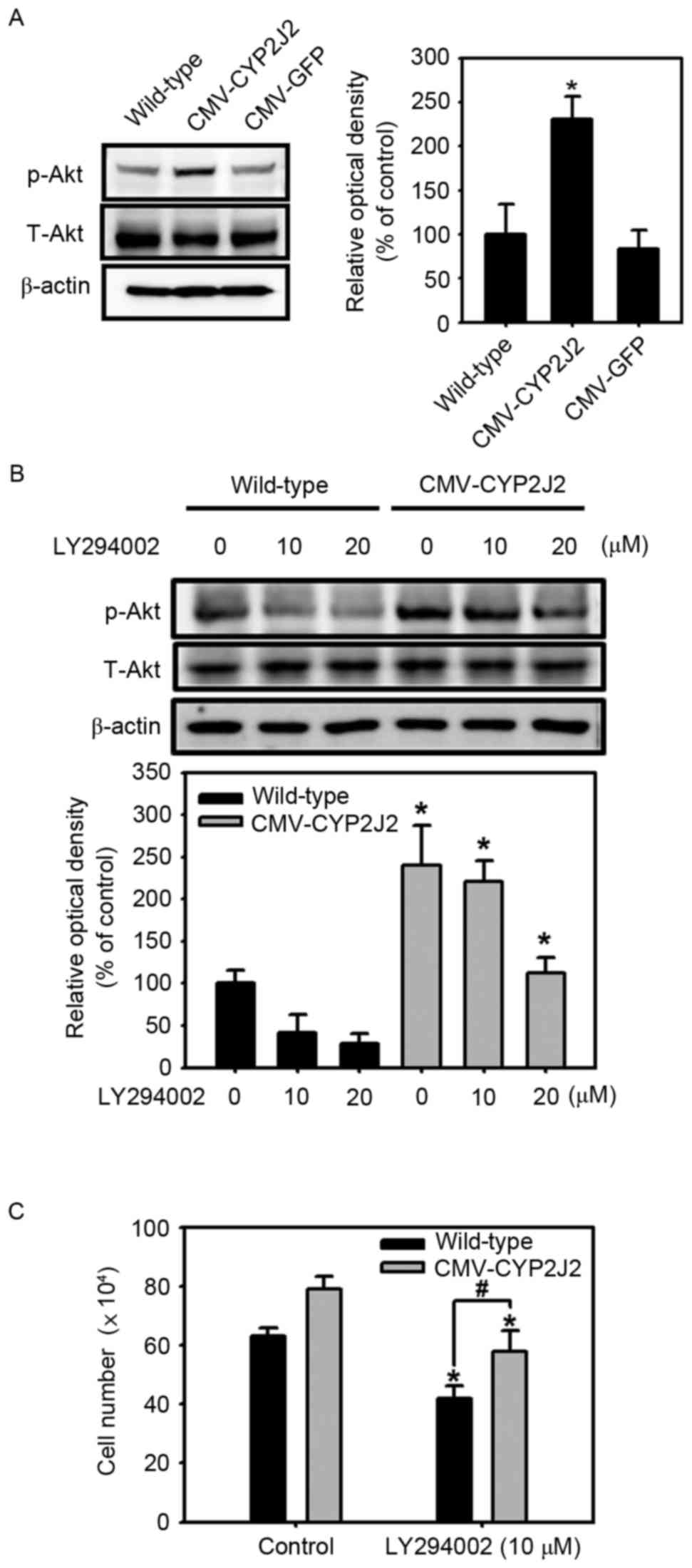 | Figure 3.Involvement of the Akt signaling
pathway in CYP2J2-induced cell proliferation. (A) The expression
levels of phosphorylation of Akt (Ser473) in wild-type,
CMV-CYP2J2- and CMV-GFP-transfected HepG2 cells were assessed by
western blot analysis. Each of the examples shown is representative
of three experiments. The graphs denote the mean ± SEM of three
independent experiments for each condition, as determined from
densitometry analysis relative to β-actin. *P<0.05 vs.
wild-type. (B) Wild-type and CMV-CYP2J2-transfected HepG2 cells
were treated with LY294002 (0–20 µM) for 8 h, and the
phosphorylation of Akt was then detected by western blot analysis.
Each of the examples shown is representative of three experiments.
The graphs denote the mean ± SEM of three independent experiments
for each condition, as determined from densitometry analysis
relative to β-actin. *P<0.05 vs. wild-type. (C) Wild-type and
CMV-CYP2J2-transfected HepG2 cells were cultured for 48 h, and the
cells were then cultured with or without 10 µM LY294002 for
additional 24 h. Cell proliferation rates in wild-type and
CMV-CYP2J2-transfected cells were assessed by direct cell counting.
Error bars denote the mean ± SEM of three independent experiments
levels. *P<0.05 vs. control; #P<0.05 vs.
wild-type. CYP2J2, cytochrome P450 2J2; SEM, standard error of the
mean; GFP, green fluorescent protein; CMV, cytomegalovirus; Cdk,
cyclin-dependent kinase; p, phosphorylated; T, total. |
Effect of CYP2J2 overexpression on
resistance to an anticancer agent
To investigate whether overexpression of CYP2J2
affects resistance to an anticancer agent, the present study
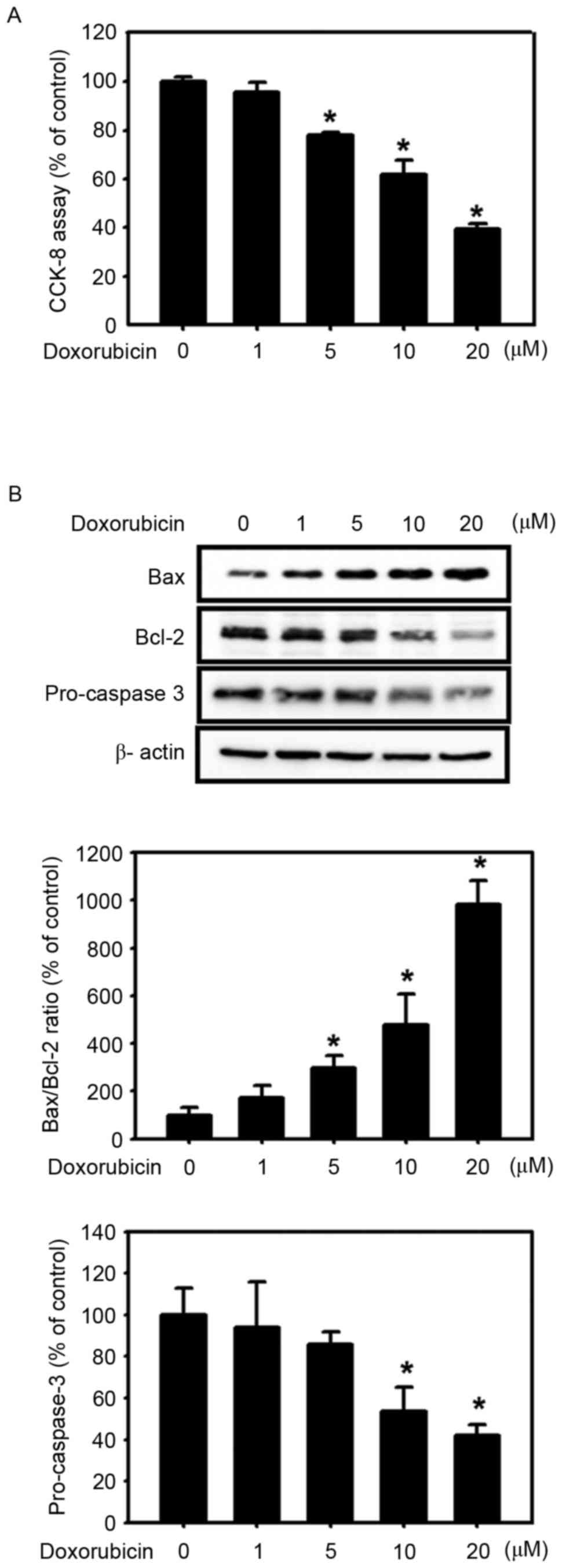
subsequently examined cell viability in the presence of doxorubicin
using the CCK-8 assay. Doxorubicin (adriamycin), an anticancer
agent used in the treatment of advanced hepatocellular carcinoma
due to its antitumor action (28),
induces apoptotic cell death in various types of cells, including
cancer cells (29). Doxorubicin
reduced significantly the cell viability of wild-type HepG2 cells
in a dose-dependent manner (0–20 µM; Fig.
4A). To examine whether the apoptotic pathway is involved in
doxorubicin-induced cytotoxicity in HepG2 cells, the expression
levels of apoptosis-associated proteins, including Bax, Bcl-2 and
pro-caspase-3, were analyzed. Incubation of wild-type HepG2 cells
with doxorubicin (0–20 µM) for 24 h increased the expression of
Bax, decreased the expression of Bcl-2, enhanced the Bax/Bcl-2
ratio and diminished the expression levels of pro-caspase-3 in a
dose-dependent manner (Fig. 4B).
These results suggest that doxorubicin-induced cytotoxicity is
mediated by the apoptotic pathway. Inhibition of apoptosis is
generally considered to be a major determinant of resistance to
chemotherapy (29). To compare the
cytotoxic effect of doxorubicin in wild-type and
CMV-CYP2J2-transfected HepG2 cells, these cell lines were treated
with 10 µM doxorubicin for 24 h, and cell viability was determined
using the CCK-8 assay. As shown in Fig.
5A, doxorubicin significantly decreased cell viability in both
cell lines. Notably, the doxorubicin-induced reduction of cell
viability was significantly attenuated in CMV-CYP2J2-transfected
HepG2 cells compared with that in wild-type HepG2 cells.
Additionally, doxorubicin induced a significant increase in the
Bax/Bcl-2 ratio and decreased pro-caspase-3 levels in both cell
lines; however, the increase in the Bax/Bcl-2 ratio and the
decrease in pro-caspase-3 levels were inhibited in
CMV-CYP2J2-transfected HepG2 cells in comparison with those in
wild-type HepG2 cells. These results suggest that the cytotoxic
effect of doxorubicin is significantly attenuated by CYP2J2
overexpression; in other words, CYP2J2 overexpression confers
resistance to doxorubicin in HepG2 cells.
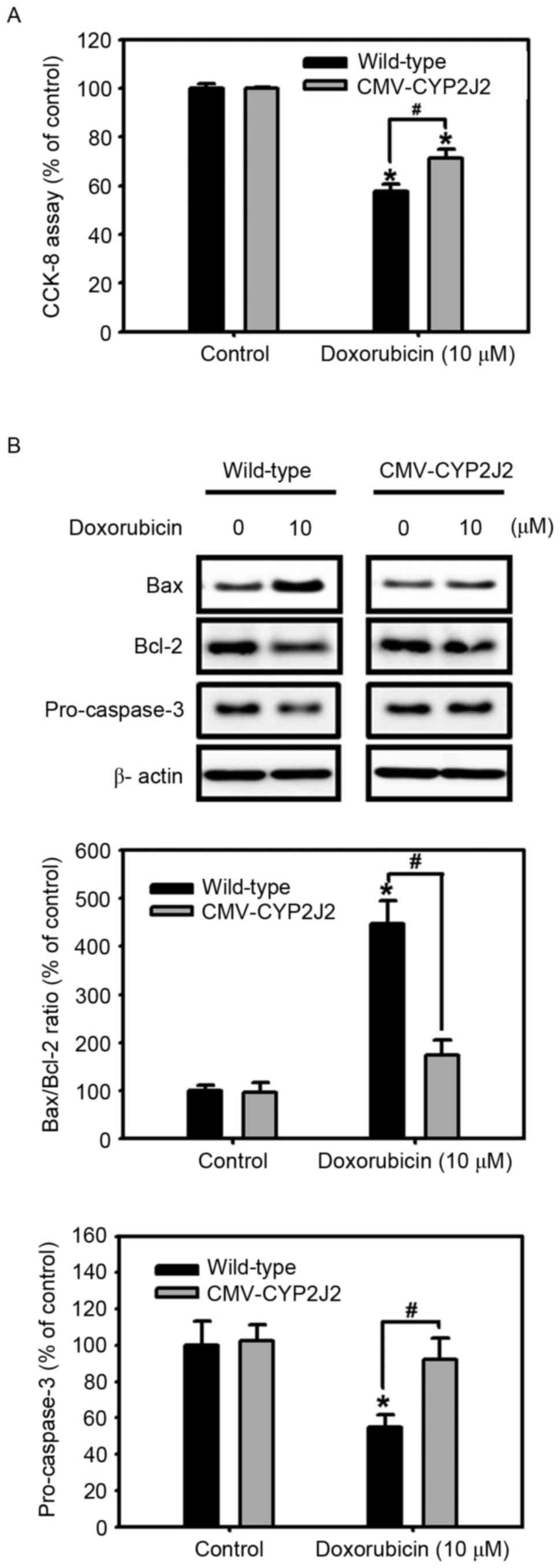 | Figure 5.Effect of CYP2J2 overexpression on the
resistance to doxorubicin. (A) Wild-type and CMV-CYP2J2-transfected
HepG2 cells were cultured with or without 10 µM doxorubicin for 24
h, and cell viability was measured using a CCK-8 reduction assay.
Values are expressed as the mean ± SEM of three experiments with
triplicate dishes. *P<0.05 vs. control; #P<0.05
vs. wild-type. (B) Wild-type and CMV-CYP2J2-transfected HepG2 cells
were cultured under the indicated conditions for 24 h. Cell lysates
were subjected to western blot analysis using anti-Bax, anti-Bcl-2
and anti-pro-caspase-3-specific antibodies. Each of the examples
shown is representative of three experiments. The intensities of
the Bax, Bcl-2 and pro-caspase-3 bands were determined by
densitometry analysis relative to β-actin, and the Bax/Bcl-2 ratio
was calculated. Values are expressed as the mean ± SEM of three
independent experiments. *P<0.05 vs. control;
#P<0.05 vs. wild-type. CCK, Cell Counting kit; Bcl,
B-cell lymphoma; Bax, Bcl-2 associated X protein; SEM, standard
error of the mean; CYP2J2, cytochrome P450 2J2; SEM, standard error
of the mean; CMV, cytomegalovirus. |
In conclusion, the present study has demonstrated
that CYP2J2 promotes cell proliferation and drug resistance to an
anticancer agent in HepG2 cells. CYP2J2-mediated cell proliferation
requires the activity of Akt, which is known to be frequently
dysregulated in numerous types of cancer (24). Additionally, overexpression of CYP2J2
decreased the apoptotic cell death caused by the above anticancer
agent. Robust cell proliferative capacity and resistance to
anticancer agents are major hurdles to the development of efficient
chemotherapy, and abnormal cell proliferation and apoptotic cell
death have been extensively studied to identify potential
therapeutic targets against human cancer (30). Various cellular components, including
hypoxia-inducible factor-1α (31),
transducin β-like protein 1-related protein (32) and fibroblast growth factor receptor 4
(33), have been shown to contribute
to cancer cell proliferation, and due to their anti-apoptotic
properties, they have been studied as putative targets for cancer
therapy (31–33). The results of the present study
revealed that CYP2J2 also serves important roles in inducing cell
proliferation and inhibiting the cell death induced by anticancer
agents. Therefore, CYP2J2 can be a potential target to reduce
cancer cell proliferation and resistance to chemotherapy.
Acknowledgements
The present study was supported by the National
Research Foundation, which is funded by the Ministry of Education
of the Korean Government (grant no. 2014R1A1A2056042;
NRF-2016R1D1A1A02936940), and by the Ministry of Science,
Information Communication Technology and Future Planning of the
Korean Government (grant no. 2012R1A4A1028835).
References
|
1
|
Capdevila JH, Falck JR and Harris RC:
Cytochrome P450 and arachidonic acid bioactivation. Molecular and
functional properties of the arachidonate monooxygenase. J Lipid
Res. 41:163–181. 2000.PubMed/NCBI
|
|
2
|
Enayetallah AE, French RA, Thibodeau MS
and Grant DF: Distribution of soluble epoxide hydrolase and of
cytochrome P450 2C8, 2C9 and 2J2 in human tissues. J Histochem
Cytochem. 52:447–454. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fisslthaler B, Popp R, Kiss L, Potente M,
Harder DR, Fleming I and Busse R: Cytochrome P450 2C is an EDHF
synthase in coronary arteries. Nature. 401:493–497. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rosolowsky M and Campbell WB: Synthesis of
hydroxyeicosatetraenoic (HETEs) and epoxyeicosatrienoic acids
(EETs) by cultured bovine coronary artery endothelial cells.
Biochim Biophys Acta. 1299:267–277. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee HC, Lu T, Weintraub NL, VanRollins M,
Spector AA and Shibata EF: Effects of epoxyeicosatrienoic acids on
the cardiac sodium channels in isolated rat ventricular myocytes. J
Physiol. 1:153–168. 1999. View Article : Google Scholar
|
|
6
|
Node K, Huo Y, Ruan X, Yang B, Spiecker M,
Ley K, Zeldin DC and Liao JK: Anti-inflammatory properties of
cytochrome P450 epoxygenase-derived eicosanoids. Science.
285:1276–1279. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen JK, Capdevila J and Harris RC:
Cytochrome p450 epoxygenase metabolism of arachidonic acid inhibits
apoptosis. Mol Cell Biol. 21:6322–6331. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang B, Graham L, Dikalov S, Mason RP,
Falck JR, Liao JK and Zeldin DC: Overexpression of cytochrome P450
CYP2J2 protects against hypoxia-reoxygenation injury in cultured
bovine aortic endothelial cells. Mol Pharmacol. 60:310–320.
2001.PubMed/NCBI
|
|
9
|
Jiang JG, Chen CL, Card JW, Yang S, Chen
JX, Fu XN, Ning YG, Xiao X, Zeldin DC and Wang DW: Cytochrome P450
2J2 promotes the neoplastic phenotype of carcinoma cells and is
up-regulated in human tumors. Cancer Res. 65:4707–4715. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang SM, Mishina YM, Liu S, Cheung A,
Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner
S, et al: Tankyrase inhibition stabilizes axin and antagonizes Wnt
signalling. Nature. 461:614–620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen C, Wei X, Rao X, Wu J, Yang S, Chen
F, Ma D, Zhou J, Dackor RT, Zeldin DC and Wang DW: Cytochrome P450
2J2 is highly expressed in hematologic malignant diseases and
promotes tumor cell growth. J Pharmacol Exp Ther. 336:344–355.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Freedman RS, Wang E, Voiculescu S, Patenia
R, Bassett RL Jr, Deavers M, Marincola FM, Yang P and Newman RA:
Comparative analysis of peritoneum and tumor eicosanoids and
pathways in advanced ovarian cancer. Clin Cancer Res. 13:5736–5744.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang JG, Ning YG, Chen C, Ma D, Liu ZJ,
Yang S, Zhou J, Xiao X, Zhang XA, Edin ML, et al: Cytochrome p450
epoxygenase promotes human cancer metastasis. Cancer Res.
67:6665–6674. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Critelli RM, De Maria N and Villa E:
Biology of Hepatocellular Carcinoma. Dig Dis. 33:635–641. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kroetz DL and Zeldin DC: Cytochrome P450
pathways of arachidonic acid metabolism. Curr Opin Lipidol.
13:273–283. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Manchado E, Guillamot M and Malumbres M:
Killing cells by targeting mitosis. Cell Death Differ. 19:369–377.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Williams GH and Stoeber K: The cell cycle
and cancer. J Pathol. 226:352–364. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gruh I, Wunderlich S, Winkler M, Schwanke
K, Heinke J, Blömer U, Ruhparwar A, Rohde B, Li RK, Haverich A and
Martin U: Human CMV immediate-early enhancer: A useful tool to
enhance cell-type-specific expression from lentiviral vectors. J
Gene Med. 10:21–32. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stead E, White J, Faast R, Conn S,
Goldstone S, Rathjen J, Dhingra U, Rathjen P, Walker D and Dalton
S: Pluripotent cell division cycles are driven by ectopic Cdk2,
cyclin A/E and E2F activities. Oncogene. 21:8320–8333. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee MY, Lim HW, Lee SH and Han HJ: Smad,
PI3K/Akt and Wnt-dependent signaling pathways are involved in
BMP-4-induced ESC self-renewal. Stem Cells. 27:1858–1868. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mitsiades CS, Mitsiades N and Koutsilieris
M: The Akt pathway: Molecular targets for anti-cancer drug
development. Curr Cancer Drug Targets. 4:235–256. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Joh EH, Hollenbaugh JA, Kim B and Kim DH:
Pleckstrin homology domain of Akt kinase: A proof of principle for
highly specific and effective non-enzymatic anti-cancer target.
PLoS One. 7:e504242012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Milburn CC, Deak M, Kelly SM, Price NC,
Alessi DR and Van Aalten DM: Binding of phosphatidylinositol
3,4,5-trisphosphate to the pleckstrin homology domain of protein
kinase B induces a conformational change. Biochem J. 375:531–538.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yeo W, Mok TS, Zee B, Leung TW, Lai PB,
Lau WY, Koh J, Mo FK, Yu SC, Chan AT, et al: A randomized phase III
study of doxorubicin versus cisplatin/interferon
alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy
for unresectable hepatocellular carcinoma. J Natl Cancer Inst.
97:1532–1538. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rebbaa A, Zheng X, Chou PM and Mirkin BL:
Caspase inhibition switches doxorubicin-induced apoptosis to
senescence. Oncogene. 22:2805–2811. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takasaki C, Kobayashi M, Ishibashi H,
Akashi T and Okubo K: Expression of hypoxia-inducible factor-1α
affects tumor proliferation and antiapoptosis in surgically
resected lung cancer. Mol Clin Oncol. 5:295–300. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo Y, Wang J, Zhang L, Shen S, Guo R,
Yang Y, Chen W, Wang Y, Chen G and Shuai X: Theranostical
nanosystem-mediated identification of an oncogene and highly
effective therapy in hepatocellular carcinoma. Hepatology.
63:1240–1255. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ye YW, Zhou Y, Yuan L, Wang CM, Du CY,
Zhou XY, Zheng BQ, Cao X, Sun MH, Fu H and Shi YQ: Fibroblast
growth factor receptor 4 regulates proliferation and antiapoptosis
during gastric cancer progression. Cancer. 117:5304–5313. 2011.
View Article : Google Scholar : PubMed/NCBI
|