Introduction
Prostate cancer is known as a main cancer type among
men worldwide, and causes a high rate of death at the advanced
stage of disease (1). Since
understanding that abnormal activation of prostate-specific
antigen/androgen receptor signaling promotes the aggressiveness of
prostate cancer (2), androgen
deprivation therapy has been applied as the main treatment for
prostate cancer (3). However,
patients with hormone-independent prostate cancer (HIPC) have a
poor prognosis subsequent to androgen deprivation. Gemcitabine, a
deoxycytidine analog, has been used as a first-line drug for HIPC
(4). Gemcitabine exhibits anti-cancer
activity in numerous cancer types by disturbing DNA biosynthesis
(5). In addition, gemcitabine
resistance has become a major obstacle for its application as a
treatment for HIPC. However, the underlying mechanisms of
resistance to gemcitabine remain unclear.
As a non-chromosomal DNA binding protein, high
mobility group box1 (HMGB1) was described in numerous cellular
biological progressions, including cell differentiation, cell
autophagy and cell migration (6,7). Different
functions of HMGB1 depend on its various target genes or pathways
in different tissues. For example, HMGB1 could participate in
inflammatory reactions by regulating nuclear factor-κB expression
(8), while HMGB1 induced upregulation
of c-myc contributes to tumor malignancies. In autophagy, HMGB1 is
involved at several levels. HMGB1 regulates the expression of heat
shock protein β-1 (HSPB1), which is important for dynamic
intracellular trafficking during autophagy and mitophagy (9). Additionally, HMGB1 could disturb the
Beclin-1-B cell lymphoma-2 (BCL-2) complex and then activate
autophagy through interacting with Beclin-1 (10). A number of studies have demonstrated a
crucial role for autophagy in cancer development and therapy
(11,12). Notably, HMGB1 exhibits a significant
function in facilitating autophagy in response to cytotoxic insults
(7,13). Therefore, HMGB1-mediated autophagy is
worthy of investigating drug resistance.
In the present study, gemcitabine was shown to
induce the expression of HMGB1, which in turn decreased the
sensitivity to this drug in HIPC cells. Further experiments
indicated that HMGB1 upregulation enhances the expression of HSPB1,
which may function as an effector targeted by HMGB1 in gemcitabine
induced autophagy progression. Thus, the present findings provide
potential targets for the treatment of HIPC.
Materials and methods
Cell culture and chemicals
The human prostate cancer cell line PC-3 was
purchased from the American Type Culture Collection (Manassas, VA,
USA) and cultured in RPMI-1640 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) containing 10% FBS in a 37°C incubator in 5%
CO2. Gemcitabine or 3-methyladenosine (3-MA) was
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and
dissolved in dimethyl sulfoxide.
Antibodies and shRNA
Antibodies against light chain 3 (LC3)-I/II (catalog
no. sc-16756), sequestosome 1 (SQSTM1) (catalog no. sc-25575),
Beclin-1 (catalog no. sc-11427), HSPB1 (catalog no. sc-1048), HMGB1
(catalog no. sc-56698) and GAPDH (catalog no. sc-25778) were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
IRDye® 800CW- or IRDye® 680RD-conjugated
secondary antibodies (926–32210; 926-68071; 926-32211; 926-68070)
were purchased from LI-COR Biosciences (Lincoln, NE, USA).
HMGB1-shRNA (Sigma-Aldrich; Merck KGaA) and pcDNA3.1-HMGB1
(self-constructed; HMGB1 CDS; CCDS9335.1; GenBank; National
Institute of Health, Bethesda, MD, USA) were stably transfected
with the Lipofectamine 2000 Transfection Reagent (Invitrogen;
Thermo Fisher Scientific, Inc.), according the manufacturer's
instructions.
Western blot analysis
The whole cell lysates from PC-3 or shHMGB1 PC-3
cells treated with 10 µg/ml gemcitabine were prepared with cold
cell radioimmunoprecipitation assay lysis buffer (20–188; EMD
Millipore, Billerica, MA, USA) and the total protein concentration
was measured using a Bradford assay (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Acquired proteins (35 µg/lane) were separated
by 10% SDS-PAGE gel and then transferred to a nitrocellulose
membrane. The membrane was blocked with 5% milk for 2 h at room
temperature and incubated with primary antibodies against LC3-I/II
(dilution, 1:1,000), SQSTM1 (dilution, 1:1,000), HMGB1 (dilution,
1:1,000), Beclin-1 (dilution, 1:500), HSPB1 (dilution, 1:500) and
GAPDH antibody (dilution, 1:2,000) at 4°C overnight. Subsequently,
the membranes were incubated with appropriate secondary antibodies
at 1:10,000 dilution at room temperature for 1 h. IRDye®
800CW- or IRDye® 680-conjugated secondary antibodies
(IRDye® 800CW goat anti-Mouse IgG (H + L), cat. no.
926-32210; IRDye® 680RD goat anti-Rabbit IgG (H + L),
cat. no. 926-68071; IRDye® 800CW goat anti-Rabbit IgG (H
+ L), cat. no. 926-32211; IRDye® 680RD goat anti-Mouse
IgG (H + L), cat. no. 926-68070) (LI-COR Biosciences, Lincoln, NE,
USA) were used for staining, and then the secondary antibodies were
detected by an Odyssey® infrared imaging system (LI-COR
Biosciences).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from the PC-3 cells with or without
gemcitabine treatment was obtained with the RNAiso™ Plus reagent
(Takara Bio, Inc., Otsu, Japan) and reverse-transcribed by a
PrimeScript™ RT reagent kit (Takara Bio, Inc.). qPCR was performed
using SYBR Green mix (Roche Diagnostics, Basel, Switzerland),
according to the manufacturer's instructions. β-actin was used as a
loading control. The thermocycling conditions for RT-qPCR reactions
were as follows: 95°C 5 min, followed by 40 cycles of 95°C 45 sec,
annealing at 55°C 45 sec and extension 72°C 1 min. The following
primers were used: HSPB1 forward, 5′-GCGTGTCCCTGGATGTCAAC-3′; HSPB1
reverse, 5′-TGTATTTCCGCGTGAAGCAC-3′; Beclin1 forward,
5′-TGTGAGGAATGCACAGATAC-3′; Beclin1 reverse,
5′-TGTCCACTGTGCCAGATGT-3′; HMGB1 forward,
5′-TGTCCATTGGTGATGTTGCG-3′; HMGB1 reverse,
5′-GGACAGGGCTATCTAAAGACACA-3′; β-actin forward,
5′-CTCCATCCTGGCCTCGCTGT-3′; β-actin reverse,
5′-GCTGTCACCTTCACCGTTCC-3′. The relative mRNA level was calculated
using the 2−ΔΔCq method (14).
Cell viability assay
Cell viability of PC-3 treated with 2, 4, 8, 16, 32
or 64 µg/ml of gemcitabine for 48 h was detected using the Cell
Counting Kit-8 (CCK8) assay (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan), according to the manufacturer's protocol.
Absorbance was then measured by a microplate reader at 450 nm. Data
were obtained from at least three separate experiments performedin
triplicate.
Flow cytometry
PC-3 cells were seeded into 6-well plates
(5×105 cells/well) and pre-treated with 2 mM 3-MA or
DMSO for 24 h. The PC-3 cells treated with 3-MA or stably
transfected with shHMGB1 were stimulated with 10 µg/ml gemcitabine
for 48 h. The cells were then collected and stained using the
Annexin-V-FLUOS staining kit (Roche Diagnostics), according to the
manufacturer's protocol. The stained cells were analyzed
immediately on a FACSCalibur flow cytometer (BD Biosciences, San
Jose, CA, USA) using the CellQuest 3.0 software system (BD
Biosciences).
Statistical analysis
SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA)
was used in statistical analyses. Group distributions were
performed with the Student's t-test or one-way and two-way analysis
of variance (Post-hoc test one-way, Tukey's test; post-hoc test
two-way, Sidak's test). P<0.05 was considered to indicate a
statistically significant difference.
Results
Autophagy was induced by gemcitabine
treatment in HIPC cells
The human hormone-independent prostate cancer PC3
cell line was used to determine whether autophagy could be induced
by gemcitabine. Firstly, the sensitivity to different
concentrations of gemcitabine was examined using the CCK8 cell
viability assay in PC3 cells. The results indicated that
gemcitabine suppressed PC3 cell viability in a
concentration-dependent manner, with a 50% inhibitory concentration
(IC50) of 17.82 µg/ml (Fig. 1A).
Based on the cell viability curve of PC3 to gemcitabine, a
nonlethal concentration of 10 µg/ml was adopted as the treatment
concentration of gemcitabine to induce autophagy in subsequent
experiments. Western blot analysis subsequently showed that the
conversion of LC3-I to LC3-II was increased and the SQSTM1 level
was decreased, which suggested the role of gemcitabine in promoting
autophagy in PC3 cells (Fig. 1B).
Considering the upregulation of HMGB1 subsequent to gemcitabine
treatment (Fig. 1B), HMGB1-dependent
autophagy was detected by testing the expression levels of its
effectors, HSPB1 and Beclin-1, in PC3 cells. The present findings
demonstrated that upregulation of HSPB1 and Beclin-1 occurred at
both mRNA and protein levels in PC3 cells with gemcitabine
stimulation (Fig. 1C and D). Overall,
the results showed that the chemotherapeutic agent gemcitabine
could activate autophagy in HIPC cells.
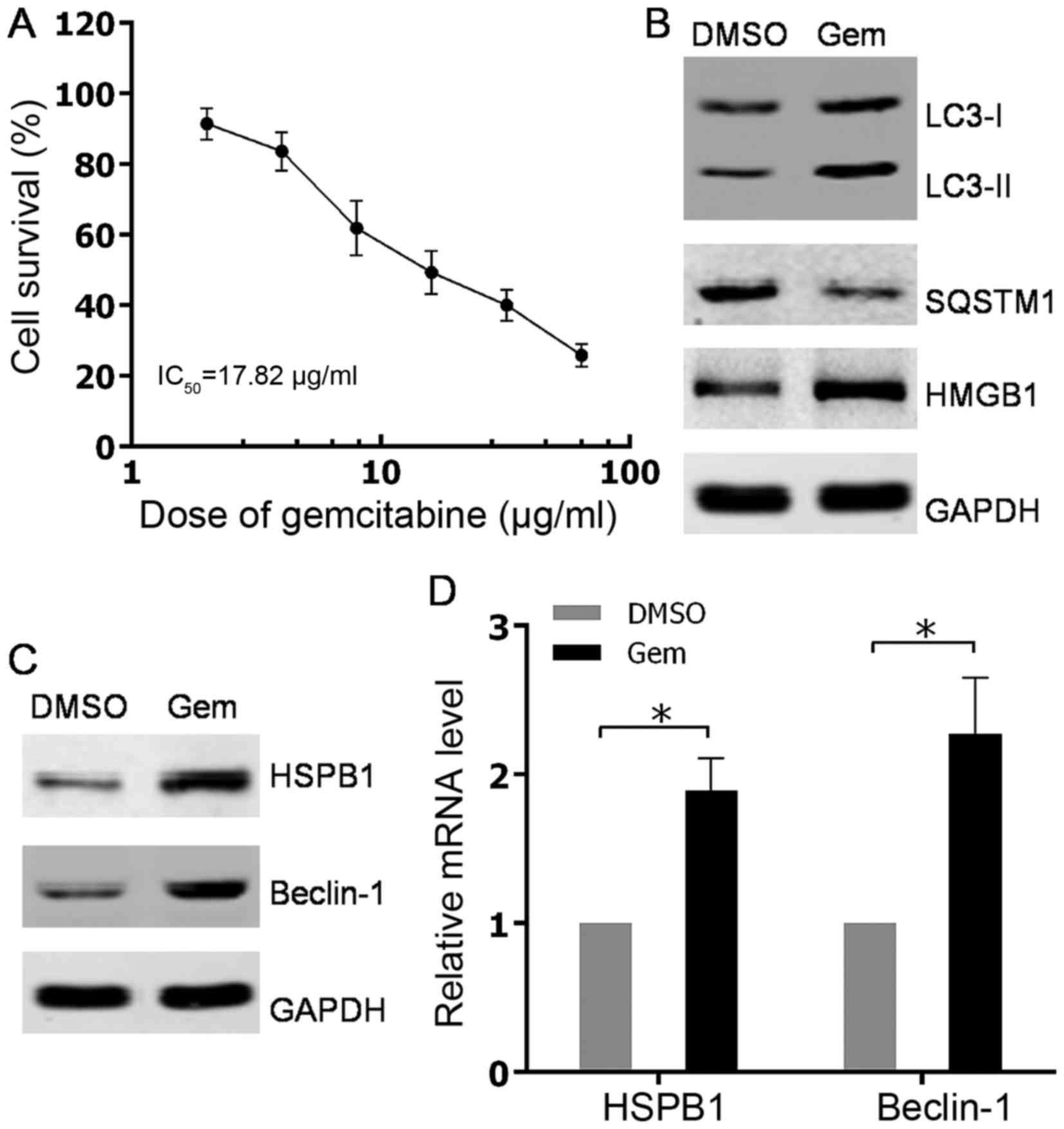 | Figure 1.Autophagy was induced by gemcitabine
treatment in HIPC cells. (A) PC-3 cells were treated with 2, 4, 8,
16, 32 or 64 µg/ml gemcitabine for 48 h. Cell survival was
determined using Cell Counting Kit-8 assays. All experiments were
independently repeated at least three times. *P<0.05. (B and C)
PC3 cells were treated with 10 µg/ml gemcitabine or DMSO for 48 h.
The expression levels of indicated proteins were analyzed via
western blotting. (D) PC3 cells were treated with 10 µg/ml
gemcitabine or DMSO for 48 h. HSPB1 and Beclin-1 mRNA levels were
measured with quantitative PCR. All experiments were independently
repeated at least three times. *P<0.05. DMSO, dimethyl
sulfoxide; Gem, gemcitabine; LC3, light chain 3; SQSTM1,
sequestosome 1; HMGB1, high mobility group box 1; HSPB1, heat shock
protein β-1. |
Gemcitabine induced upregulation of
HMGB1 contributes to autophagy in HIPC cells
To understand the underlying mechanisms for
gemcitabine-induced autophagy in HIPC cells, the roles of the
important regulator of autophagy HMGB1 and its associated factors
in gemcitabine-induced autophagy progression were investigated. The
present data showed that the protein level of HMGB1was
significantly increased in a dose-dependent manner in PC3 cells
after treated with gemcitabine (Fig.
2A). Consistently, the elevated expression of HMGB1 was also
verified at mRNA level after gemcitabine addition (Fig. 2B). Additionally, through examining
autophagy-associated markers, it was found that gemcitabine-induced
autophagy in PC3 cells was attenuated by stable knockdown of HMGB1
using shRNA (Fig. 2C). However,
stably ectopic expression of HMGB1 in PC3 cells evidently promoted
autophagy progression compared with parental PC3 cells (Fig. 2D). Notably, data indicated that
abrogation of HMGB1 impaired the induction of HSPB1, but not
Beclin-1 at both mRNA and protein levels subsequent to gemcitabine
treatment (Fig. 2C, E and F). As
expected, overexpression of HMGB1 raised the protein level of
HSPB1, while no significant alterations of Beclin-1 expression were
found in PC3 cells with stably expressing HMGB1 (Fig. 2D). Thus, the aforementioned findings
revealed that HMGB1 may mediate gemcitabine-induced autophagy in
PC3 cells via its target gene, HSPB1.
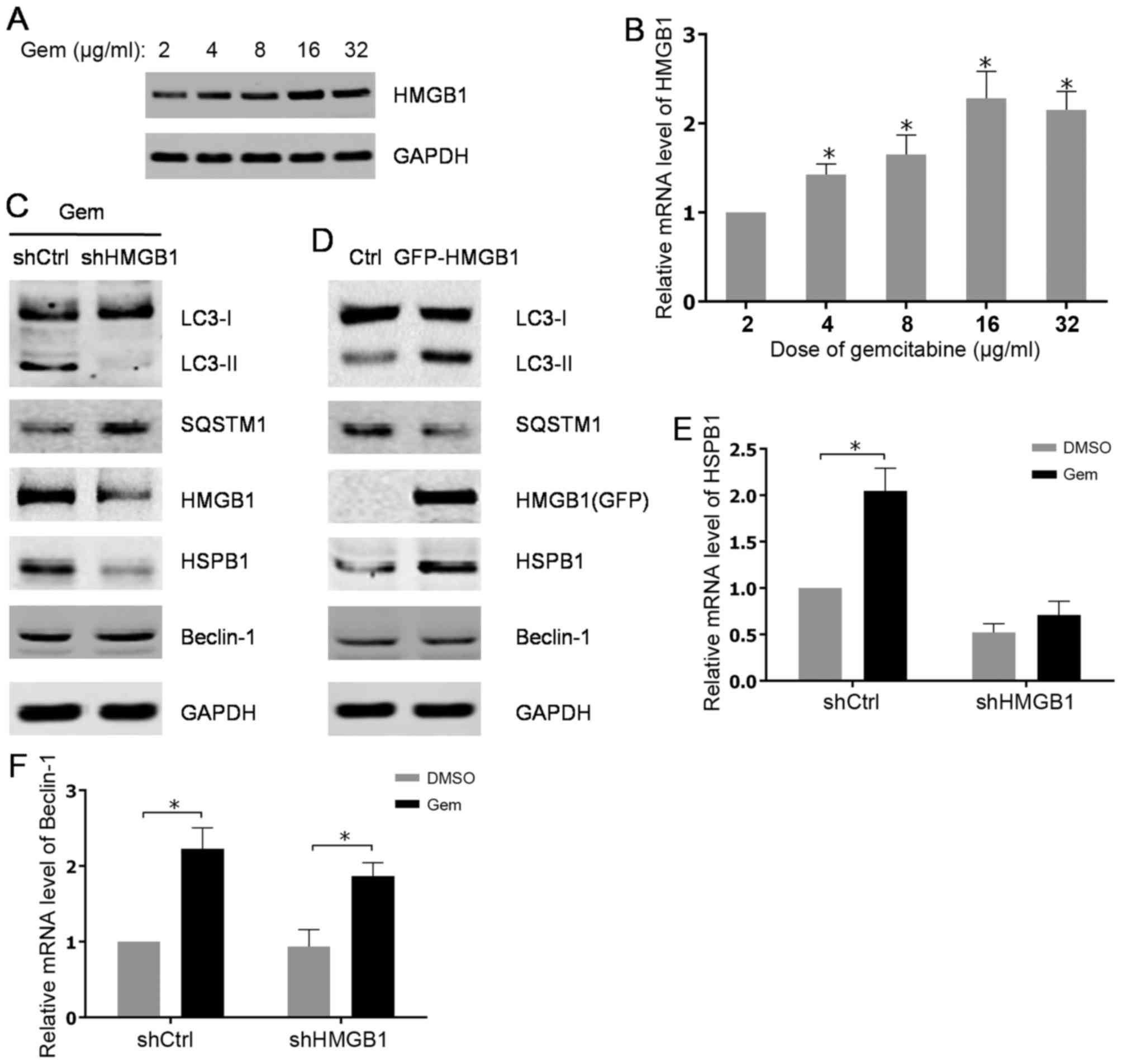 | Figure 2.Gemcitabine-induced upregulation of
HMGB1 contributes to autophagy in HIPC cells. (A) PC-3 cells were
treated with 2, 4, 8, 16 or 32 µg/ml of gemcitabine for 48 h. The
expression levels of HMGB1 were analyzed by western blot analysis.
(B) HMGB1 mRNA levels were measured using qPCR. All experiments
were independently repeated at least three times. *P<0.05. (C
and D) PC3 cells were transfected with shRNA or pcDNA3.1-HMGB1 and
screened with G418. The PC3 cells transfected with shRNA were
treated with 10 µg/ml gemcitabine for 48 h. The expression levels
of indicated proteins were analyzed by western blot analysis. (E
and F) The transfected PC3 cells were treated with 10 µg/ml
gemcitabine or DMSO for 48 h. HSPB1 and Beclin-1mRNA levels were
measured with qPCR. All experiments were independently repeated at
least three times. *P<0.05. qPCR, quantitative polymerase chain
reaction; Gem, gemcitabine; Ctrl, control; LC3, light chain 3;
SQSTM1, sequestosome 1; HMGB1, high mobility group box1; HSPB1,
heat shock protein β-1; shCtrl, control short hairpin RNA; shHMGB1,
short hairpin RNA against HMGB1. |
Autophagy induced by gemcitabine
protects HIPC cells from the cytotoxicity of gemcitabine
To determine the function of autophagy in response
to gemcitabine, the sensitivity to gemcitabine in HIPC cells with
different treatments was measured by cell viability assay. The
results suggested that elimination of HMGB1 by shRNA in PC3 cells
resulted in an apparent reduction of IC50 for gemcitabine treatment
(Fig. 3A). Additionally, PC3 cells
were either treated with gemcitabine alone or co-treated with
gemcitabine and the inhibitor of autophagy 3-MA. The present
findings showed that the group treated with only gemcitabine was
more resistant to gemcitabine compared to the group that received
co-treatment, indicating the protective role of autophagy in HIPC
cells exposed to gemcitabine (Fig.
3B). Additionally, consistent with the aforementioned results,
inhibition of autophagy in PC3 cells by HMGB1 shRNA or
3-MA-enhanced cell apoptosis induced by gemcitabine (Fig. 3C and D). Therefore, these data
elucidated that HMGB1-mediated autophagy reduced the sensitivity of
HIPC cells to the drug gemcitabine.
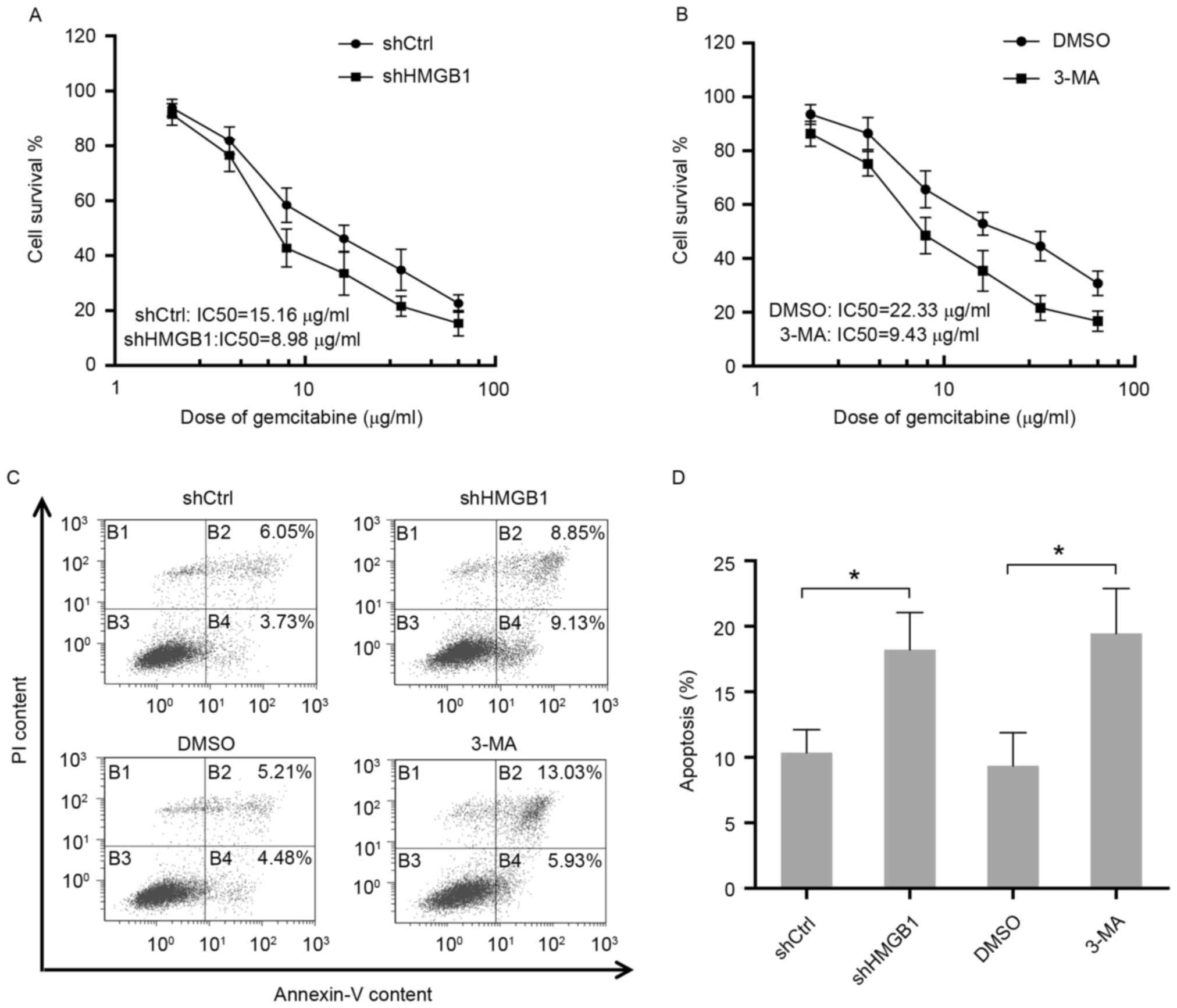 | Figure 3.Autophagy induced by gemcitabine
protects HIPC cells from the cytotoxicity of gemcitabine. (A) PC-3
cells stably transfected with HMGB1-shRNA or Ctrl-shRNA were
treated with 2, 4, 8, 16, 32 or 64 µg/ml of gemcitabine for 48 h.
Cell survival was determined using CCK-8 assays. All experiments
were independently repeated at least three times. (B) PC-3 cells
pre-treated with 2 mM 3-MA or DMSO for 24 h were then treated with
2, 4, 8, 16, 32 or 64 µg/ml gemcitabine for 48 h. Cell survival was
determined using CCK-8 assays. All experiments were independently
repeated at least three times. (C) Apoptosis in transfected or
treated PC3 cells were analyzed by flow cytometry. (D) Quantitation
of apoptosis data. Gem, gemcitabine; Ctrl, control; LC3, light
chain 3; SQSTM1, sequestosome 1; HMGB1, high mobility group box1;
HSPB1, heat shock protein β-1; shCtrl, control short hairpin RNA;
shHMGB1, short hairpin RNA against HMGB1; 3-MA, 3-methyladenosine;
PI, propidium iodide; IC50, 50% inhibitory concentration. |
Discussion
Autophagy is a biological process through which
cells use the lysosome to degrade damaged organelles and
macromolecules. Therefore, autophagy may prevent the accumulation
of damaged or useless components in cells, which confers cell
survival under unfavorable conditions. The dysregulation of
autophagy has been associated with numerous cancer types, and
enhanced autophagy following chemotherapy and radiotherapy was
observed in various cancer cells (15–17).
Previous studies have indicated that acquired autophagy is closely
associated with multidrug resistance by decreasing the permeability
of cell membranes or slowing down cell metabolism (18). In the present study, using LC3-II and
SQSTM1 (p62) as markers of autophagy, it was found that the
first-line chemotherapeutic gemcitabine induced autophagy in HIPC
cells. Additionally, disruption of autophagy by the autophagy
inhibitor 3-MA increased the sensitivity to gemcitabine in HIPC
cells. Thus, the induction of autophagy has an important role in
gemcitabine resistance of HIPC cells. Consistently, it has been
reported that induced autophagy modulates sensitivity of colorectal
cancer cells to oxaliplatin (19) and
mediates multi-drug resistance in osteosarcoma (20).
Autophagy is a complex progression associated with
various factors and signaling pathways. Among these factors, HMGB1
is a critical regulator of autophagy, which was the focus of the
present study. The results showed that the mRNA and protein levels
of HMGB1 were increased in a dose-dependent manner following
gemcitabine treatment. In addition, stable knockdown of HMGB1 by
shRNA reversed the autophagy induced by gemcitabine in HIPC cells.
Stable overexpression of HMGB1 in HIPC cells without treatment also
enhanced the autophagy progress, which indicated an increased level
of LC3-II and a reduction of SQSTM1. In addition, attenuation of
HMGB1 weakened the resistance to gemcitabine in HIPC cells.
Therefore, it was hypothesized that gemcitabine resistance in HIPC
cells results from HMGB1-associated autophagy.
The roles of HMGB1 in autophagy are performed via
two well-known factors, HSPB1 and Beclin-1. Nuclear localization of
HMGB1 activates HSPB1 expression (21), which affects dynamic intracellular
trafficking during autophagy (22).
Additionally, cytosolic localization of HMGB1 interacts with
Beclin-1 and dissolved it from BCL-2 (10). Released Beclin-1 regains its ability
to promote autophagy (23). Notably,
exposure to gemcitabine stimulates the expressions of HSPB1 and
Beclin-1 in PC3 cells. However, alterations of HMGB1 level by
knockdown or overexpression had an effect on the level of HSPB1,
but not Beclin-1, in PC3 cells. This is coincident with previous
studies that HSPB1 is tightly regulated by HMGB1 (24). Therefore, upregulation of Beclin-1 in
response to gemcitabine may be activated by other
autophagy-associated factors or pathways, and the functions of
HSPB1 and Beclin-1 in gemcitabine-induced autophagy of HIPC cells
require additional investigation.
In summary, it was demonstrated that HMGB1
expression is increased in the HIPC PC3 cell line after gemcitabine
treatment. Subsequently, the increasing HMGB1 expression causes
autophagy and decreases the sensitivity to gemcitabine in HIPC
cells. Therefore, the present findings provided a novel mechanism
for gemcitabine resistance in HIPC cells, which could be used as a
target for the diagnosis and treatment of patients with HIPC.
Acknowledgements
The present study was supported by the Shenzhen
Science and innovation Commission Research Foundation (grant no.
JCYJ20150403101028172), Guangdong Provincial Medical Scientific
Research Foundation (grant no. A2014634) and Shenzhen Health and
Family Planning Commission Research Foundation (grant no.
201401002).
References
|
1
|
Trewartha D and Carter K: Advances in
prostate cancer treatment. Nat Rev Drug Discov. 12:823–824. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lonergan PE and Tindall DJ: Androgen
receptor signaling in prostate cancer development and progression.
J Carcinog. 10:202011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Madan RA and Arlen PM: Recent advances
revolutionize treatment of metastatic prostate cancer. Future
Oncol. 9:1133–1144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
el-Rayes BF, Shields AF, Vaitkevicius V
and Philip PA: Developments in the systemic therapy of pancreatic
cancer. Cancer Invest. 21:73–86. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gray SG, Baird AM, O'Kelly F, Nikolaidis
G, Almgren M, Meunier A, Dockry E, Hollywood D, Ekström TJ, Perry
AS and O'Byrne KJ: Gemcitabine reactivates epigenetically silenced
genes and functions as a DNA methyltransferase inhibitor. Int J Mol
Med. 30:1505–1511. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Srinivasan M, Banerjee S, Palmer A, Zheng
G, Chen A, Bosland MC, Kajdacsy-Balla A, Kalyanasundaram R and
Munirathinam G: HMGB1 in hormone-related cancer: A potential
therapeutic target. Horm Cancer. 5:127–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang D, Kang R, Cheh CW, Livesey KM, Liang
X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et
al: HMGB1 release and redox regulates autophagy and apoptosis in
cancer cells. Oncogene. 29:5299–5310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bierhaus A, Schiekofer S, Schwaninger M,
Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Klöting
I, et al: Diabetes-associated sustained activation of the
transcription factor nuclear factor-kappaB. Diabetes. 50:2792–2808.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tang D, Kang R, Livesey KM, Kroemer G,
Billiar TR, Van Houten B, Zeh HJ III and Lotze MT: High-mobility
group box 1 is essential for mitochondrial quality control. Cell
Metab. 13:701–711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang D, Kang R, Livesey KM, Cheh CW,
Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ III
and Lotze MT: Endogenous HMGB1 regulates autophagy. J Cell Biol.
190:881–892. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livesey KM, Tang D, Zeh HJ and Lotze MT:
Autophagy inhibition in combination cancer treatment. Curr Opin
Investig Drugs. 10:1269–1279. 2009.PubMed/NCBI
|
|
12
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Lotze MT, et al: HMGB1-induced
autophagy promotes chemotherapy resistance in leukemia cells.
Leukemia. 25:23–31. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qian HR and Yang Y: Functional role of
autophagy in gastric cancer. Oncotarget. 7:17641–17651. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mancias JD and Kimmelman AC: Mechanisms of
selective autophagy in normal physiology and cancer. J Mol Biol.
428:1659–1680. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen K and Shi W: Autophagy regulates
resistance of non-small cell lung cancer cells to paclitaxel.
Tumour Biol. 37:10539–10544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rocchi A and He C: Emerging roles of
autophagy in metabolism and metabolic disorders. Front Biol
(Beijing). 10:154–164. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu W, Zhang Z, Zhang Y, Chen X, Guo S,
Lei Y, Xu Y, Ji C, Bi Z and Wang K: HMGB1-mediated autophagy
modulates sensitivity of colorectal cancer cells to oxaliplatin via
MEK/ERK signaling pathway. Cancer Biol Ther. 16:511–517. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R,
Vernon P, Cao L and Tang D: HMGB1 promotes drug resistance in
osteosarcoma. Cancer Res. 72:230–238. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Narumi T, Shishido T, Otaki Y, Kadowaki S,
Honda Y, Funayama A, Honda S, Hasegawa H, Kinoshita D, Yokoyama M,
et al: High-mobility group box 1-mediated heat shock protein beta 1
expression attenuates mitochondrial dysfunction and apoptosis. J
Mol Cell Cardiol. 82:1–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Acunzo J, Katsogiannou M and Rocchi P:
Small heat shock proteins HSP27 (HspB1), αB-crystallin (HspB5) and
HSP22 (HspB8) as regulators of cell death. Int J Biochem Cell Biol.
44:1622–1631. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wirth M, Joachim J and Tooze SA:
Autophagosome formation-the role of ULK1 and Beclin1-PI3KC3
complexes in setting the stage. Semin Cancer Biol. 23:301–309.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kang R, Livesey KM, Zeh HJ III, Lotze MT
and Tang D: Metabolic regulation by HMGB1-mediated autophagy and
mitophagy. Autophagy. 7:1256–1258. 2011. View Article : Google Scholar : PubMed/NCBI
|














