Introduction
Histone deacetylases (HDACs) are enzymes that remove
acetyl groups from histones and a number of non-histone proteins,
leading to chromatin condensation and transcription repression
(1). To date, 18 HDAC enzymes have
been identified in humans, which have been categorized into four
classes (2). Class I HDACs including
HDAC1, 2, 3 and 8 have been reported to be overexpressed in several
cancers, including gastric (3),
esophageal (4), colorectal (5), prostate (6) and lung (7)
cancer. Aberrant HDAC activity has been detected in a number of
types of human cancer, thereby contributing to tumor initiation and
progression (2,8). Targeting HDACs is a novel strategy in
the development of anticancer drugs (2).
Small molecular HDAC inhibitors (HDACis) are a
promising new class of anticancer drugs (9). The USA Food and Drug Administration
(FDA) has approved three HDACis: Vorinostat and romidepsin, for the
treatment of cutaneous T cell lymphoma, and belinostat, for the
treatment of relapsed or refractory peripheral T cell lymphoma
(10–12). Over 20 chemically distinct HDACis are
currently in clinical trials for the treatment of various types of
hematological malignancy and solid tumor (13).
Chidamide, a new HDACi, was approved for the
treatment of recurrent or refractory peripheral T cell lymphoma in
December 2014 by the Chinese FDA (14). Chidamide selectively inhibits HDAC1,
2, 3 and 10, the HDAC isotypes documented to be associated with
malignant phenotypes (15). Chidamide
has been applied in clinical trials for various types of
hematological malignancy and solid tumor (14,16).
Several in vitro studies reported that chidamide alone
induced apoptosis, and a combination of chidamide with other
chemotherapeutic drugs enhanced cell apoptosis in cancer cells
(17,18). In addition, chidamide was demonstrated
to induce cell apoptosis, cell cycle arrest and cell growth
inhibition (17–19).
Acquired resistance to anticancer agents is common
in cancer therapy. Previous studies have revealed that the acquired
resistance to the HDACi vorinostat is associated with a lack of G2
checkpoint activation and a lack of HDAC6 expression, with an
increased level of HDAC1, 2 and 4 expression (20,21).
Another HDACi, romidepsin, may cause the reversible induction of
multidrug resistance protein expression in tumor cells, leading to
transient resistance (22,23). Resistance following chronic treatment
with the HDACi valproic acid is associated with elevated Akt
activation in renal cell carcinoma in vivo (24).
In the present study, an acquired
chidamide-resistant A549-CHI-R cell line was established, with the
aim of characterizing in detail the mechanism of chidamide
resistance. In addition, the possible cross-resistance to other
chemotherapeutic drugs was investigated.
Materials and methods
Chemicals and reagents
Chidamide was supplied by Shenzhen ChipScreen
Biosciences, Ltd., (Shenzhen, China), and was dissolved in dimethyl
sulfoxide (DMSO). Cisplatin (CDDP) was obtained from Qilu
Pharmaceutical Co. Ltd., (Jinan, China). Vinorelbine (VNR) and
gemcitabine (GEM) were purchased from Jiangsu Hansoh Pharmaceutical
Co. Ltd., (Jiangsu, China). Paclitaxel (TAX) was obtained from
Bristol-Myers Squibb (New York, NY, USA). 5-fluorouracil (5-FU) was
obtained from Tianjin Jinyao Amino Acid Co. Ltd., (Tianjin, China).
Cycloheximide (CHX) was obtained from Beyotime Institute of
Biotechnology (Jiangsu, China). MTT was purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). RPMI-1640 medium
was purchased from Beijing Xigong Biotechnology Co. Ltd., (Being,
China). Fetal bovine serum was obtained from Shanghai Ex Cell
Biology Inc., (Shanghai, China).
Cell culture and establishment of
chidamide-resistant cell lines
The human non-small cell lung cancer (NSCLC) A549
cell line was purchased from the Cell Bank of the Cancer Institute,
Chinese Academy of Medical Science (Beijing, China). Cells were
cultured in RPMI-1640 medium containing 10% (v/v) fetal bovine
serum at 37°C in a humidified atmosphere with 5% (v/v)
CO2. A549 cells were exposed to gradually increasing
chidamide concentrations of 4, 8, 16, 32 and 64 µM for ~6 months,
and a chidamide-resistant lung cancer cell line was established,
designated A549-CHI-R.
Growth inhibition
Cell viability was evaluated using an MTT assay.
Growing cells (5×103 cells/well) were seeded on 96-well
plates with 100 µl medium. To assess cell viability, 100 µl medium
containing serial dilutions (Table I)
of chidamide, 5-FU, cisplatin, GEM, VNR or TAX was added, and the
cultures were incubated at 37°C. At 72 h, the medium was discarded,
20 µl saline containing 100 µg MTT was added to each well and the
cells were incubated at 37°C for 4 h. The supernatant was removed
and 150 µl DMSO was added to each well. The absorbance was measured
at 570 nm using a plate reader.
 | Table I.Drug dilutions in growth inhibition
assay. |
Table I.
Drug dilutions in growth inhibition
assay.
| Items | Chidamid (µM) | 5-FU (µM) | CDDP (µM) | GEM (µM) | VNR (nM) | TAX (nM) |
|---|
| A549 | 0/2.5/5/10/20 | 0/2.5/5/7.5/10 | 0/2.5/5/10/20 |
0/0.01/0.1/0.5/1 | 0/1/10/100/500 | 0/0.01/0.1/1/5 |
| A549-CHI-R | 0/25/50/75/100 | 0/2.5/5/7.5/10 | 0/2.5/5/10/20 |
0/0.01/0.1/1/10 |
0/1/10/100/1000 | 0/1/10/100/500 |
The surviving cell fraction was calculated using the
following formula: [(Mean absorbance of test cells-mean absorbance
of background)/(mean absorbance of control cells-mean absorbance of
background)] ×100%. The IC50 was determined by plotting
the logarithm of the drug concentration vs. the percentage of
surviving cells. Each assay was performed in quadruplicate at least
three times, and the mean and standard deviation were calculated.
Percentage inhibition values of compounds were calculated by
comparison with DMSO-treated control wells.
Clone formation assay
A total of 800 cells were plated on 6-well plates.
At 24 h, 5 µM chidamide was added, and the cells were allowed to
proliferate. The cell culture medium was replaced when necessary.
At ~10 days, when the differences in the growth of colonies had
appeared, the cells were washed with saline, fixed with 100%
methanol at room temperature for approximately 5 min and dyed with
0.005% crystal violet (Sigma-Aldrich; Merck KGaA), a
chromatin-binding stain, at room temperature for approximately 20
min. The colony formation rates were calculated using the following
formula: Number of clones formed/number of seeding cells ×100%.
HDAC1 gene transfection and
knockout
Prior to transfection, 2×106 cells were
seeded into 6-well plates. When cells reached ~70% confluence, they
were transiently transfected with a 2 µg human HDAC1 plasmid
(Invitrogen; Thermo Fisher Scientific Inc., Waltham, MA, USA) or
crisper/cas9 HDAC1-knockout plasmid (Viewsolid Biotech Co. Ltd,
Beijing, China). Lipofectamine® 3000 transfection
reagent (Thermo Fisher Scientific Inc.) was used for cell
transfection, according to the manufacturer's protocol. At 48 h
post-transfection, total protein or nuclear protein was extracted
as described in the subsequent sections, or the cells were seeded
at 5,000 per well in 96-well tissue culture plates, and several
concentrations (0 and 5 µM for A549 or 0 and 50 µM for A549-CHI-R)
of chidamide were added to assess cell viability.
Western blot analysis
The cells were plated on 6-well plates, allowed to
attach for 24 h and then treated with 5 µM chidamide for 72 h, or
10 µg/ml CHX for 0, 4, 8, 12 or 24 h. The cells were washed with
saline and lysed with radioimmunoprecipitation assay lysis buffer
(20 mM Tris-HCl, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA,
1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM
β-glycerophosphate, 1 mM Na3VO4, 1 µg/ml
leupeptin and 1X protease inhibitor cocktail). A total of 50 µg
protein samples (the protein was quantified using the BSA method)
were separated on 10% SDS-PAGE, and were transferred
electrophoretically to a nitrocellulose membrane (GE Healthcare,
Chicago, IL, USA). Blocking was performed using skimmed milk at
room temperature for 2 h. Membranes were then incubated with the
following primary antibodies: β-actin (cat. no. A5441;
Sigma-Aldrich; Merck KGaA; 1:5,000), HDAC1, 2 and 3 (HDAC antibody
sampler kit, cat. no. 9928; dilutions 1:1,000, 1:1,000 and 1:500,
respectively; Cell Signaling Technology, Inc., Danvers, MA, USA)
and HDAC10 (1:500; cat. no. BS1161; Bioworld Technology Inc., St.
Louis Park, MN, USA) at 4°C overnight. Following incubation with a
horseradish peroxidase (HRP)-conjugated secondary antibody (OriGene
Technologies Inc., Rockville, MD, USA), including Anti-Mouse
IgG/HRP (cat. no. TA130004; 1:3,000) or Anti-Rabbit IgG/HRP (cat.
no. TA140003; 1:3,000) at room temperature for 2 h, the membranes
were developed using a luminol chemiluminescence detection kit
(Santa Cruz Biotechnology Inc., Dallas, TX, USA) according to the
manufacturer's protocol.
HDAC activity assay
HDAC activity was measured with the fluorometric
HDAC Activity Assay kit (cat. no., ab156064; Abcam, Cambridge, UK)
according to the manufacturer's protocol. Briefly, HDAC assay
buffer containing a substrate peptide was incubated with HeLa
nuclear extract (supplied with the kit) as a positive control, or
A549 or A549-CHI-R nuclear extract, in a microtiter plate.
Trichostatin A (supplied with the kit) was then added to the
inhibitor control assay wells. At 20 min, 20 µl developing solution
was added to each well for a further 20 min. Finally, 5 µl stop
buffer was added to every well and incubated for 30 min at room
temperature. At the end of the treatment, plates were detected
using fluorescence filters (excitation, 355 nm; emission, 480
nm).
Cell cycle arrest
Cells were then plated onto 6-well plates, allowed
to attach for 24 h and then treated with 1 nM TAX or 20 nM VNR. At
72 h, the cells were harvested and washed with saline then
centrifuged at 335 × g for 5 min at 4°C. The pellets were
re-suspended in 300 µl of saline and were fixed by adding 700 µl of
cold absolute ethanol and incubating at −20°C overnight. The next
day, the cells were centrifuged at 335 × g for 5 min at 4°C and the
supernatant was removed. The cells were washed with cold saline
twice, stained with propidium iodide on ice for 20 min and analyzed
with a flow cytometer (BD LSR II system; BD Biosciences, Franklin
Lakes, NJ, USA).
Statistical analysis
Assays were performed in triplicate, and results are
presented as the mean ± standard deviation. Statistical difference
was assessed using Student's t-test (GraphPad_Prism V5.0; GraphPad
Software Inc., La Jolla, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Establishment of a chidamide-resistant
non-small cell lung cancer cell line
To investigate the acquired resistance to chidamide
in cancer therapy, a chidamide-resistant lung cancer cell line,
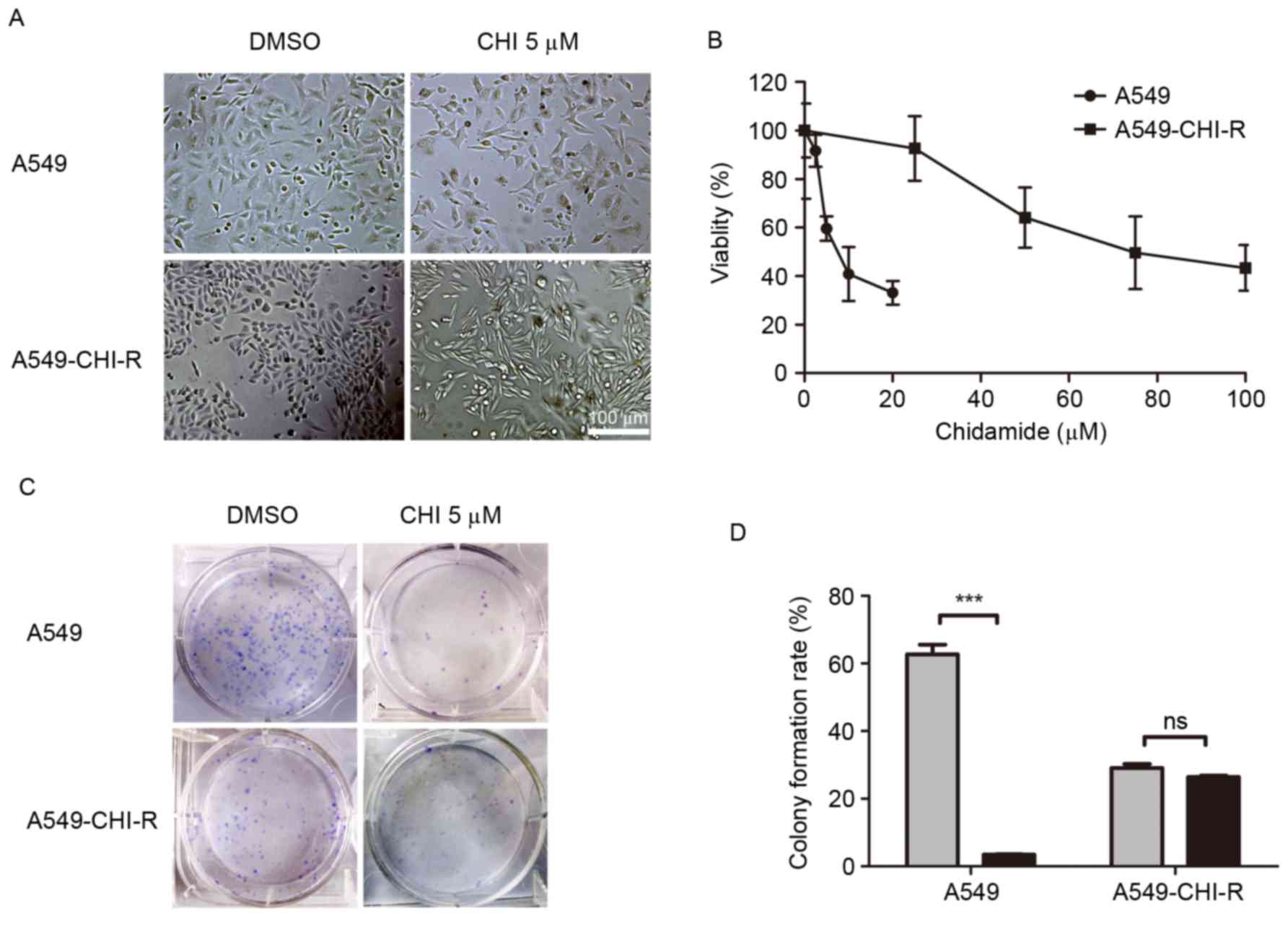
A549-CHI-R, was established (Fig. 1).
When treated with 5 µM chidamide, the morphology of A549-CHI-R
cells became elongated and thin (Fig.
1A). The IC50 values of chidamide for A549-CHI-R and
parental A549 cells were 78.34 and 9.07, respectively. A549-CHI-R
was ~9-fold more resistant to chidamide compared with the parental
A549 cells (Fig. 1B). Following 10
days of chidamide treatment, parental A549 cells exhibited markedly
decreased colony formation rates (P<0.001) compared with
untreated controls, whereas colony formation inhibition was not
observed in the A549-CHI-R cell line (Fig. 1C). The colony formation rates for
A549/CHI 5 µM, A549/DMSO, A549-CHI-R/CHI 5 µM and A549-CHI-R/DMSO
cells were 3.5, 56.9, 26.5 and 26.9%, respectively (Fig. 1D).
HDAC1 protein degradation is inhibited
in chidamide-resistant lung cancer cells
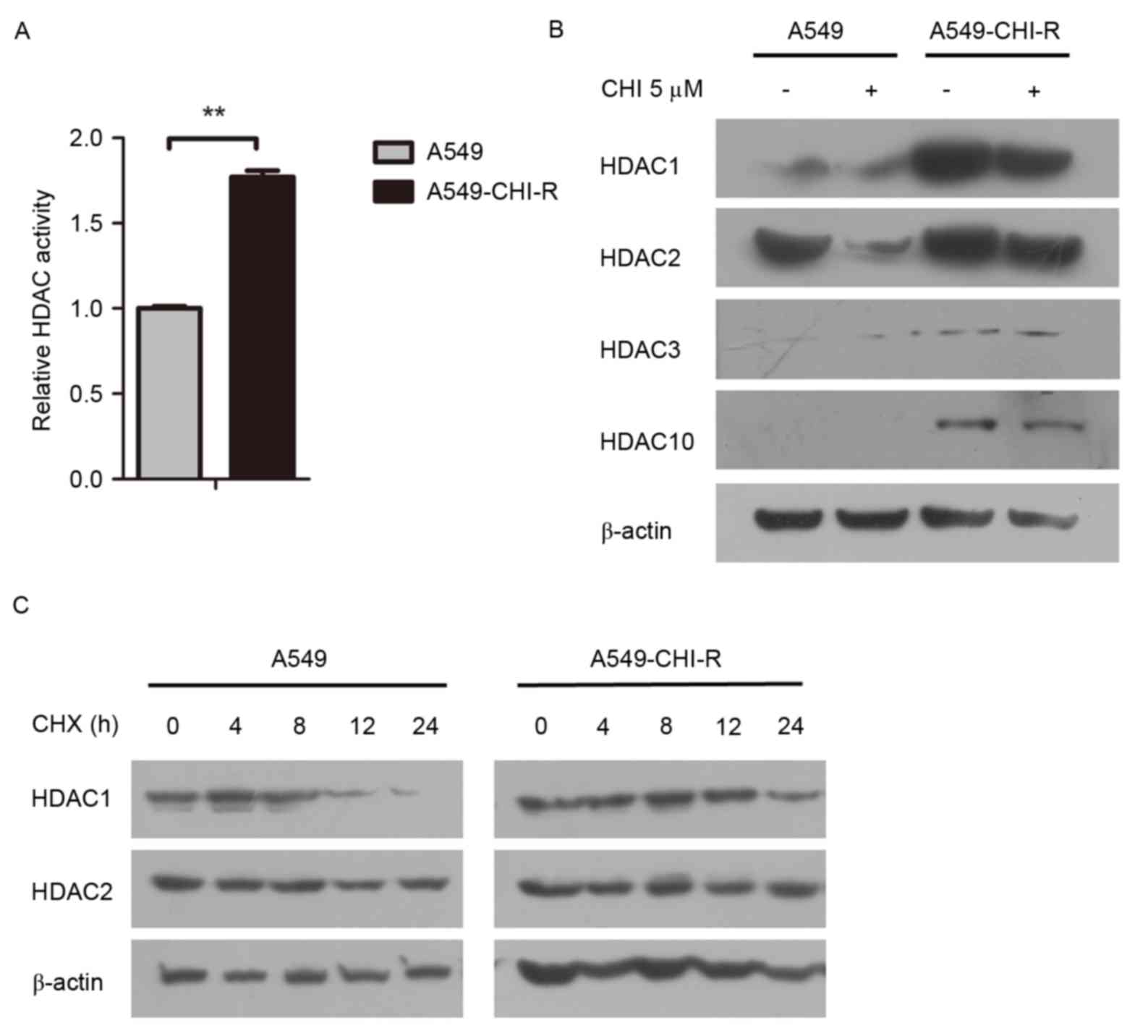
Intracellular HDAC activity was examined in the
parental and resistant cell lines. The HDAC activity in the
A549-CHI-R cells was 1.77-fold higher than in the parental A549
cells (Fig. 2A). Western blot
analysis revealed that HDAC protein expression was markedly
increased in the A549-CHI-R cells compared with the parental A549
cells (Fig. 2B). To analyze the
mechanisms of HDAC activity increase in A549-CHI-R cells, protein
synthesis was first inhibited by CHX. HDAC1 protein expression
became negligible after 24 h in parental A549 cells (Fig. 2C, left panel), but slightly decreased
after 24 h in A549-CHI-R cells (Fig.
2C, right panel). However, HDAC2 protein was slightly increased
in parental A549 and A549-CHI-R cells after 24 h. These results
indicated that the increased HDAC1 activity in A549-CHI-R cells was
not induced by protein synthesis, but by the inhibition of protein
degradation.
HDAC1 contributes to chidamide
resistance in lung cancer cells
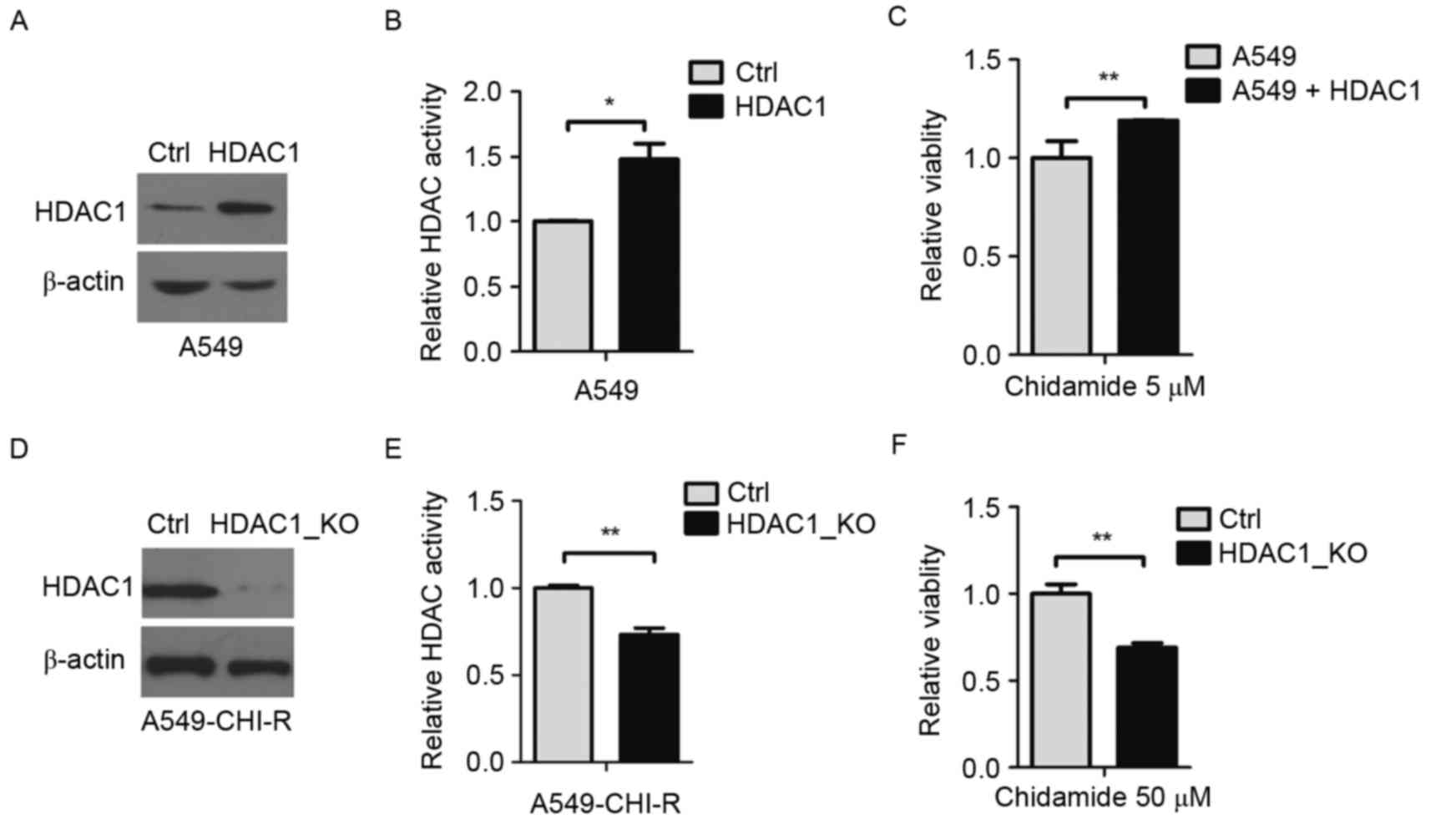
To investigate the pivotal role of HDAC1 in
chidamide resistance, HDAC1 was overexpressed in A549 cells. The
protein overexpression was confirmed by western blot analysis
(Fig. 3A). Compared with parental
cells, HDAC1-overexpressing cells exhibited ~1.5-fold increased
intracellular HDAC activity (P<0.05; Fig. 3B). Following 5 µM of chidamide
treatment, the survival rate of HDAC1-overexpressing A549 cells was
1.2-fold higher than the parental A549 cells (P<0.01; Fig. 3C), indicating that the overexpression
of HDAC1 contributed to chidamide resistance.
HDAC1 was then knocked out in the
chidamide-resistant A549-CHI-R cell line. HDAC1 protein decreased
in HDAC1-knockout A549-CHI-R cells (Fig.
3D). Compared with A549-CHI-R cells, intracellular HDAC
activity rate was decreased to 0.27-fold following HDAC1-knockout
(Fig. 3E). When treated with 50 µM
chidamide, the survival rate of HDAC1-knockout A549-CHI-R cells was
0.32-fold lower than the A549-CHI-R cells (P<0.01; Fig. 3F).
Cross-resistance of the
chidamide-resistant lung cancer cell line
It was investigated whether the chidamide-resistant
A549-CHI-R cell line was cross-resistant to other chemotherapeutic
drugs. IC50 values revealed that A549-CHI-R cells
remained sensitive to CDDP and 5-FU. However, compared with the
parental A549 cell line, the A549-CHI-R cells were 10.55-, 13.23-
and >100-fold (P<0.01) more resistant to GEM, VNR and TAX,
respectively (Table II).
| Table II.Drug sensitivity in parental A549 and
A549-CHI-R cells. |
Table II.
Drug sensitivity in parental A549 and
A549-CHI-R cells.
|
| A549 | A549-CHI-R |
|
|---|
|
|
|
|
|
|---|
| Drug |
IC50 | 95% CI |
IC50 | 95% CI | Resistance
index |
|---|
| Chidamide | 9.07 | 6.00–11.32 | 78.34 | 51.30–111.87 | 8.64 |
| 5-fluorouracil | 4.03 | 2.09–7.75 | 5.17 | 2.77–7.58 | 1.28 |
| Cisplatin | 9.13 | 8.90–9.37 | 7.87 | 5.75–10.76 | 0.86 |
| Gemcitabine | 0.21 | 0.01–3.65 | 2.25 | 1.67–3.03 | 10.55 |
| Vinorelbine | 31.06 | 11.32–85.17 | 411.00 | 282.10–598.80 | 13.23 |
| Paclitaxel | 0.89 | 0.11–1.98 | 123.40 | 6.00–260.00 | 123.40 |
The cell cycle of A549-CHI-R and A549 cells was
analyzed by flow cytometry. Cells were incubated with 1 nM TAX or
20 nM VNR for 72 h. Compared with untreated cells, the percentage
of A549 cells in the G2/M phase markedly increased, from 28.2% to
77.7 (TAX) and 79.4% (VNR). However, following the same treatment,
compared with control cells, the percentage of A549-CHI-R cells in
the G2/M phase increased from 30.5 to 52.1 and 42.3%. Therefore,
A549-CHI-R cells were more resistant to G2/M arrest caused by TAX
and VNR (Table III).
| Table III.Effects of TAX or VNR on the cell
cycle distribution in parental A549 and A549-CHI-R cells. |
Table III.
Effects of TAX or VNR on the cell
cycle distribution in parental A549 and A549-CHI-R cells.
|
| A549 cells, % | A549-CHI-R cells,
% |
|---|
|
|
|
|
|---|
| Cell cycle
phase | Ctrl | TAX | VNR | Ctrl | TAX | VNR |
|---|
| Sub-G1 | 1.7 | 0.3 | 0.5 | 0.2 | 0.1 | 0.1 |
| G1 | 60.2 | 15.1 | 10.8 | 53.4 | 34.8 | 41.7 |
| S | 9.9 | 6.9 | 9.3 | 15.9 | 13.0 | 15.9 |
| G2/M | 28.2 | 77.7 | 79.4 | 30.5 | 52.1 | 42.3 |
Discussion
In the present study, a chidamide-resistant NSCLC
cell line, A549-CHI-R, was established. With growth inhibition and
colony formation assays, it was revealed that A549 and A549-CHI-R
cell lines exhibited a ~9-fold difference in sensitivity to
chidamide (IC50 of A549 cell line, 9.07 µM;
IC50 of the A549-CHI-R cell line, 78.34 µM). Compared
with parental A549 cells, A549-CHI-R cells exhibited a slower
growth rate and a reduced colony formation rate. A previous study
demonstrated that HL-60/LR cells, human acute myeloid leukemia
cells resistant to LAQ824 (a hydroxamic acid analog pan-HDACi),
exhibited a markedly higher growth compared with parental HL-60
cells (21). A vorinostat-induced
subline, HCT116/vorinostat, exhibited a slightly slower growth rate
compared with the parental HCT116 cell line (20). The different results indicate that the
growth rate of HDACi-resistant cell lines may be drug-specific,
cell type-specific or even case-specific.
A number of HDACi-resistant cancer cell lines have
already been reported (20,21,25). The
mechanisms of HDACi resistance include the upregulation of
P-glycoprotein, other ATP-binding cassette transporters, cell cycle
proteins and signaling proteins, alterations to HDAC protein level,
increases in thioredoxin level, nuclear factor-κB activation and
anti-apoptotic/prosurvival mechanisms (26). The molecular mechanism for acquired
resistance varies in different HDACi-resistant cells. The acquired
resistance of HCT116/VOR cells was associated with a reduction in
histone acetylation, G2/M checkpoint activation and apoptosis
susceptibility (20).
Pan-HDACi-resistant HL-60/LR cells expressed higher levels of
HDAC1, 2 and 4, but lacked expression of HDAC6, with concomitant
hyper-acetylation of heat shock protein 90 (21).
Chidamide was revealed to be a low nanomolar
inhibitor of HDAC1, 2, 3 and 10 (15). HDAC1 belongs to class I HDACs, which
are the most closely associated with malignant phenotypes (27). HDAC1 overexpression has been reported
to be positively associated with cell division, differentiation and
tumorigenesis (28–30). Loss of HDAC1 resulted in a 60%
reduction in total HDAC activity and a loss of stem cell viability
(28). Consistently, total HDAC
activity was also elevated in A549-CHI-R and A549
HDAC1-overexpressed cells. Similarly, HDAC1 was also accumulated in
chidamide-resistant A549-CHI-R cells, consistent with other
HDACi-resistant cells (23).
Post-translational modifications of HDAC have been
demonstrated to perform pivotal roles in the regulation of gene
expression. HDACs modified by ubiquitination are targeted for
degradation (31). Certain chemicals
can target HDAC1 to induce proteasome-mediated degradation
(32). Specifically, treatment with
diesel exhaust particulate induced degradation of HDAC1 in a human
bronchial epithelial cell line, BEAS-2B (33). The present study reported a decreased
degradation of HDAC1 in A549-CHI-R cells. Overexpression of HDAC1
in cervical cancer cells restrained cell proliferation and induced
premature senescence (34). Taken
together, HDAC1 accumulation may be a predictive marker for the
resistance to chidamide.
Cross-resistance is a common response to
chemotherapy. HDACi-resistant cell lines, HL-60/LR and
HCT116/vorinostat, exhibited cross-resistance to other HDACis
(20,21). Pemetrexed-resistant NSCLC cell lines
exhibited cross-resistance to CDDP (35). A549-CHI-R cells, which exhibit
enhanced HDAC activity, demonstrated cross-resistance to GEM, TAX
and VNR in the present study. Consistent with this,
paclitaxel-resistant NSCLC cells exhibited enhanced HDAC activity
and tumorigenicity (36). Increased
HDAC1 expression in NSCLC tissue predicted a poor prognosis for
patients treated with paclitaxel (36). TAX and VNR are microtubule-targeting
drugs; their most potent cytotoxic action is the suppression of
microtubule dynamics, leading to mitotic arrest and subsequent cell
death (37). Gong et al
(38) demonstrated that chidamide
inhibits cell proliferation by inducing cell cycle arrest. The
present study revealed that G2/M arrest decreased in A549-CHI-R
cells compared with parental A549 cells following treatment with
TAX or VNR. However, the chidamide-resistant cells retained
sensitivity and susceptibility to the drugs CDDP and 5-FU. These
drugs induce cell death by inhibiting DNA synthesis (39,40). A
combination of CDDP or 5-FU with chidamide may synergistically
induce apoptosis (17,18). The results of the present study are
consistent with these previous studies.
In conclusion, a chidamide-resistant cell line was
established, and it was proposed that HDAC1 accumulation may
contribute to chidamide resistance. In addition, the
chidamide-resistant cell line remained sensitive to 5-FU and CDDP,
but cross-resistant to TAX and VNR, indicating a potential strategy
for cancer therapy.
Acknowledgements
The present study was supported by the National
Natural Science Foundation (grant nos. 81321091, 81452761 and
31171322) and the National Basic Research Program of China (grant
no. 2011CB910700704). The authors thank Shenzhen ChipScreen
Biosciences Ltd., (Shenzhen, China) for providing chidamide used in
the present study.
Glossary
Abbreviations
Abbreviations:
|
HDAC
|
histone deacetylase
|
|
A549-CHI-R
|
chidamide-resistant A549 cells
|
|
CDDP
|
cisplatin
|
|
GEM
|
gemcitabine
|
|
5-FU
|
5-fluorouracil
|
|
TAX
|
paclitaxel
|
|
VNR
|
vinorelbine
|
|
NSCLC
|
non-small cell lung cancer
|
References
|
1
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Glozak MA and Seto E: Histone deacetylases
and cancer. Oncogene. 26:5420–5432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Song J, Noh JH, Lee JH, Eun JW, Ahn YM,
Kim SY, Lee SH, Park WS, Yoo NJ, Lee JY and Nam SW: Increased
expression of histone deacetylase 2 is found in human gastric
cancer. APMIS. 113:264–268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Toh Y, Yamamoto M, Endo K, Ikeda Y, Baba
H, Kohnoe S, Yonemasu H, Hachitanda Y, Okamura T and Sugimachi K:
Histone H4 acetylation and histone deacetylase 1 expression in
esophageal squamous cell carcinoma. Oncol Rep. 10:333–338.
2003.PubMed/NCBI
|
|
5
|
Giannini R and Cavallini A: Expression
analysis of a subset of coregulators and three nuclear receptors in
human colorectal carcinoma. Anticancer Res. 25:4287–4292.
2005.PubMed/NCBI
|
|
6
|
Waltregny D, North B, Van Mellaert F, de
Leval J, Verdin E and Castronovo V: Screening of histone
deacetylases (HDAC) expression in human prostate cancer reveals
distinct class I HDAC profiles between epithelial and stromal
cells. Eur J Histochem. 48:273–290. 2004.PubMed/NCBI
|
|
7
|
Bartling B, Hofmann HS, Boettger T, Hansen
G, Burdach S, Silber RE and Simm A: Comparative application of
antibody and gene array for expression profiling in human squamous
cell lung carcinoma. Lung Cancer. 49:145–154. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sharma S, Kelly TK and Jones PA:
Epigenetics in cancer. Carcinogenesis. 31:27–36. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Garber K: Purchase of Aton spotlights HDAC
inhibitors. Nat Biotechnol. 22:364–365. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duvic M and Vu J: Vorinostat: A new oral
histone deacetylase inhibitor approved for cutaneous T-cell
lymphoma. Expert Opin Investig Drugs. 16:1111–1120. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Grant C, Rahman F, Piekarz R, Peer C, Frye
R, Robey RW, Gardner ER, Figg WD and Bates SE: Romidepsin: A new
therapy for cutaneous T-cell lymphoma and a potential therapy for
solid tumors. Expert Rev Anticancer Ther. 10:997–1008. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Poole RM: Belinostat: First global
approval. Drugs. 74:1543–1554. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee JH, Choy ML and Marks PA: Mechanisms
of resistance to histone deacetylase inhibitors. Adv Cancer Res.
116:39–86. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi Y, Dong M, Hong X, Zhang W, Feng J,
Zhu J, Yu L, Ke X, Huang H, Shen Z, et al: Results from a
multicenter, open-label, pivotal phase II study of chidamide in
relapsed or refractory peripheral T-cell lymphoma. Ann Oncol.
26:1766–1771. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ning ZQ, Li ZB, Newman MJ, Shan S, Wang
XH, Pan DS, Zhang J, Dong M, Du X and Lu XP: Chidamide
(CS055/HBI-8000): A new histone deacetylase inhibitor of the
benzamide class with antitumor activity and the ability to enhance
immune cell-mediated tumor cell cytotoxicity. Cancer Chemother
Pharmacol. 69:901–909. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dong M, Ning ZQ, Xing PY, Xu JL, Cao HX,
Dou GF, Meng ZY, Shi YK, Lu XP and Feng FY: Phase I study of
chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in
patients with advanced solid tumors and lymphomas. Cancer Chemother
Pharmacol. 69:1413–1422. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou Y, Pan DS, Shan S, Zhu JZ, Zhang K,
Yue XP, Nie LP, Wan J, Lu XP, Zhang W and Ning ZQ: Non-toxic dose
chidamide synergistically enhances platinum-induced DNA damage
responses and apoptosis in non-small-cell lung cancer cells. Biomed
Pharmacother. 68:483–491. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu L, Qiu S, Liu Y, Liu Z, Zheng Y, Su X,
Chen B and Chen H: Chidamide and 5-flurouracil show a synergistic
antitumor effect on human colon cancer xenografts in nude mice.
Neoplasma. 63:193–200. 2016.PubMed/NCBI
|
|
19
|
Wang H, Guo Y, Fu M, Liang X, Zhang X,
Wang R, Lin C and Qian H: Antitumor activity of Chidamide in
hepatocellular carcinoma cell lines. Mol Med Rep. 5:1503–1508.
2012.PubMed/NCBI
|
|
20
|
Dedes KJ, Dedes I, Imesch P, von Bueren
AO, Fink D and Fedier A: Acquired vorinostat resistance shows
partial cross-resistance to ‘second-generation’ HDAC inhibitors and
correlates with loss of histone acetylation and apoptosis but not
with altered HDAC and HAT activities. Anticancer Drugs. 20:321–333.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fiskus W, Rao R, Fernandez P, Herger B,
Yang Y, Chen J, Kolhe R, Mandawat A, Wang Y, Joshi R, et al:
Molecular and biologic characterization and drug sensitivity of
pan-histone deacetylase inhibitor-resistant acute myeloid leukemia
cells. Blood. 112:2896–2905. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiao JJ, Huang Y, Dai Z, Sadée W, Chen J,
Liu S, Marcucci G, Byrd J, Covey JM, Wright J, et al:
Chemoresistance to depsipeptide FK228
[(E)-(1S,4S,10S,21R)-7-[(Z)-ethylidene]-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8,7,6]-tricos-16-ene-3,6,9,22-pentanone]
is mediated by reversible MDR1 induction in human cancer cell
lines. J Pharmacol Exp Ther. 314:467–475. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamada H, Arakawa Y, Saito S, Agawa M,
Kano Y and Horiguchi-Yamada J: Depsipeptide-resistant KU812 cells
show reversible P-glycoprotein expression, hyper-acetylated
histones, and modulated gene expression profile. Leukemia Res.
30:723–734. 2006. View Article : Google Scholar
|
|
24
|
Juengel E, Makarević J, Tsaur I, Bartsch
G, Nelson K, Haferkamp A and Blaheta RA: Resistance after chronic
application of the HDAC-inhibitor valproic acid is associated with
elevated Akt activation in renal cell carcinoma in vivo. PLoS One.
8:e531002013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu Y, Das K, Wu J, Lee MH and Tan P: RNH1
regulation of reactive oxygen species contributes to histone
deacetylase inhibitor resistance in gastric cancer cells. Oncogene.
33:1527–1537. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Robey RW, Chakraborty AR, Basseville A,
Luchenko V, Bahr J, Zhan Z and Bates SE: Histone deacetylase
inhibitors: Emerging mechanisms of resistance. Mol Pharm.
8:2021–2031. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakagawa M, Oda Y, Eguchi T, Aishima S,
Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M and Tsuneyoshi
M: Expression profile of class I histone deacetylases in human
cancer tissues. Oncol Rep. 18:769–774. 2007.PubMed/NCBI
|
|
28
|
Jamaladdin S, Kelly RD, O'Regan L, Dovey
OM, Hodson GE, Millard CJ, Portolano N, Fry AM, Schwabe JW and
Cowley SM: Histone deacetylase (HDAC) 1 and 2 are essential for
accurate cell division and the pluripotency of embryonic stem
cells. Proc Natl Acad Sci USA. 111:pp. 9840–9845. 2014, View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Choi JH, Kwon HJ, Yoon BI, Kim JH, Han SU,
Joo HJ and Kim DY: Expression profile of histone deacetylase 1 in
gastric cancer tissues. Jpn J Cancer Res. 92:1300–1304. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Korfei M, Skwarna S, Henneke I, MacKenzie
B, Klymenko O, Saito S, Ruppert C, von der Beck D, Mahavadi P,
Klepetko W, et al: Aberrant expression and activity of histone
deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax.
70:1022–1032. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Seigneurin-Berny D, Verdel A, Curtet S,
Lemercier C, Garin J, Rousseaux S and Khochbin S: Identification of
components of the murine histone deacetylase 6 complex: Link
between acetylation and ubiquitination signaling pathways. Mol Cell
Biol. 21:8035–8044. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu D, Zhou P, Zhang L, Zheng Y and He F:
HPV16 activates the promoter of Oct4 gene by sequestering HDAC1
from repressor complex to target it to proteasomal degradation. Med
Hypotheses. 79:531–534. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cao D, Bromberg PA and Samet JM: COX-2
expression induced by diesel particles involves chromatin
modification and degradation of HDAC1. Am J Respir Cell Mol Biol.
37:232–239. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chuang JY and Hung JJ: Overexpression of
HDAC1 induces cellular senescence by Sp1/PP2A/pRb pathway. Biochem
Biophys Res Commun. 407:587–592. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang D, Ochi N, Takigawa N, Tanimoto Y,
Chen Y, Ichihara E, Hotta K, Tabata M, Tanimoto M and Kiura K:
Establishment of pemetrexed-resistant non-small cell lung cancer
cell lines. Cancer Lett. 309:228–235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang L, Li H, Ren Y, Zou S, Fang W, Jiang
X, Jia L, Li M, Liu X, Yuan X, et al: Targeting HDAC with a novel
inhibitor effectively reverses paclitaxel resistance in non-small
cell lung cancer via multiple mechanisms. Cell Death Dis.
7:e20632016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jordan MA and Wilson L: Microtubules as a
target for anticancer drugs. Nat Rev Cancer. 4:253–265. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gong K, Xie J, Yi H and Li W: CS055
(Chidamide/HBI-8000), a novel histone deacetylase inhibitor,
induces G1 arrest, ROS-dependent apoptosis and differentiation in
human leukaemia cells. Biochem J. 443:735–746. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sorenson CM and Eastman A: Mechanism of
cis-diamminedichloroplatinum(II)-induced cytotoxicity: Role of G2
arrest and DNA double-strand breaks. Cancer Res. 48:4484–4488.
1988.PubMed/NCBI
|
|
40
|
Santi DV, McHenry CS and Sommer H:
Mechanism of interaction of thymidylate synthetase with
5-fluorodeoxyuridylate. Biochemistry. 13:471–481. 1974. View Article : Google Scholar : PubMed/NCBI
|